

Abstract
Non-heme manganese catalases are widely distributed over microbial life and represent an environmentally important alternative to heme-containing catalases in antioxidant defense. Manganese catalases contain a binuclear manganese complex as their catalytic active site rather than a heme, and cycle between Mn2(II,II) and Mn2(III,III) states during turnover. X-ray crystallography has revealed the key structural elements of the binuclear manganese active site complex that can serve as the starting point for computational studies on the protein. Four manganese catalase enzymes have been isolated and characterized, and the enzyme appears to have a broad phylogenetic distribution including both bacteria and archae. More than 100 manganese catalase genes have been annotated in genomic databases, although the assignment of many of these putative manganese catalases needs to be experimentally verified. Iron limitation, exposure to low levels of peroxide stress, thermostability and cyanide resistance may provide the biological and environmental context for the occurrence of manganese catalases.
Keywords: catalase, manganese, peroxide, antioxidant, oxidative stress, reactive oxygen species
Introduction
Catalases serve as the first line of defense against hydrogen peroxide [1,2], a reactive oxygen species variously formed as a cell signaling agent [3], a weapon of intercellular warfare [4], or a by-product of aerobic metabolism [5,6]. All catalases catalyze the redox disproportionation of hydrogen peroxide:
| (1) |
The reaction is highly exergonic (ΔH° = −47 kcal/mol; ΔS° = +34 kcal/mol·K; ΔG° = −57 kcal/mol for the reaction stoichiometry shown in Eqn. 1)[7], and is very favorable under physiological conditions (pH 7, 37°C). This intrinsic instability contributed to the discovery of catalase catalysis in 1818, soon after Thenard developed a synthesis for hydrogen peroxide, based on the observation that dioxygen is vigorously liberated when animal tissues are treated with a dilute solution of H2O2 without the tissue being consumed [8]. It was, of course, the heme-containing catalase enzyme of animal tissues that was responsible for these effects, the enzyme name reflecting its important role in the discovery of biological catalysis. The effervescence resulting from hydrogen peroxide decomposition has been widely used as a qualitative test for catalase activity [9], more recently leading to the discovery of alternative, non-heme catalases in lactic acid bacteria. Since many of these bacteria lack heme and are devoid of cytochromes, the observation of even weak catalase activity was notable, and has been shown to be associated with a class of catalase that is independent of a heme requirement. The non-heme catalase responsible for this activity was found to be widespread among strains of Pediococci [10] and Lactobacilli [11], and the manganese metal specificity for the reaction was ultimately demonstrated by characterization of the purified protein from Lactobacillus plantarum [12].
2. Structural Studies
Several manganese catalases have been isolated from native sources (Lactobacillus plantarum [12], Thermus thermophilus [13], Thermoleophilum album [14]) or cloned for heterologous recombinant expression (Pyrobaculum caldifontis manganese catalase [15]). The enzymes from L. plantarum (LPC) and T. thermophilus (TTC), which have been most extensively studied [16], have both been crystallized and their structures solved by X-ray diffraction [17,18]. Both enzymes contain a binuclear manganese catalytic active site that supports a two-electron oxidation-reduction cycle during turnover, alternating between reduced bis-manganese(II) [Mn2(II,II)] and oxidized bis-manganese(III) [Mn2(III,III)] states.
2.1 Lactobacillus plantarum manganese catalase [17]
Lactobacillus plantarum manganese catalase is a homohexameric protein, with six equivalent subunits assembled into a globular shell surrounding a solvent-filled central cavity (Fig. 1). The solvent cavity is connected to the bulk solvent through pores that lie along the three-fold axis of the hexamer.
Figure 1.

Lactobacillus plantarum manganese catalase. A single subunit of LPC is rendered as ribbons embedded in the surface-rendered globular hexameric holoenzyme. Based on PDB ID: 1JKU.
2.1.1 Protein fold
The overall protein fold of LPC (Fig. 2A) is characteristic of the ferritin superfamily of proteins (which includes ferritin [19], ribonucleotide reductase [20], and methane monooxygenase [21]) that contain a bis-μ-oxo-bridged binuclear metal complex in a four-helix bundle coiled-coil domain [22]. These proteins typically form a diiron complex, whereas LPC binds manganese. The central catalytic core domain of LPC exhibits a four-helix bundle architecture, formed from a pair of antiparallel α-helical hairpins, with a left-handed crossover connection between helices 2 and 3 that reverses the orientation of the C-terminal helices. The binuclear manganese active site is bound in the middle of this coiled-coil domain in a cluster of glutamate and histidine residues arising from the α-helical chains. The catalytic core domain represents approximately 50% of the polypeptide, and the remainder of the protein contains relatively little well-defined secondary structure. An N-terminal extension from the bundle core forms an antiparallel β-sheet linkage with a neighboring subunit, stabilizing the multimer. A much longer C-terminal extension includes a binding site for a divalent cation (Ca2+) that has a structural rather than catalytic role.
Figure 2.

Structure of manganese catalase subunits. Secondary structural elements of (A) LPC (based on PDB ID: 1JKU) and (B) TTC (based on PDB ID: 2V8U) polypeptides is indicated: α-helix, red; β-sheet, yellow; turn, blue. Manganese ions are rendered as magenta spheres. N- and C-terminal regions are noted.
2.1.2 Active site
The binuclear manganese metal complex in the oxidized Mn2(III,III) state is comprised of an Mn2(O)(OH) solvent-bridged core, with an additional solvent molecule coordinated in an apical position on Mn1 and a complement of protein-derived ligands to complete the coordination sphere of each ion (Fig. 3). One glutamate carboxylate (Glu66) bridges (μ1,3) between the metal centers, defining a plane that bisects the Mn2O2 core. Two other glutamates are bound to the cluster on the same (“distal”) side of the bisecting plane. One residue (Glu35) is bound in a monodentate fashion to Mn1, and the other carboxylate (Glu148) adopts a bidentate chelating coordination mode at Mn2. Two histidine ligands (His69 and His181) are coordinated on the “proximal” side of the cluster. The arrangement of the ligands produces roughly octahedral six-coordination at both metal centers. Two of the metal ligands (Glu66 and His69) arise from the same helix and are separated by two residues in the polypeptide sequence, forming an EXXH metal binding motif. This sequence pattern reflects a basic feature of protein anatomy: the geometry of the α-helix (3.6 residue/turn) requires that two residues arising from the same helix and oriented in the same direction in space must lie 4 residues apart in sequence. The EXXH motif is a common feature of proteins containing a binuclear metal complex, and is present in all of the approximately 100 sequences of putative proteins assigned as manganese catalases in genomic databases (Sect.4). A non-coordinated glutamic acid residue (Glu178) lies above the Mn2O2 core, forming a hydrogen bond with the terminally coordinated solvent molecule attached to Mn1. Its headgroup appears to be conformationally constrained by buttressing interactions with surrounding side chains, preventing it from coordinating directly to the metal center, as the corresponding group in diiron clusters does [19–21]. The structure of the complex itself does not give any clues to the origin of the metal binding selectivity of the protein, although the only metal present in the purified protein to any significant extent is manganese.
