

Abstract
Recently, there has been a paradigm shift away from an emphasis on the role of the endocrine (circulating) renin−angiotensin system (RAS) in the regulation of the sodium and extracellular fluid balance, blood pressure, and the pathophysiology of hypertensive organ damage toward a focus on the role of tissue RAS found in many organs, including kidney. A tissue RAS implies that RAS components necessary for the production of angiotensin II (Ang II) reside within the tissue and its production is regulated within the tissue, independent of the circulating RAS. Locally produced Ang II plays a role in many physiological and pathophysiological processes such as hypertension, inflammation, oxidative stress, and tissue fibrosis. Both glomerular and tubular compartments of the kidney have the characteristics of a tissue RAS. The purpose of this article is to review the recent advances in tissue RAS research with a particular focus on the role of the glomerular RAS in the progression of renal disease.
Keywords: Renin−angiotensin system, Angiotensin II, TGF-β, Tissue fibrosis, Glomerulosclerosis
Introduction
Since the discovery of kidney renin by Tigerstedt and Bergman [1], the renin−angiotensin system (RAS) has been established as an endocrine (circulating) system that plays a role in several organs to maintain the sodium and extracellular fluid balance, and thereby regulate blood pressure (BP). Angiotensin II (Ang II) is the most powerful biological product of this system and its action is transmitted by two main G-protein-coupled receptors with seven-transmembrane domains—Ang II type 1 receptor and type 2 receptor (AT1R and AT2R). Recently, the landscape of this system has become more complex with the discovery of new peptides, new proteins, new enzymatic pathways, new functions of RAS, and a tissue Ang II-generating system, a so-called ‘local’ or ‘tissue’ RAS, that acts at the tissue level in a paracrine and autocrine manner [2, 3]. A growing body of evidence from clinical and experimental studies has further highlighted the role that the tissue RAS plays in various disease conditions such as hypertension, inflammation, oxidative stress and tissue fibrosis in many organs [4–7]. It is now well known that the kidney contains all of the elements of the RAS, and locally produced Ang II contributes to not only kidney ontogeny but also to the regulation of BP and progression of chronic kidney disease (CKD) [6–8]. The objective of this review is to explain the role of the renal tissue RAS, with particular focus on the role of the glomerular RAS in disease progression based on recent data. The presence and role of the tubular RAS in the kidney have been extensively reviewed by Kobori et al. [7] and will not be discussed here.
Recent advances in RAS biology
Traditionally, the circulating RAS is known to regulate BP, sodium balance and fluid homeostasis (Fig. 1). Briefly, renin (protease) secreted from the granular cells of the juxtaglomerular apparatus reacts with angiotensinogen (AGT) produced by the liver to release Ang I (1–10), which is further cleaved by a dipeptidyl carboxypeptidase, angiotensin-converting enzyme (ACE), released from capillary endothelial cells of the lung, to convert Ang I to Ang II (1–8). Ang II is considered the major physiologically active component of RAS. The biological actions of Ang II are transmitted by two seven-transmembrane G-protein-coupled receptors—AT1R and AT2R. Most of the physiological effects of Ang II are conveyed by AT1R. AT1R activation induces an increase in blood volume and BP by stimulating vasoconstriction, along with adrenal aldosterone secretion, renal sodium reabsorption and sympathetic neurotransmission. This classical view of the RAS has been significantly expanded by more recent findings that increased the complexity of the system [9, 10]. Ang II is now considered to play a role in cell proliferation, hypertrophy, superoxide production, inflammation and extracellular matrix (ECM) production through the induction of cytokines, chemokines and growth factors [11]. Furthermore, accumulating evidence indicates that other biologically active peptides [Ang (1–7), Ang III and Ang IV] besides Ang II are generated via the activity of ACE2, a homolog of ACE, and several peptidases such as neprilysin (NEP), aminopeptidase A (AP-A) and AP-N. ACE2 is a monocarboxypeptidase that catalyzes the conversion of Ang I to ng (1–9) or the conversion of Ang II to Ang (1–7). The action of Ang (2–10) derived from Ang I via AP-A is still not definitively characterized, but has been implicated in the modulation of vasopressor responses in hypertensive rats [12]. Additionally, new receptors such as Mas receptor, AT4R and prorenin/renin receptor (PRR) have been identified [13–15]. The binding of prorenin to PRR leads to the activation of prorenin to active renin by displacement of the prosegment. Interestingly, stimulation of the PRR activates intracellular signaling, thus upregulating the expression of profibrotic proteins [16]. The recent characterization of the ACE2/Ang (1–7)/Mas receptor axis provides a new pathway for inducing vasodilatory, antiproliferative, and antifibrotic actions, which counteract the classical ACE/Ang II/AT1R axis of the RAS [17]. In parallel with the recognition of new RAS components and activation pathways, the concept of a tissue RAS has emerged with the support of tremendous clinical and experimental research. The functional aspects of tissue RAS actions are based on the tissue-based synthesis of ANG II, independent of the circulating RAS.