Figure 3.
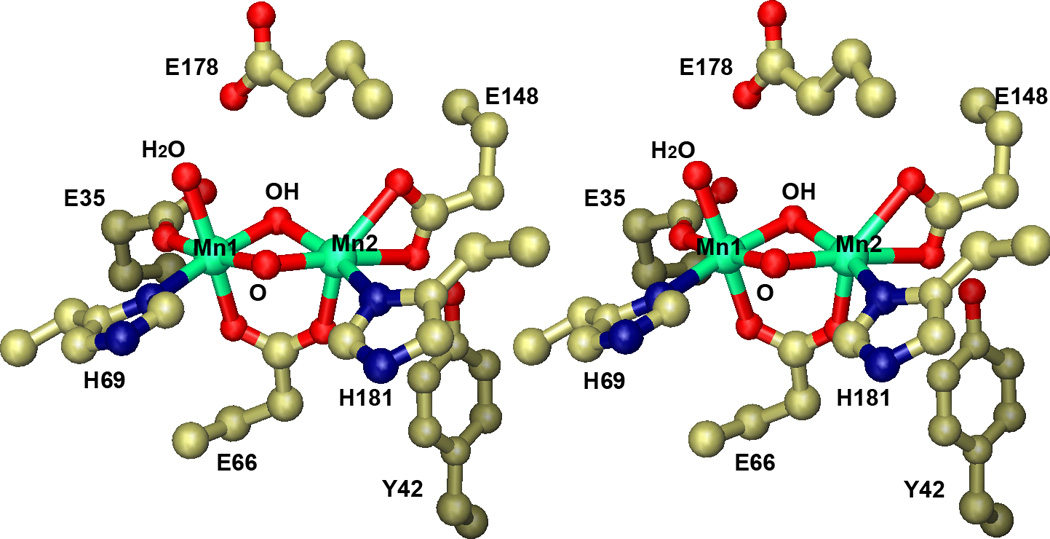
Active site of native Mn(III,III) Lactobacillus plantarum manganese catalase. Stereoscopic view of the binuclear manganese complex in the oxidized Mn2(III,III) state. Based on PDB ID: 1JKU.
A bulge formed by a pair of glycine residues in the middle of helix A (Gly30,Gly31) creates an access channel connecting the catalytic manganese cluster to the solvent-filled core of the protein. The channel is lined by hydrophilic groups including both acidic and basic side chains and the path can be traced by a chain of crystallographically defined solvent molecules buried in the protein interior. The presence of both positively and negatively charged groups in the channel is expected to create a kind of mixed-bed ion exchange surface along the path that may serve to restrict entry by ions, while allowing access to neutral species (including H2O2, H2O, and O2).
2.1.3 Extended active site environment
The active site is embedded in the protein interior near the middle of the four helix bundle, and connected to all four helices via hydrogen bonding networks radiating from the metal cluster. One hydrogen bonding network extends out from the two histidine ligands (His69 and His181), and is anchored to the carboxylate headgroup of Glu65, which in turn forms a salt bridge with Arg177. Gln9 also appears to contribute to this network. Another hydrogen bonding web extends out on the opposite side of the cluster, through Arg147. The Nε of the guanidinium group of Arg147 is hydrogen-bonded to the phenolic group of Tyr42, which in turn interacts with the coordinating carboxylate of Glu148. Tyrosine hydrogen bonding to Glu148 may play a role in modulating the coordination chemistry at Mn2, and a conformational switch between alternative monodentate binding modes. Together, the hydrogen bonding networks organize the active site environment, and may play a role in delocalizing charge that may accumulate on the metal complex during turnover, lowering activation barriers for catalysis.
The tyrosine residue in the outer sphere of the LPC metal complex (Tyr42) is a highly conserved feature found in many redox-active metalloenzymes. Although in most cases its role is not clear, in some enzymes it performs a well-defined function. For example, the corresponding tyrosine residues in ribonucleotide reductase (Y122) [23] and the oxygen-evolving complex of photosystem II (YZ) [24] form stable protein free radicals that serve as storage sites for oxidizing equivalents. Tyr42 may have a similar function in t he LPC site, protecting the metal complex from oxidative damage by forming a transient phenoxyl radical, although it also has a structural role, and substitution by phenylalanine disrupts the bonding of the Mn2O2 metallocluster core [25].
2.1.4 Metal-center reactivity
The active site metal complex is able to bind small inorganic anions (azide and fluoride) [12,26,27]. Both of these anions are conjugate bases of weak acids with pKa values near the physiological pH range, and both exhibit pH-dependent binding and inhibition, indicating that it is the neutral (protonated) species is able to reach the active site. Azide is a peroxide analog that acts as a competitive inhibitor for manganese catalase turnover (KI = 5 mM at pH 5.5, 80 mM at pH 7), binding to both oxidized and reduced manganese complexes. A crystal structure has been solved for azide bound to the oxidized Mn2(III,III) form of the enzyme [17], showing the anion coordinating to Mn1, substituting for the terminally bound solvent molecule (Fig. 4). Since the molecular orbitals of the linear azide molecule closely resemble the hydrogen peroxide orbitals that are involved in metal interactions, the binding mode of the azide inhibitor may indicate how substrate binds in the LPC active site. The azide is roughly aligned with the entrance channel over the Glu35 ligand, and extends away from the metallocluster into a pocket formed by amino acid side chains, including Ser29 and Leu174. The pendant Glu178 headgroup is within hydrogen-bonding distance (2.8 Å) of the coordinating azide N1 atom. This complex may represent the bonding pattern involved in oxidation of hydrogen peroxide by the metallocluster and formation of the dioxygen product.
Figure 4.
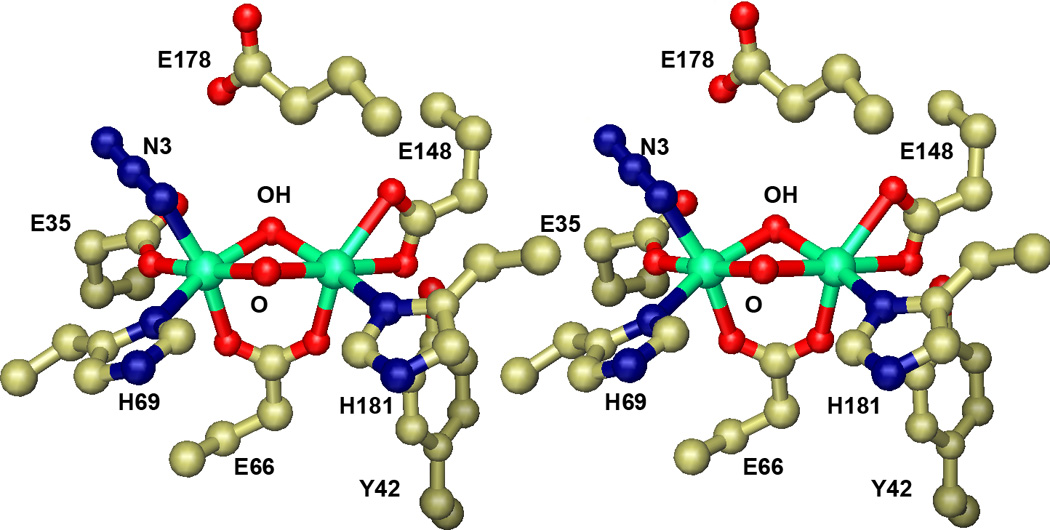
Active site of azide-bound Lactobacillus plantarum manganese catalase. Stereoscopic view of the binuclear manganese complex in the oxidized Mn2(III,III) state with azide bound. Based on PDB ID: 1JKV.
Fluoride binding is associated with a distinct pattern of cluster interactions. Fluoride traps the complex in the reduced Mn2(II,II) state when the enzyme is exposed to the ion during turnover, suggesting that the most stable complex is formed with the Mn2(II,II) metallocluster. Since fluoride interactions dramatically perturb the electronic coupling within the complex [28,29] a μ-bridging coordination mode has been proposed, with fluoride inserting into the cluster core and replacing one of the bridging solvent molecules [28]. This implies that the bridging solvent molecules are relatively labile in the reduced complex, and suggests that substrate may insert into the bridge positions in the reductive half-reaction during turnover.
Other than azide and fluoride, very few other ions interact with the LPC metal centers. Unlike heme-containing catalases, which are inhibited by cyanide at µM concentrations [30,31], manganese catalase is insensitive to cyanide inhibition [12]. The contrast in cyanide sensitivity between the two enzymes is so great that cyanide insensitive catalase activity has been cited as evidence for nonheme catalase in microbiological extracts.
The inorganic chemistry of manganese includes multiple oxidation states of the metal ion, including higher oxidation states (IV,V) which have also been observed in some biological complexes. The rich oxidation chemistry of manganese is reflected in the chemistry of manganese catalase, and other oxidation states of the binuclear cluster may be prepared, in addition to the Mn2(III,III) and Mn2(II,II) forms. In particular, a “superoxidized” Mn2(III,IV) state is generated when hydroxylamine is present during turnover [32]. This mixed valent complex is catalytically inactive, but reduction to the Mn2(II,II) form by reaction with excess hydroxylamine restores activity. The superoxidized Mn2(III,IV) state of manganese catalase has been investigated as a model for the S2-state of the photosynthetic water-splitting active site, a tetra-manganese metallocluster that contains a mixed valent Mn(III)Mn(IV) pair [33].
2.1.5 Catalytic mechanism
The overall turnover reaction (Eqn. 1) involves transfer of two electrons and two protons (a hydrogen molecule equivalent) from one molecule of hydrogen peroxide to another. The large activation barrier associated with the uncatalyzed process (which may be estimated at 18 kcal/mol based on reaction kinetics) reflects the difficulty of accomplishing this intermolecular redox reaction in a simple bimolecular collision. The binuclear manganese active site of manganese catalase is well-suited to its catalytic function, serving as a storage site for electrons and protons during catalysis. The two metal centers can each perform a one-electron redox cycle (involving Mn(III) and Mn(II) oxidation states), storing reducing equivalents during the reaction. The change in metal oxidation state (and formal charge) in turn alters the basicity of the bridging solvent molecules, creating sites for proton storage during turnover. The enzyme separates the overall chemistry into two half reactions (Equations 2 & 3):
| (2) |
| (3) |
The turnover reaction is comprised of an oxidative half-reaction (Eqn. 2) in which the substrate peroxide is oxidized to dioxygen, coupled to reduction of the manganese cluster, and a reductive half-reaction (Eqn. 3) in which the reduced metallocluster delivers the electrons and protons to a second molecule of substrate, resulting in O-O bond cleavage and formation of two molecules of water. In contrast to the nearly diffusion-controlled turnover rate that is characteristic of heme-containing catalases (kcat = 4×107 s−1) [34], non-heme manganese catalases exhibit much slower kinetics. For LPC, kcat = 2×105 s−1 and KM = 350 mM at 25°C [12]. Similar values have been reported for TTC (kcat = 2.6×105 s−1 and KM = 80 mM at 65°C)[35].
A molecular mechanism for manganese catalase turnover has been proposed based on structural and biochemical data [17], in which the neutral hydrogen peroxide substrate reaches the deeply buried active site via the access channel and coordinates to Mn1, displacing the terminally bound solvent in the oxidized Mn2(III,III) complex (Fig. 5, Complex 2). The pendant Glu178 may play a role in facilitating movement of protons from the bound substrate to the bridging solvents as electrons are transferred to the Mn(III) centers, lowering the activation barrier for these processes. This proton-coupled electron transfer would generate the dioxygen product of the oxidative half-reaction, and leave the enzyme in the reduced Mn2(II,II) state (Fig. 5, Complex 3), with each of the solvent bridges binding one of the protons. Upon dissociation, the dioxygen molecule could exit from the active site either though the access channel or by diffusion through the protein, with solvent replacing dioxygen in the axial position of Mn1 (Fig. 5, Complex 4). Subsequently, a second molecule of hydrogen peroxide would enter the active site and bind terminally to Mn1 (Fig. 5, Complex 5a), or, alternatively, insert into the cluster core to form a μ1,1 bridging peroxy complex (Fig. 5, Complex 5b) resulting in activation of the O-O bond. Reductive cleavage of the O-O bond would be coupled to oxidation of the two metal centers to the Mn(III) state, completing the catalytic cycle (Fig. 5, Complex 1).
Figure 5.