Fig. 1.
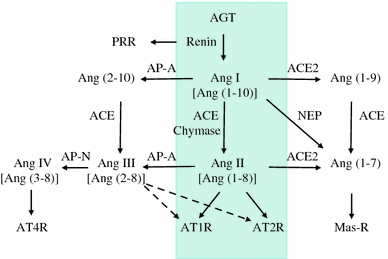
Overview of the renin−angiotensin system (RAS). The schematic shows the circulating RAS (inside the four-sided line) as well as newly recognized enzymatic pathways that lead to the formation and metabolism of products derived from angiotensinogen (AGT). PRR prorenin/renin receptor, ACE angiotensin-converting enzyme, ACE2 angiotensin-converting enzyme 2, AP-A aminopeptidase A, AP-N aminopeptidase N, NEP neprilysin, Ang I angiotensin I, Ang II angiotensin II, AT1R angiotensin II type I receptor, AT2R angiotensin II type 2 receptor, AT4R angiotensin II type 4 receptor. Modified from Refs. [9, 10]
Ang II as a central mediator in progressive glomerular injury
Most CKD that progresses into renal failure begins at the glomerulus. A relentless glomerular injury usually induces glomerulosclerosis characterized by the massive accumulation of ECM, local tuft adhesion to Bowman’s capsule and/or crescent formation [18, 19]. Ang II has emerged as a crucial mediator in progressive glomerular diseases through the induction of glomerular hypertension as well as nonhemodynamic effects that include the production of reactive oxygen species (ROS), up-regulation of profibrotic growth factors (platelet-derived growth factor, transforming growth factor-β [TGF-β], tumor necrosis factor-α), and macrophage activation and infiltration [11, 20]. These injurious actions induced by Ang II affect the behaviors of all four types of glomerular cells [mesangial cells (MC), endothelial cells (GEC), and visceral and parietal epithelial cells (POD and PEC, respectively)] that are involved in severe pathological alterations and constitute a vicious cycle that leads to nephron loss for disease progression (Fig. 2). Extensive studies in various human diseases and in animal models have shown that ACE inhibitors (ACEIs) and/or AT1R blockers (ARBs) are superior to other antihypertensive agents for protecting the kidney against progressive glomerular deterioration, which supports the concept that Ang II is a local paracrine/autocrine effector for the progression of glomerular injury [21, 22].
Fig. 2.
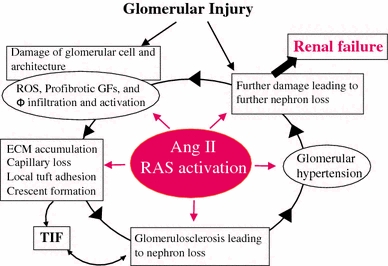
The central role of angiotensin II (RAS activation) in progressive glomerular injury. ROS reactive oxygen species, GFs growth factors, Φ macrophage, TIF tubulo-interstitial fibrosis; ECM, extracellular matrix. Modified from Refs. [18, 20]
Involvement of the glomerular RAS in disease progression
A growing body of evidence demonstrates that all of the components of the RAS are present within the glomerulus and the resultant product, Ang II, regulates glomerular capillary blood flow and capillary wall permeability, and contributes to the development and progression of glomerular diseases as described above. Seminal studies by Seikaly et al. [23] with micropuncture methods showed that the concentrations of total immunoreactive Ang (reflecting Ang II and lesser amounts of three fragments) in rat glomerular filtrate averaged 32 nM compared with 32 pM in systemic plasma, indicating that the Ang II concentration in Bowman’s space is 1000-fold higher than that in the systemic circulation. They subsequently demonstrated for the first time that isolated rat glomeruli can produce Ang II independent of neural innervation, vascular attachment, or exogenous influences. These findings firmly support the glomerulus-based synthesis of Ang II [24]. Many studies using immunohistochemical and in situ hybridization techniques have reported that RAS components such as AGT, ACE, ACE2, Ang II, AT1R and AT2R can be detected in normal and diseased glomeruli in both rats and humans, and a parallel increase in AGT and Ang II, with inconsistent findings regarding the remaining RAS components, is seen in diseased glomeruli from several types of glomerulopathy in rats and humans [25–30]. In genetically manipulated animals, rat glomeruli that have been modified with the human renin and AGT genes developed glomerular sclerosis and showed MC activation (α-smooth muscle actin-positive) [31]. Upstream stimulatory factor 2 transgenic mice show increased renin expression and enhanced renin activity in the kidney, which stimulates the generation of glomerular Ang II which leads to glomerular hypertrophy and ECM accumulation accompanied by enhanced TGF-β expression and albuminuria [32]. Furthermore, recent biochemical analyses of isolated glomeruli have revealed that, in diabetic rats, the level of glomerular Ang II peptide is increased due to an increased level of AGT protein and an increase in the formation of Ang II via an unidentified enzymatic pathway that does not involve ACE within glomeruli [33].