Proposed catalytic cycle for Lactobacillus plantarum manganese catalase. Oxidative half-reaction: (1) Resting oxidized Mn2(III,III) state. (2) Terminal peroxide adduct. (3) Terminal dioxygen adduct. Reductive half-reaction: (4) Resting reduced Mn2(II,II) state. (5) Activated hydrogen peroxide complexes: (a) terminally bound substrate; (b) μ1,1 bridging substrate.
2.2 Thermus thermophilus manganese catalase [18]
The manganese catalase from the thermophilic eubacterium Thermus thermophilus (TTC) adopts a protein fold similar to LPC (Fig. 2), but assembles into a more compact hexamer (Fig. 6). TTC is also more highly ordered in the lattice and exhibits smaller B-factors (temperature factors) compared to LPC. The average crystallographic B-factor for TTC backbone atoms (2V8U; 7.1 Å), compared to 16 Å for LPC (1JKU), yielding higher crystallographic resolution (1 Å) for the thermophilic protein.
Figure 6.

Thermus thermophilus manganese catalase. A single subunit of TTC is rendered as ribbons embedded in the surface-rendered globular hexameric holoenzyme. Based on PDB ID: 2V8U.
2.2.1 Protein fold
The catalytic core of TTC is a four-helix bundle coiled-coil domain that includes an unusually long (55 residue) left-handed crossover between the two hairpins (Fig. 2B). The active site is buried in the interior of the four-helix bundle, cross-linking the α-helical rods. As in LPC, N- and C-terminal extensions from the central catalytic domain form connections with neighboring subunits in the multimer.
2.2.2 Active site
The binuclear manganese active site of TTC closely resembles the LPC cluster, with a solvent-bridged Mn2O2 core (Fig. 7). A μ1,3 bridging carboxylate (Glu70) spans the core and a histidine/carboxylate ligand set completes the coordination environment of the metal ions, with Glu and His ligands respectively binding on proximal and distal sides of the cluster. In TTC, Mn1 is coordinated by Glu36 and His73, while Mn2 is coordinated by Glu155 and His188. The TTC structure suggests somewhat greater conformational freedom for the non-bridging carboxylates in the active site compared to LPC, and Glu36 appears to be able to bind in a bidentate chelating mode which displaces the axial solvent in at Mn1. In this alternate conformation, a crystallographically defined solvent molecule is present in the region above the solvent core. In place of the pendant carboxylate (Glu178) in LPC, there is a lysine (Lys162) in the corresponding position in TTC. The structural correspondence suggests that Lys162 may play a role in proton shuttling in the TTC active site, as proposed for Glu178 in LPC. The side chain amino group Nε hydrogen of Lys162 is hydrogen bonded to the terminally coordinated solvent molecule on Mn1. The lysine side chain also exhibits conformational heterogeneity in the crystal structure, suggesting a degree of flexibility in the active site.
Figure 7.

Active site of native Mn(III,III) Thermus thermophilus manganese catalase. Stereoscopic view of the binuclear manganese complex in the oxidized Mn2(III,III) state. Based on PDB ID: 2V8U.
In contrast to the unique access channel in LPC, the active site of TTC has two channels extending out from the deeply buried metal complex. One is a relatively narrow 16 Å long channel connecting the active site to the central chamber of the hexamer, entering the active site near Glu36, similar to the access channel found in LPC. The channel is associated with a loop that disrupts helix A, starting at a pair of glycine residues (Gly31,Gly32). This disruption of the regular helical pattern creates a channel that allows access into the protein interior. The other channel is relatively broad and 22 Å long, extending out from the opposite end of the cluster, passing over Glu 155 to open to the protein surface, directly connecting the active site to the bulk solvent (Fig. 8). This second channel is also associated with a glycine pair (Gly185–Gly186) in helix D, which maintains the α-helical backbone structure but breaks the pattern of side chain packing between the helices. While some charged residues are present in this second channel, the path is largely neutral and hydrophilic.
Figure 8.

The 22 Å long access channel in TTC. A cross-section of the TTC four-helix bundle is shown, near the level of the binuclear manganese active site (red spheres). The protein is surface-rendered and solvent molecules within the channel appear as cyan spheres. Based on PDB ID: 2V8U.
2.2.3 Extended active site environment
The manganese cluster in TTC is coupled to a hydrogen bonding web that extends from the coordinating histidines (His73,His188) through a salt bridge (Glu69-Arg184) and including Glu9 and Tyr180. The organization of this hydrogen bonding web is remarkably conserved between TTC and LPC, supporting a role for this element of the protein structure in the function of the catalytic complex. An outer sphere tyrosine (Tyr43) forms a hydrogen bond to the carboxylate headgroup of the Glu155 ligand.
2.2.4 Metal-centered reactivity
The TTC active site shares with the LPC manganese complex the basic oxidation-reduction chemistry that supports the turnover reactions (Eq. 2 & 3). However, the active site is readily accessible to a wide range of ions, and the coordination chemistry of the TTC metal complex is much more extensive than seen for LPC. Complexes of TTC with large ions (including phosphate) have been reported [36]. The oxidation chemistry of the TTC cluster is also more extensive than observed for LPC. In addition to the Mn2(II,II) and Mn2(III,III) states, mixed valent states (Mn2(II,III) and Mn2(III,IV)) have been prepared. Each of these complexes forms adducts with exogenous ions.
The accessibility of the TTC active site to a variety of exogenous molecules contrasts with the restricted access to the LPC site. This difference has led to a proposal that TTC may have additional catalytic functions beyond the catalase activity previously characterized. The possibility that TTC may be a bifunctional catalase-peroxidase is particularly intriguing [18]. Peroxidase reactivity would imply that a reducing substrate (AH2) other than hydrogen peroxide could serve to drive the oxidative half-reaction during turnover, with hydrogen peroxide reduction completing the cycle (Equations 4 & 5):
| (4) |
| (5) |
However, there is no experimental evidence for peroxidase activity of the TTC enzyme, and identifying the (presently unknown) co-substrate for this reaction could prove to be challenging.
3. Computational Approaches
A variety of powerful computational approaches have been used to investigate the structure and reactivity of manganese catalase, including density functional theory (DFT) ab initio electronic structure calculations and molecular dynamics simulations.
3.1 Ab initio reaction profile [37]
The reaction coordinate for manganese catalase turnover has been explored to map the energy profile and to identify possible reaction intermediates involved in the turnover reaction. The calculations have been based on a simplified model of the active site in which the coordinating amino acid side chains have been truncated to formate (for Glu) and imidazole (for His) (Fig. 9). The geometry of the model is roughly based on the crystal structure of the oxidized active site for TTC. Constrained geometry optimization was used to minimize the energy of the reactive complexes, beginning with the fully reduced Mn2(II,II) form and progressing through reductive and oxidative half-reactions (Fig. 10).
Figure 9.

Active site model used for ab initio calculation of reaction energy profile. [Based on Ref. 37]
Figure 10.

Manganese catalase turnover reaction energy profile. [Based on Ref. 37]
The initial complex is formed by insertion of substrate into the labile Mn2(II,II) core, substituting for a bridging water molecule, and forming a μ1,1-bridging peroxide adduct. This coordination mode contributes to activation of the O-O bond of the bound peroxide, facilitating cleavage of the substrate (Fig. 10, Step 1). The reaction is nominally spin-forbidden, and is associated with a spin-crossing evolution of the ground state of the cluster. Transfer of an electron from one of the Mn(II) ions to the substrate populates an anti-bonding orbital on the bound peroxide, further weakening the O-O bond. The subsequent highly exergonic bond-breaking step correlates the ground state of the substrate complex to a charge-transfer excited state product complex in which a hydroxyl free radical is bound to a Mn(II) metal center (Fig. 10, Step 2), which relaxes to di-Mn(III) at the end of the oxidative half-reaction (Fig. 10, Step 3) with dissociation of the product water. In this way the free radical is trapped by the metal complex, avoiding Fenton chemistry, an important feature of catalase turnover. Both metal cores contract with the increase in metal oxidation state along a breathing mode of the complex. The barrier height for the oxidative half-reaction is estimated to be 15.4 kcal/mol based on this calculation, representing the largest barrier in the catalytic reaction profile. It is, in fact, 3–5 kcal/mol larger than the barrier height predicted by the experimental rate of the catalyzed reaction. This discrepancy might reflect stabilizing contributions from residues in the extended outer sphere environment of the active site, for example proton-donating groups, or may simply reflect overestimation of the barrier height by the BLY3P functional used in the calculation.
The oxidative half-reaction is initiated by outer-sphere association of a second molecule of substrate with the Mn2(III,III) form (Fig. 10, Step 4), stabilized by formation of a short (1.39 Å) strong (4.4 kcal/mol) hydrogen bond with the μ-bridging oxo group. Proton transfer to the bridging oxygen deprotonates the substrate, which coordinates end-on to an open position on Mn(III) (Fig. 10, Step 5). Completion of the turnover cycle involves proton transfer from the distal oxygen of the coordinated substrate to doubly protonate one of the bridges, coupled to electron transfer to the Mn center (proton coupled electron transfer, PCET) (Fig. 10, Step 6). The oxidation proceeds through two sequential one-electron one-proton PCET reactions, and an intermediate complex is formed containing a mixed valence Mn2(II,III) core with a terminally bound superoxide. Internal reorganization of the metal oxidation states is followed by formation of a dioxygen adduct (Fig. 10, Step 7) and dissociation of the O2 product. This process restores the active site to the initial Mn(II,II) complex that served as the starting point for the cycle.
3.2 Ab initio electronic structure calculations [38]
Broken-symmetry (BS) DFT calculations have been used to investigate the electronic structural details of the dimanganese cluster in the superoxidized Mn2(III,IV) oxidation state. Based on computational analysis of alternative valence assignments and protonation states, it has been concluded that both bridging solvents are deprotonated in the Mn2(III,IV) complex, consistent with chemical intuition for the effect of the higher Mn oxidation state on the ligand acidity and preserving the overall charge of the cluster core. The two solvent bridges support a significant electronic coupling within the cluster (Heisenberg exchange coupling constant, Jexch = 95 cm−1) that may contribute to efficient electron transfer between the two metal ions.
3.2 Molecular dynamics simulations [39]
Global features of manganese catalase protein motion have been investigated by molecular dynamics simulation. The LPC model used for the calculations was built by applying the parm99 force field to the native LPC X-ray coordinate set (PDB ID:1JKU) to form a system of 124,179 atoms including the hexameric protein, 6 dimanganese active sites, 6 calcium ions, and 48 potassium counterions solvated by 3.7 attoliters of TIP3P water. The formal charge on the active site is predicted to be zero, consistent with the large Coulomb penalty for charge burial in proteins, implying that the non-coordinated Glu178 is in the protonated state in the native structure. Molecular trajectories were calculated for this model for up to 12 nsec to investigate protein dynamics.
Based on the simulations, it is predicted that the rigidity of the dimanganese core will degrade the mobility of the helices, particularly near the middle of the helix bundle where the active site effectively cross-links the coiled-coil domain. In contrast, the ends of the helix bundle exhibit much greater flexibility, as do the extended loop regions. Overall, the patterns of rms fluctuation calculated for the protein closely resemble the experimental B-factors derived from the crystallographic analysis. Within the active site, the calculations show a rapid exchange between monodentate and bidentate coordination modes for Glu148, and facile flipping of the protonated headgroup of Glu178. The hydrogen bonding network extending from the two histidine ligands (His69 and His181), noted in the crystal structure, is not very rigid and appears to flex with the helix bundle, forming a loose web of weak hydrogen bonds that ruptures and reforms as the protein moves.
4. Phylogenetic distribution of manganese catalase [40]
The explosive growth of genomic data in recent years has resulted in identification of nearly 100 putative manganese catalase genes in a wide range of organisms, including both prokaryotes and crenarchaeota (Fig. 11, Supplementary Fig. 1) [40,41]. Notably, no eukaryotes are represented in this group, and to date only a small number of these putative proteins have actually been isolated and the manganese catalase assignment experimentally verified.
Figure 11.

Radial phylogenetic tree for putative manganese catalases in genomic databases. (1) Lactobacillus plantarum manganese catalase (LPC); (2) Thermus thermophilus manganese catalase (TTC); (3) Pyrobaculum caldifontis manganese catalase; (4) Salmonella enterica katNp; (5) Escherichia coli O157:H7 Z1921p. Generated from the predicted protein sequences using Phylip cluster analysis [42] and Dendroscope-3 tree builder [43].
Protein sequence analysis sorts the group into at least four deeply rooted clades (Fig. 11). The radial phylogenetic tree diagram illustrates the sequence distance between the members of the group. While some of the sequences appear to cluster along classic taxonomic divisions, others clearly show a level of protein similarity that is inconsistent with the large evolutionary separation between source organisms, strongly suggesting a horizontal mode of genetic transmission. This idea is supported by the observation that the hypothetical manganese catalase gene is located in a prophage island in at least one genome (E. coli O157:H7) [44].