AGT is the only known substrate for renin, the rate-limiting enzyme of the RAS, and the amount of AGT is therefore an essential determinant for the amount of tissue-based Ang II production and tissue RAS activity [7]. However, the specific cellular origins of AGT and the activation mode of the RAS that leads to Ang II formation within the glomerulus remain to be fully elucidated. A remarkable study by Lee et al. using a rat remnant model reported that, as a result of hemodynamic changes, injured or activated GEC synthesizes AGT, which triggers a cascade from the glomerular generation of Ang II–TGF-β and ECM protein gene expression, which results in the development of segmental glomerular sclerotic lesions [34]. This pathological progression can be prevented by ARB, which indicates that Ang II–AT1R signaling plays a central role in disease progression in this rat model. We have investigated the potential role of the glomerular Ang II–TGF-β pathway (profibrotic pathway) in pathological glomerular ECM remodeling during the progression of CKD [35–38]. Recently, we performed an immunohistochemical analysis using renal biopsy samples of immunoglobulin A nephropathy (IgAN) and minor glomerular abnormalities (MGA) that were found in the early stage of disease by a school urinary screening system in Japan [30]. Glomerular AGT is weakly expressed by GEC in MGA patients, but strongly induced at endocapillary sites including GEC and MC in IgAN patients, although they have normal glomerular filtration rate and BP (Fig. 3). The level of glomerular AGT parallels the levels of glomerular Ang II and TGF-β expression in diseased glomeruli. The level of glomerular injury, such as cell proliferation, ECM accumulation and proteinuria, is also closely correlated with the levels of AGT and Ang II. Additionally, the AGT level seems to determine the Ang II level in nephritic glomeruli. Furthermore, several cell culture studies, including ours, have shown that Ang II can stimulate AGT mRNA and AGT protein biosynthesis by renal cells, suggesting that its action might constitute an auto-amplifying loop of the activity of the intrarenal RAS [7, 30]. Therefore, we postulate that even in the early stage of IgAN, glomerular RAS activation seems to occur as a result of increased GEC- and MC-AGT expression and promotes to the enhanced local generation of Ang II, which leads to clinical and pathological abnormalities. A glomerular Ang II–AGT-positive feedback loop might drive RAS activation for further glomerular injury. The substantial association between glomerular RAS activity (Ang II generation) and glomerular TGF-β, ROS generation and pathological alterations was then investigated using a rat model of crescentic glomerulonephritis (GN) in combination with treatment with ARB (candesartan) [39]. For the ROS-generating system, we focused on the expression of Nox2, a major component of NAD(P)H oxidase, which is well known to be a major source of ROS in the diseased kidney and is activated by Ang II stimulation [37]. Vehicle-treated nephritic rats showed significant proteinuria and severe crescentic nephritis while treatment with ARB significantly attenuated proteinuria, glomerular Ang II accumulation, superoxide production and associated pathological alterations (Fig. 4). Consistent with these histological findings, a biochemical analysis using isolated glomeruli revealed that glomerular production of AGT, Ang II, and TGF-β and Nox2 was enhanced in nephritic rats while treatment with ARB significantly reduced the production of each of these in glomeruli to close to the control level (Fig. 5). These results suggest that a glomerular Ang II-generating system works in vivo and the produced Ang II induces TGF-β expression and superoxide, and finally contributes to the development of crescentic GN in rats. Similarly, Nakamura et al. [40] reported that salt-sensitive glomerular injury in eNOS−/− mice was mediated by the enhancement of glomerular GEC-AGT-derived Ang II, through the augmentation of glomerular oxidative stress. Taken together, these findings suggest that GEC-AGT expression plays a key role in glomerular RAS activation followed by glomerular pathological alterations in CKD.
Fig. 3.
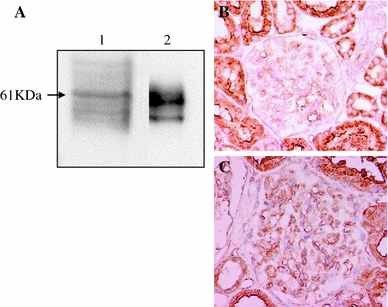
Protein expression of angiotensinogen (AGT) in isolated human glomeruli and immunohistochemical staining of AGT in patients with minor glomerular abnormalities (MGA) or IgA nephropathy (IgAN). a Western blot analysis was performed using samples of isolated human glomeruli (lane 1) and purified human AGT (lane 2), respectively. Anti-human AGT antibody reacted with a 61 kDa band in each sample. b In patients with MGA, AGT was strongly expressed in proximal tubules and weakly detected in glomerular endothelial cells. c In patients with IgAN, AGT expression was strongly induced by glomerular endothelial cells and mesangial cells. Modified from Ref. [30]
Fig. 4.
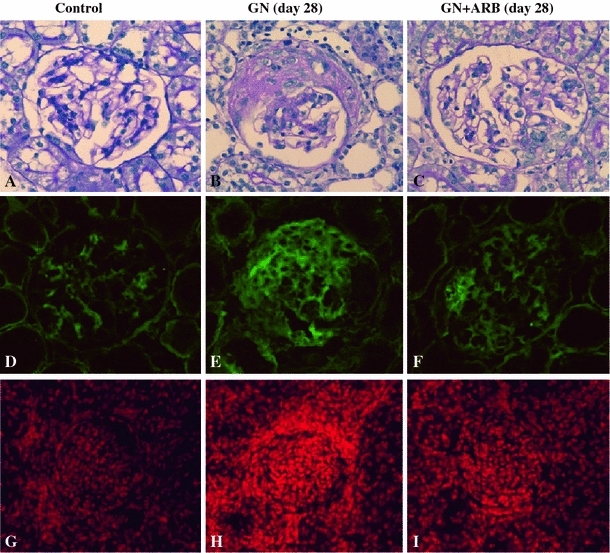
Effects of the ARB candesartan in anti-GBM antibody-induced nephritic rats. Nephritic rats were treated with or without candesartan, sacrificed on day 28, and then subjected to an immunohistochemical examination. Light microscopic examination showed that severe crescentic nephritis had developed by day 28 (b) but was significantly attenuated by treatment with ARB (c). PBS-injected rats were used as normal control rats (a, d, g). Immunostaining revealed that nephritic rats showed diffuse and strong glomerular Ang II staining (e), while ARB treated-nephritic rats showed segmental accentuated staining of Ang II (f). Control rats showed weak positive Ang II staining (d). Strong superoxide production (DHE dye) was detected in nephritic rats (h) compared with control rats (g), but was significantly attenuated in ARB treated-nephritic rats (i). Modified from Ref. [39]
Fig. 5.
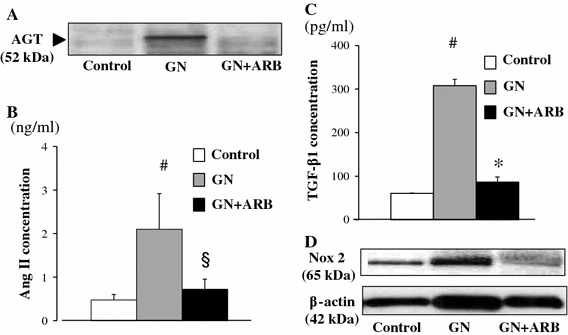
Biochemical analysis of nephritic rats on day 28 with or without treatment with ARB. Samples from isolated glomeruli from either control rats, day 28 nephritic rats or ARB-treated day 28 nephritic rats were subjected to Western blot analysis using anti-AGT antibody (a), Ang II measurement using ELISA (b), TGF-β measurement using ELISA (c) and Western blot analysis using anti-Nox2 antibody (d). Control control rats, GN nephritic rats without ARB treatment, GN + ARB nephritic rats with ARB treatment. # p < 0.01 versus control; § p < 0.05 versus GN; *p < 0.01 versus GN. Modified from Ref. [39]
Glomerular Ang II production is also regulated by the expression ratio of ACE to ACE2 within the glomerulus [27]. ACE2 plays a primary role in converting Ang II to Ang (1–7), which mediates vasodilation, antiproliferative, and antifibrotic actions via Mas receptor, and therefore has the potential to counterbalance the effects of ACEs [17]. ACE2 is now considered to be an endogenous ACEI [41]. In the diabetic mouse and human kidney, increased ACE expression and decreased ACE2 expression is observed in damaged glomeruli and indicates that the glomerular ACE/ACE2 balance plays a role in mediating glomerular injury, possibly by increasing the glomerular accumulation of Ang II [27, 42]. Notably, male ACE2 mutant (ACE2−/y) mice with an increase in the renal tissue Ang II level develop glomerulosclerosis [41]. Sensitive indicators of ROS production, lipid peroxidation products and the glomerulosclerosis score were markedly enhanced in those mice while ARB prevented these increases, which strongly supports the notion that ACE2 plays a role in Ang II-induced glomerular injury. More recently, a similar relationship between ACE2 and ACE expression in diseased glomeruli was reported even in patients with IgAN [43].