The occurrence of putative manganese catalases in a number of organisms typically described as obligate anaerobes is an unexpected result of genomic data mining. While some of these organisms (e.g. Desulfobacterium spp.) are catalase-positive, others (e.g. Clostridium spp., Anaerofustis spp.) have been historically described as catalase-negative. Although these organisms are unable to grow in the presence of oxygen, many of them are able to mount defenses against oxidative stress associated with the microaerophilic boundaries of their environment. The clade of obligately anaerobic Firmicutes is one of the more homogeneous and deeply rooted groups represented in the dendrogram, which may suggest an evolutionary source for a manganese catalase that was subsequently transferred to lactic acid bacteria (e.g. Lactobacillus plantarum, Fig. 11, 1) and other facultative anaerobes whose niches overlap in the environment.
Another deeply rooted clade includes the manganese catalase from the thermophilic eubacterium Thermus thermophilus (Fig. 11, 2) and the enzyme from the hyperthermophilic crenarchaeon Pyrobaculum caldifontis (Fig. 11, 3), which have both been experimentally verified. This branch of the sequence tree also includes a predicted manganese catalase from Bacillus anthracis (Supplementary Fig. 1), and defines the T-catalase subfamily of non-heme catalases, which shows the greatest sequence divergence from other family members.
Genomic data mining has also uncovered the occurrence of putative manganese catalase genes in a number of pathogenic bacteria. These include Salmonella enterica subsp. enterica serovar Typhimurium (katN) [45], enterohemorrhagic, Escherichia coli O157:H7 (locus ID Z1921) [44], and Pseudomonas aeruginosa (katN) [46]. Knock-out experiments have been used to demonstrate a possible role for KatN in the stress response mechanisms of Salmonella enterica [47,48]. Recently, the manganese catalase assignments of E. coli O157:H7 Z1921p and S. enterica katN have been further investigated by expressing the recombinant proteins in E. coli1. The purified Z1921p protein is a soluble hexameric species containing approximately 1 equivalent of iron, but no appreciable manganese is bound (Table 1). Even when the protein is expressed with manganese supplementation under iron-limiting conditions, almost no Mn is bound. Surprisingly, no catalase activity has been detected for either of the purified proteins, regardless of the metal content. The absence of any detectable catalase activity in the purified recombinant proteins, even when manganese is bound, is consistent with an earlier observation that a Salmonella strain lacking either of the major catalases (HPI and HPII) exhibits no detectable catalase activity [48]. These results indicate that at least some of the assignments listed in Fig. 11 may be incorrect, and that additional functional studies will be needed to clarify the assignments.
Table 1.
Characterization of recombinant KatN protein
| Source organism | Mass (ESI- MS) |
Mass (calc) | Mr (SEC) | Fe (g- atom/mol) |
Mn (g- atom/mol) |
Catalase activity (U/mg) |
|---|---|---|---|---|---|---|
| E. coli O157:H7a | 32168 | 32169 | 2×105 | 1.0 | 0.0 | 0.0 |
| 0.34c | 0.14c | 0.0 | ||||
| Salmonella entericab | – | – | – | 0.89 | 0.55 | 0.0 |
Expressed in Escherichia coli BL21 Star DE3 expression host.
Expressed in Escherichia coli BL21-AI expression host.
Chemically defined MOPS minimal expression medium containing 1 mM MnCl2.
5. Biological significance
Organisms that contain manganese catalase play important roles in human health and disease as endogenous microflora, inhibiting the growth of microbial pathogens [49], but may also include the pathogens themselves. The biological role of manganese catalase in some of these organisms has been explored by in vivo studies, including analysis of the effects of manganese catalase expression and over-expression. In Lactobacillus casei, a strain that is normally catalase-negative, heterologous over-expression of manganese catalase enhances viability following treatment with hydrogen peroxide, which may contribute to construction of more robust probiotic bacteria [50]. On the other hand, over-expression of manganese catalase by an anti-inflammatory probiotic strain (Lactobacillus casei BL23) did not increase their ability to protect against epithelial damage from reactive oxygen species in a model for inflammatory bowel disease [51].
Four environmental contexts that may be important for manganese catalase selection are iron limitation, microaerophilic oxidative stress, thermostability, and cyanide resistance:
Iron limitation
Iron is an essential inorganic nutrient for many organisms. Bacteria adapt to environments that restrict iron availability by a variety of mechanisms, and manganese catalase may represent such an adaptation. Lactic acid bacteria have adapted to iron limitation in the environment by adopting a lifestyle of “total iron abstinence” lacking any iron metalloprotein (including cytochromes), and using other metal ions to perform essential functions. Pathogenic bacteria also experience iron limitation during infection, and may benefit from the availability of a nonheme catalase.
Microaerophilic oxidative stress
A relatively low level of hydrogen peroxide production may be associated with a microaerophilic environment. This low steady-state level of hydrogen peroxide can be efficiently dealt with by manganese catalase, whose active site is indefinitely stable in both oxidized and reduced states. In contrast, heme-containing catalases form a high-potential (Compound I) intermediate during the reductive turnover half-reaction, which can oxidize protein side chains if there is insufficient hydrogen peroxide to continue the reaction. Based on this consideration, manganese catalase may provide an advantage at low hydrogen peroxide levels.
Thermostability
The ability of non-heme manganese catalases to function in extreme environments (including hot springs and volcanic sea vents) may indicate a special role for this family of enzymes in the biosphere. The four-helix bundle protein architecture is very robust, and the relative inertness of the manganese complexes in all catalytic states contrasts with the exceptional reactivity of the Compound I intermediate formed during heme catalase turnover. These considerations suggest that manganese catalase may provide a selective advantage at elevated temperatures.
Cyanide resistance
Some pathogenic microorganisms produce hydrogen cyanide during invasion of the host [52], and Pseudomonas aeruginosa has been shown to kill C. elegans by releasing cyanide [53]. The potent inhibition of heme catalase by cyanide suggests a possible role for manganese catalase during infection, when the pathogen must simultaneously mount an antioxidant defense and produce cyanide.
6. Conclusions
Manganese catalases are widespread in the environment, supporting essential antioxidant functions independent of iron or heme requirements. The role of manganese catalases in health and disease are just beginning to be investigated.
Supplementary Material
Cladogram for putative manganese catalases reported in genomic databases. Nodes in the hierarchical clustering tree are labeled with the sequence distance metric. The entries are labeled with the Peroxibase (http://peroxibase.toulouse.inra.fr) accession code and the taxonomic name of the source organism. Generated from the predicted protein sequences using Phylip cluster analysis and Dendroscope-3 tree builder.
ACKNOWLEDGEMENTS
Support from the National Institutes of Health (grant GM42680 to J.W. W.) is gratefully acknowledged.
ABBREVIATIONS
- LPC
Lactobacillus plantarum manganese catalase
- TTC
Thermus thermophilus manganese catalase
- DFT
density functional theory
- PCET
proton coupled electron transfer
- BS
broken symmetry
- ESI-MS
electrospray-mass spectrometry
- SEC
size exclusion chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
M.M. Whittaker and J.W. Whittaker, unpublished results
References
- 1.Beyer WF, Fridovich I. Catalases—with and without heme. Basic Life Sci. 1988;49:651–661. [PubMed] [Google Scholar]
- 2.Chelikani P, Fita I, Loewen PC. Diversity of structures and properties among catalases. Cell Mol Life Sci. 2004;61:192–208. doi: 10.1007/s00018-003-3206-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Molecular Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Fang FC. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2004;2:820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 5.González-Flecha B, Demple B. Metabolic sources of hydrogen peroxide in aerobically growing Escherichia coli. J. Biol. Chem. 1995;270:13681–13687. doi: 10.1074/jbc.270.23.13681. [DOI] [PubMed] [Google Scholar]
- 6.Korshunov S, Imlay JA. Two sources of endogenous hydrogen peroxide in Escherichia coli. Mol. Micro. 2010;75:1389–1401. doi: 10.1111/j.1365-2958.2010.07059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson DP, Kiesow LA. Enthalpy of decomposition of hydrogen peroxide by catalase at 25°C. Anal. Biochem. 1972;49:474–478. doi: 10.1016/0003-2697(72)90451-4. [DOI] [PubMed] [Google Scholar]
- 8.Thénard LJ. Mémoire sur la combinaison de l’oxygène avec l’eau, et sur les propriétés extraordinaires que possède l’eau oxigéée, Mémoires de l’Académie royale des sciences de l’Instiut de France, année 1818. 1820;3:385–488. [Google Scholar]
- 9.Gagnon M, Hunting W, Esselen WB. A new method for catalase determination. Anal. Chem. 1959;31:144. [Google Scholar]
- 10.Delwiche EA. Catalase of Pedicoccus cerevisiae. J. Bacteriol. 1961;81:416–418. doi: 10.1128/jb.81.3.416-418.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnston MA, Delwiche EA. Isolation and Characterization of the cyanide- and azide-resistant catalase of Lactobacillus plantarum. J. Bacteriol. 1965;90:352–356. doi: 10.1128/jb.90.2.352-356.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kono Y, Fridovich I. Isolation and characterization of the pseudocatalase of Lactobacillus plantarum. J. Biol. Chem. 1983;258:6015–6019. [PubMed] [Google Scholar]
- 13.Barynin VV, Grebenko AI. T-catalase is nonheme catalase of the extremely thermophilic bacterium Thermus thermophilus HB8. Dokl. Akad. Nauk SSSR. 1986;286:461–464. [Google Scholar]
- 14.Allgood GS, Perry JJ. Characterization of a manganese-containing catalase from the obligate thermophile Thermoleophilum album. J. Bacteriol. 1986;168:563–567. doi: 10.1128/jb.168.2.563-567.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amo T, Atomi H, Imanaka T. Unique Presence of a Manganese Catalase in a Hyperthermophilic Archaeon, Pyrobaculum calidifontis VA1. J. Bacteriol. 2002;184:3305–3312. doi: 10.1128/JB.184.12.3305-3312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barynin VV, Whittaker JW. Manganese catalase Handbook of metalloproteins online [Google Scholar]
- 17.Barynin VV, Whittaker MM, Antonyuk SV, Lamzin VS, Harrison PM, Artymiuk PJ, Whittaker JW. Crystal structure of manganese catalase from Lactobacillus plantarum. Structure. 2001;9:725–738. doi: 10.1016/s0969-2126(01)00628-1. [DOI] [PubMed] [Google Scholar]
- 18.Antonyuk SV, Melik-Adman VR, Popov AN, Lamzin VS, Hempstead PD, Harrison PM, Artymyuk PJ, Barynin VV. Three-dimensional structure of the enzyme dimanganese catalase from Thermus thermophilus at 1 Å resolution. Crystallography Reports. 2000;45:105–113. [Google Scholar]
- 19.Trikha J, Theil EC, Allewell NM. High resolution crystal structures of amphibian red-cell L ferritin. J. Mol. Biol. 1995;248:949–967. doi: 10.1006/jmbi.1995.0274. [DOI] [PubMed] [Google Scholar]
- 20.Nordlund P, Eklund H. Structure and function of the Escherichia coli ribonucleotide reductase protein R2. J. Mol. Biol. 1993;232:123–164. doi: 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- 21.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 22.Summa CM, Lombardi A, Lewis M, DeGrado WF. Tertiary templates for the design of diiron proteins. Curr. Opin. Struct. Biol. 1999;9:500–508. doi: 10.1016/S0959-440X(99)80071-2. [DOI] [PubMed] [Google Scholar]
- 23.Larsson A, Sjöberg BM. Identification of the stable free radical tyrosine residue in ribonucleotide reductase. EMBO J. 1986;5:2037–2040. doi: 10.1002/j.1460-2075.1986.tb04461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 25.Whittaker MM, Igarashi T, Barynin VV, Whittaker JW. Outer sphere mutagenesis of manganese catalase from Lactobacillus plantarum disrupts the cluster core. Eur. J. Biochem. 2003;270:1102–1116. doi: 10.1046/j.1432-1033.2003.03459.x. [DOI] [PubMed] [Google Scholar]
- 26.Waldo GS, Penner-Hahn JE. Mechanism of manganese catalase peroxide disproportionation: determination of manganese oxidation states during turnover. Biochemistry. 1995;34:1507–1512. doi: 10.1021/bi00005a006. [DOI] [PubMed] [Google Scholar]
- 27.Whittaker MM, Barynin VV, Antonyuk SV, Whittaker JW. The oxidized (3,3) state of manganese catalase. Comparison of enzymes from Thermus thermophilus and Lactobacillus plantarum. Biochemistry. 1999;38:9126–9136. doi: 10.1021/bi990499d. [DOI] [PubMed] [Google Scholar]
- 28.Meier AE, Whittaker MM, Whittaker JW. EPR polarization studies on Mn catalase from Lactobacillus plantarum. Biochemistry. 1996;35:348–360. doi: 10.1021/bi952126s. [DOI] [PubMed] [Google Scholar]
- 29.Le Pape L, Perret E, Michaud-Soret I, Latour JM. Magnetization studies of the active and fluoride-inhibited derivatives of the reduced catalase of Lactobacillus plantarum: toward a general picture of the anion-inhibited and active forms of the reduced dimanganese catalases. J. Biol. Inorg. Chem. 2002;7:445–450. doi: 10.1007/s00775-001-0319-x. [DOI] [PubMed] [Google Scholar]