New approach for the analysis of Ang peptides generated by the glomerular RAS pathway
Since RAS is a far more complex and dynamic system than was originally recognized, assays that are more selective, sensitive, and rapid than conventional radioimmunoassay and high-performance liquid chromatographic separation of peptide products are needed for the identification of RAS components and peptide-enzymatic cascades in RAS. The emergence of matrix-assisted laser desorption ionization time of flight (MALDI-TOF) mass spectrometry (MS) allows us to clarify Ang metabolism with more specificity and ease than with previous methods. Recently, Velez et al. examined the metabolism of Ang I in freshly isolated intact rat glomeruli using MALDI-TOF-MS [10, 44, 45]. They showed that there is prominent glomerular conversion of Ang I–Ang (2–10) and Ang (1–7), mediated by AP-A and NEP, respectively, and suspected that the formation of these alternative Ang peptides may be critical for counterbalancing the local actions of Ang II within glomeruli. They then examined the contribution of POD or GEC to Ang metabolism in glomerulus using MALDI-TOF-MS in combination with cell culture methods [45, 46]. They demonstrated that POD expressed a functional intrinsic RAS characterized by AGT, NEP, AP-A, ACE2, and renin activities, which predominantly lead to Ang (1–7) and Ang (1–9) formation, as well as Ang II degradation [45]. In contrast, GEC exhibited prominent ACE activity leading to Ang II, with the production of less Ang (1–7) and thus a lower degradative ability of Ang II [46], suggesting that injury to specific cell types in the glomeruli may lead to distinct effects on the glomerular RAS balance. In addition, many studies have reported that MC also express a functional intrinsic RAS characterized by AGT, prorenin, cathepsin B (a potential enzyme involved in renin activation), chymase, ACE, and ACE2, which primarily generates Ang II and very small amounts of Ang (1–7) and Ang (1–9) [45, 47, 48, 49]. Taken together, these findings suggest that variations in glomerular cell injury and the relative abundance of Ang I metabolites such as Ang II, Ang (1–7), Ang (1–9) and Ang (2–10) within glomeruli determine the net autocrine or paracrine effects of these Ang peptides on glomerular cells. A detailed analysis of the paramount enzymatic pathway in the RAS responsible for the conversion to these Ang peptides may provide therapeutic tools such as ACEIs for Ang II-mediated glomerular diseases.
Conclusion and future directions
There is no controversy regarding the concept that the glomerulus-based RAS plays a role in glomerular physiology and pathophysiology. Enhanced glomerular Ang II action in diseased glomeruli via ACE/Ang II/AT1R signaling promotes cell proliferation and ECM production, and decreases ECM degradation resulting in sclerotic lesions. Evidence in animal and human CKD has shown that RAS blockers such as ACEIs and ARBs are an effective and promising therapy for attenuating the progression of CKD beyond BP-lowering effect, which supports the above discussion. Several technical advances, including the use of molecular biology, peptide chemistry and the availability of transgenic and knock-out mice with altered expression of RAS components, have given us a more complex view of a glomerular RAS composed of a variety of peptidases, Ang peptides, and receptors involved in these Ang actions. The modulation of RAS pathways such as ACE2/Ang (1–7)/Mas receptor and PRR might become future therapeutic targets in CKD. Moreover, the identification of a glomerulus-specific enzymatic pathway for RAS activation could lead to a therapeutic strategy for attenuating the progression of glomerular disease in CKD.
Acknowledgments
SK is a recipient of a Grant-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan.
Conflict of interest
The author of this manuscript has no conflict of interest to disclose.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Tigerstedt R, Bergman PG. Niere und Kreislauf. Skand Arch Physiol. 1898;8:223–271. [Google Scholar]
- 2.Bader M. Tissue renin-angiotensin-aldosterone systems: targets for pharmacological therapy. Annu Rev Pharmacol Toxicol. 2010;50:439–465. doi: 10.1146/annurev.pharmtox.010909.105610. [DOI] [PubMed] [Google Scholar]
- 3.Dzau VJ. Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37:1047–1052. doi: 10.1161/01.hyp.37.4.1047. [DOI] [PubMed] [Google Scholar]
- 4.Bader M, Ganten D. Update on tissue renin-angiotensin systems. J Mol Med (Berl) 2008;86:615–621. doi: 10.1007/s00109-008-0336-0. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003;35:881–900. doi: 10.1016/S1357-2725(02)00271-6. [DOI] [PubMed] [Google Scholar]
- 6.Yosypiv IV. Renin-angiotensin system in ureteric bud branching morphogenesis: insights into the mechanisms. Pediatr Nephrol. 2011;26:1499–1512. doi: 10.1007/s00467-011-1820-2. [DOI] [PubMed] [Google Scholar]
- 7.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 8.Lai KN, Leung JC, Tang SC. The renin-angiotensin system (diabetes and the kidney). In: Lai KN, Tang SC, editors. Contributions to nephrology. Basel: Karger; 2011. vol. 170, p. 135–44. [DOI] [PubMed]
- 9.Warner FJ, Lubel JS, McCaughan GW, Angus PW. Liver fibrosis: a balance of ACEs? Clin Sci (Lond) 2007;113:109–118. doi: 10.1042/CS20070026. [DOI] [PubMed] [Google Scholar]
- 10.Velez JC. The importance of the intrarenal renin-angiotensin system. Nat Clin Pract Nephrol. 2009;5:89–100. doi: 10.1038/ncpneph1015. [DOI] [PubMed] [Google Scholar]
- 11.Rüster C, Wolf G. Angiotensin II as a morphogenic cytokine stimulating renal fibrogenesis. J Am Soc Nephrol. 2011;22:1189–1199. doi: 10.1681/ASN.2010040384. [DOI] [PubMed] [Google Scholar]
- 12.Mustafa MR, Dharmani M, Kunheen NK, Sim MK. Effects of des-aspartate-angiotensin I on the actions of angiotensin III in the renal and mesenteric vasculature of normo- and hypertensive rats. Regul Pept. 2004;120:15–22. doi: 10.1016/j.regpep.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 13.Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chai SY, Fernando R, Peck G, Ye SY, Mendelsohn FA, Jenkins TA, et al. The angiotensin IV/AT4 receptor. Cell Mol Life Sci. 2004;61:2728–2737. doi: 10.1007/s00018-004-4246-1. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–1427. doi: 10.1172/JCI14276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen G, Muller DN. The biology of the (pro)renin receptor. J Am Soc Nephrol. 2010;21:18–23. doi: 10.1681/ASN.2009030300. [DOI] [PubMed] [Google Scholar]
- 17.Ferrario CM, Varagic J. The ANG-(1–7)/ACE2/mas axis in the regulation of nephron function. Am J Physiol Ren Physiol. 2010;298:F1297–F1305. doi: 10.1152/ajprenal.00110.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases—insights from animal models. Kidney Int. 2005;67:404–419. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- 19.Qian Y, Feldman E, Pennathur S, Kretzler M, Brosius FC., 3rd From fibrosis to sclerosis: mechanisms of glomerulosclerosis in diabetic nephropathy. Diabetes. 2008;57:1439–1445. doi: 10.2337/db08-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brenner BM. Remission of renal disease: recounting the challenge, acquiring the goal. J Clin Invest. 2002;110:1753–1758. doi: 10.1172/JCI200217351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taal MW, Brenner BM. Renoprotective benefits of RAS inhibition: from ACEI to angiotensin II antagonists. Kidney Int. 2000;57:1803–1817. doi: 10.1046/j.1523-1755.2000.00031.x. [DOI] [PubMed] [Google Scholar]