- 30.Chance B. The reaction of catalase and cyanide. J. Biol. Chem. 1949;179:1299–1309. [PubMed] [Google Scholar]
- 31.Ogura Y, Yamakazi I. Steady-state kinetics of the catalase reaction in the presence of cyanide. J. Biochem. (Tokyo) 1983;94:403–408. doi: 10.1093/oxfordjournals.jbchem.a134369. [DOI] [PubMed] [Google Scholar]
- 32.Waldo GS, Fronko RM, Penner-Hahn JE. Inactivation and reactivation of manganese catalase: oxidation-state assignments using X-ray absorption spectroscopy. Biochemistry. 1991;30:10486–10490. doi: 10.1021/bi00107a017. [DOI] [PubMed] [Google Scholar]
- 33.Sproviero EM, McEvoy JP, Gascón JA, Brudvig GW, Batista VS. Computational insights into the O2-evolving complex of photosystem II. Photosynthesis Research. 2008;97:91–114. doi: 10.1007/s11120-008-9307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chance B, Greenstein DS, Roughton FJ. The mechanism of catalase action. I. Steady-state analysis. Arch Biochem Biophys. 1952;37:301–321. doi: 10.1016/0003-9861(52)90194-x. [DOI] [PubMed] [Google Scholar]
- 35.Shank M, Barynin VV, Dismukes GC. Protein coordination to manganese determines the high catalytic rate of dimanganese catalases. Comparison to functional catalase mimics. Biochemistry. 1994;33:15433–15436. doi: 10.1021/bi00255a025. [DOI] [PubMed] [Google Scholar]
- 36.Khangulov SV, Barynin VV, Antonyuk-Barynina SV. Manganese-containing catalase from Thermus thermophilus peroxide-induced redox transformation of manganese ions in presence of specific inhibitors of catalase activity. Biochim. Biophys. Acta. 1990;1020:25–33. [Google Scholar]
- 37.Siegbahn PEM. A quantum chemical study of the mechanism of manganese catalase. Theor. Chem. Acc. 2001;105:197–206. [Google Scholar]
- 38.Sinnecker S, Neese F, Lubitz W. Dimanganese catalase--spectroscopic parameters from broken-symmetry density functional theory of the superoxidized Mn(III)/Mn(IV) state. J. Biol. Inorg. Chem. 2005;10:231–238. doi: 10.1007/s00775-005-0633-9. [DOI] [PubMed] [Google Scholar]
- 39.Spiegel K, De Grado WF, Klein ML. Structural and dynamical properties of manganese catalase and the synthetic protein DF1 and their implication for reactivity from classical molecular dynamics calculations. Proteins. 2006;65:317–330. doi: 10.1002/prot.21113. [DOI] [PubMed] [Google Scholar]
- 40.Zamocky M, Furtmüller PG, Obinger C. Evolution of catalases from bacteria to humans. Antioxidants & Redox Signaling. 2008;10:1527–1547. doi: 10.1089/ars.2008.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koua D, Cerutti L, Falquet L, Sigrist CJ, Theiler G, Hulo N, Dunand C. PeroxiBase: a database with new tools for peroxidase family classification. Nucleic Acids Res. 2009;37:D261–D266. doi: 10.1093/nar/gkn680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huson DH, Richter DC, Rausch C, Dezulian T, Franz M, Rupp R. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinformatics. 2007;8:460. doi: 10.1186/1471-2105-8-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perna NT, Plunkett G, III, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Pósfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature. 2001;409:529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- 45.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, Hou S, Layman D, Leonard S, Nguyen C, Scott K, Holmes A, Grewal N, Mulvaney E, Ryan E, Sun H, Florea L, Miller W, Stoneking T, Nhan M, Waterston R, Wilson RK. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 46.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FSL, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK-S, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock REW, Lory S, Olson MV. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 47.Robbe-Saule V, Coynault C, Ibanez-Ruiz M, Hermant D, Norel F. Identification of a non-haem catalase in Salmonella and its regulation by RpoS (sigmaS) Mol. Microbiol. 2001;39:1533–1545. doi: 10.1046/j.1365-2958.2001.02340.x. [DOI] [PubMed] [Google Scholar]
- 48.Buchmeier NA, Libby SJ, Xu Y, Loewen PC, Switala J, Guiney DG, Fang FC. DNA repair is more important than catalase for Salmonella virulence in mice. J. Clin. Investigation. 1995;95:1047–1053. doi: 10.1172/JCI117750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin R, Suarez JE. Biosynthesis and degradation of H2O2 by vaginal Lactobacilli. Appl. Environ. Microbiol. 2010;76:400–405. doi: 10.1128/AEM.01631-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rochat T, Gratadoux JJ, Gruss A, Corthier G, Maguin E, Langella P, van de Guchte M. Production of a heterologous nonheme catalase by Lactobacillus casei: an efficient tool for removal of H2O2 and protection of Lactobacillus bulgaricus from oxidative stress in milk. Appl Environ Microbiol. 2006;72:5143–5149. doi: 10.1128/AEM.00482-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rochat T T, Bermúdez-Humarán L, Gratadoux JJ, Fourage C, Hoebler C, Corthier G, Langella P. Anti-inflammatory effects of Lactobacillus casei BL23 producing or not a manganese-dependant catalase on DSS-induced colitis in mice. Microb. Cell Fact. 2007;20:22. doi: 10.1186/1475-2859-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lenney W, Gilchrist FJ. Pseudomonas aeruginosa and cyanide production. Eur. Respir. J. 2011;37:482–483. doi: 10.1183/09031936.00122810. [DOI] [PubMed] [Google Scholar]
- 53.Gallagher LA, Manoil C. Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J. Bact. 2001;183:6207–6214. doi: 10.1128/JB.183.21.6207-6214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cladogram for putative manganese catalases reported in genomic databases. Nodes in the hierarchical clustering tree are labeled with the sequence distance metric. The entries are labeled with the Peroxibase (http://peroxibase.toulouse.inra.fr) accession code and the taxonomic name of the source organism. Generated from the predicted protein sequences using Phylip cluster analysis and Dendroscope-3 tree builder.