- 22.Wühl E, Schaefer F. Therapeutic strategies to slow chronic kidney disease progression. Pediatr Nephrol. 2008;23:705–716. doi: 10.1007/s00467-008-0789-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seikaly MG, Arant BS, Jr, Seney FD., Jr Endogenous angiotensin concentrations in specific intrarenal fluid compartments of the rat. J Clin Invest. 1990;86:1352–1357. doi: 10.1172/JCI114846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atiyeh BA, Arant BS, Jr, Henrich WL, Seikaly MG. In vitro production of angiotensin II by isolated glomeruli. Am J Physiol. 1995;268(2 Pt 2):F266–F272. doi: 10.1152/ajprenal.1995.268.2.F266. [DOI] [PubMed] [Google Scholar]
- 25.Lai KN, Leung JC, Lai KB, To WY, Yeung VT, Lai FM. Gene expression of the renin-angiotensin system in human kidney. J Hypertens. 1998;16:91–102. doi: 10.1097/00004872-199816010-00014. [DOI] [PubMed] [Google Scholar]
- 26.Del Prete D, Gambaro G, Lupo A, Anglani F, Brezzi B, Magistroni R, Graziotto R, Furci L, Modena F, Bernich P, Albertazzi A, D’Angelo A, Maschio G. Precocious activation of genes of the renin-angiotensin system and the fibrogenic cascade in IgA glomerulonephritis. Kidney Int. 2003;64:149–159. doi: 10.1046/j.1523-1755.2003.00065.x. [DOI] [PubMed] [Google Scholar]
- 27.Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D. Glomerular localization and expression of angiotensin-converting enzyme 2 and angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol. 2006;17:3067–3075. doi: 10.1681/ASN.2006050423. [DOI] [PubMed] [Google Scholar]
- 28.Wagner J, Gehlen F, Ciechanowicz A, Ritz E. Angiotensin II receptor type 1 gene expression in human glomerulonephritis and diabetes mellitus. J Am Soc Nephrol. 1999;10:545–551. doi: 10.1681/ASN.V103545. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki K, Han GD, Miyauchi N, Hashimoto T, Nakatsue T, Fujioka Y, et al. Angiotensin II type 1 and type 2 receptors play opposite roles in regulating the barrier function of kidney glomerular capillary wall. Am J Pathol. 2007;170:1841–1853. doi: 10.2353/ajpath.2007.060484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takamatsu M, Urushihara M, Kondo S, Shimizu M, Morioka T, Oite T, et al. Glomerular angiotensinogen protein is enhanced in pediatric IgA nephropathy. Pediatr Nephrol. 2008;23:1257–1267. doi: 10.1007/s00467-008-0801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arai M, Wada A, Isaka Y, Akagi Y, Sugiura T, Miyazaki M, et al. In vivo transfection of genes for renin and angiotensinogen into the glomerular cells induced phenotypic change of the mesangial cells and glomerular sclerosis. Biochem Biophys Res Commun. 1995;206:525–532. doi: 10.1006/bbrc.1995.1075. [DOI] [PubMed] [Google Scholar]
- 32.Shi L, Nikolic D, Liu S, Lu H, Wang S. Activation of renal renin-angiotensin system in upstream stimulatory factor 2 transgenic mice. Am J Physiol Ren Physiol. 2009;296:F257–F265. doi: 10.1152/ajprenal.90493.2008. [DOI] [PubMed] [Google Scholar]
- 33.Singh R, Singh AK, Leehey DJ. A novel mechanism for angiotensin II formation in streptozotocin-diabetic rat glomeruli. Am J Physiol Ren Physiol. 2005;288:F1183–F1190. doi: 10.1152/ajprenal.00159.2003. [DOI] [PubMed] [Google Scholar]
- 34.Lee LK, Meyer TW, Pollock AS, Lovett DH. Endothelial cell injury initiates glomerular sclerosis in the rat remnant kidney. J Clin Invest. 1995;96:953–964. doi: 10.1172/JCI118143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kagami S, Border WA, Miller DE, Noble NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-beta expression in rat glomerular mesangial cells. J Clin Invest. 1994;93:2431–2437. doi: 10.1172/JCI117251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kagami S, Kuhara T, Okada K, Kuroda Y, Border WA, Noble NA. Dual effects of angiotensin II on the plasminogen/plasmin system in rat mesangial cells. Kidney Int. 1997;51:664–671. doi: 10.1038/ki.1997.96. [DOI] [PubMed] [Google Scholar]
- 37.Kondo S, Shimizu M, Urushihara M, Tsuchiya K, Yoshizumi M, Tamaki T, et al. Addition of the antioxidant probucol to angiotensin II type I receptor antagonist arrests progressive mesangioproliferative glomerulonephritis in the rat. J Am Soc Nephrol. 2006;17:783–794. doi: 10.1681/ASN.2005050519. [DOI] [PubMed] [Google Scholar]
- 38.Urushihara M, Takamatsu M, Shimizu M, Kondo S, Kinoshita Y, Suga K, et al. ERK5 activation enhances mesangial cell viability and collagen matrix accumulation in rat progressive glomerulonephritis. Am J Physiol Ren Physiol. 2010;298:F167–F176. doi: 10.1152/ajprenal.00124.2009. [DOI] [PubMed] [Google Scholar]
- 39.Kinoshita Y, Kondo S, Urushihara M, Suga K, Matsuura S, Takamatsu M, et al. Angiotensin II type I receptor blockade suppresses glomerular renin-angiotensin system activation, oxidative stress, and progressive glomerular injury in rat anti-glomerular basement membrane glomerulonephritis. Transl Res. 2011; 158: 235−248. [DOI] [PubMed]
- 40.Nakamura T, Kataoka K, Tokutomi Y, Nako H, Toyama K, Dong YF, et al. Novel mechanism of salt-induced glomerular injury: critical role of eNOS and angiotensin II. J Hypertens. 2011;29:1528–1535. doi: 10.1097/HJH.0b013e328348ca95. [DOI] [PubMed] [Google Scholar]
- 41.Oudit GY, Herzenberg AM, Kassiri Z, Wong D, Reich H, Khokha R, et al. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am J Pathol. 2006;168:1808–1820. doi: 10.2353/ajpath.2006.051091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reich HN, Oudit GY, Penninger JM, Scholey JW, Herzenberg AM. Decreased glomerular and tubular expression of ACE2 in patients with type 2 diabetes and kidney disease. Kidney Int. 2008;74:1610–1616. doi: 10.1038/ki.2008.497. [DOI] [PubMed] [Google Scholar]
- 43.Mizuiri S, Hemmi H, Arita M, Aoki T, Ohashi Y, Miyagi M, et al. Increased ACE and decreased ACE2 expression in kidneys from patients with IgA nephropathy. Nephron Clin Pract. 2011;117:c57–c66. doi: 10.1159/000319648. [DOI] [PubMed] [Google Scholar]
- 44.Velez JC, Ryan KJ, Harbeson CE, Bland AM, Budisavljevic MN, Arthur JM, et al. Angiotensin I is largely converted to angiotensin (1–7) and angiotensin (2–10) by isolated rat glomeruli. Hypertension. 2009;53:790–797. doi: 10.1161/HYPERTENSIONAHA.109.128819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Velez JC, Bland AM, Arthur JM, Raymond JR, Janech MG. Characterization of renin-angiotensin system enzyme activities in cultured mouse podocytes. Am J Physiol Ren Physiol. 2007;293:F398–F407. doi: 10.1152/ajprenal.00050.2007. [DOI] [PubMed] [Google Scholar]
- 46.Velez JC, Janech MG, Arthur JM, Raymond JR. Cultured human glomerular endothelial cells display ACE-mediated angiotensin-II-generating capacity and limited angiotensin-II-degrading activity. Am Soc Nephrol Annual Meeting; 2010 (in abstract).
- 47.Singh R, Singh AK, Alavi N, Leehey DJ. Mechanism of increased angiotensin II levels in glomerular mesangial cells cultured in high glucose. J Am Soc Nephrol. 2003;14:873–880. doi: 10.1097/01.ASN.0000060804.40201.6E. [DOI] [PubMed] [Google Scholar]
- 48.Cristovam PC, Arnoni CP, de Andrade MC, Casarini DE, Pereira LG, Schor N, et al. ACE-dependent and chymase-dependent angiotensin II generation in normal and glucose-stimulated human mesangial cells. Exp Biol Med. 2008;233:1035–1043. doi: 10.3181/0708-RM-229. [DOI] [PubMed] [Google Scholar]
- 49.Aragão DS, Cunha TS, Arita DY, Andrade MC, Fernandes AB, Watanabe IK, et al. Purification and characterization of angiotensin converting enzyme 2 (ACE2) from murine model of mesangial cell in culture. Int J Biol Macromol. 2011;49:79–84. doi: 10.1016/j.ijbiomac.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]