

Abstract
Though nanomaterials such as carbon nanotubes have gained recent attention in biology and medicine, there are few studies at the single-molecule level that explore their interactions with disease-causing proteins. Using atomistic molecular-dynamics simulations, we have investigated the interactions of the monomeric Aβ1-42 peptide with a single-walled carbon nanotube of small diameter. Starting with peptide-nanotube complexes that delineate the interactions of different segments of the peptide, we find rapid convergence in the peptide's adsorption behavior on the nanotube surface, manifested in its arrested movement, the convergence of peptide-nanotube contact areas and approach distances, and in increased peptide wrapping around the nanotube. In systems where the N-terminal domain is initially distal from nanotube, the adsorption phenomena are initiated by interactions arising from the central hydrophobic core, and precipitated by those arising from the N-terminal residues. Our simulations and free energy calculations together demonstrate that the presence of the nanotube increases the energetic favorability of the open state. We note that the observation of peptide localization could be leveraged for site-specific drug delivery, while the decreased propensity of collapse appears promising for altering kinetics of the peptide's self-assembly.
Introduction
According to the Amyloid hypothesis, self-assembly of the 39–42 residue Amyloid beta (Aβ) peptide into insoluble aggregates is associated with the onset of the dreaded Alzheimer's disease (1). The Aβ peptide is created from exact proteolysis of the amyloidogenic sequence within the amyloid precursor protein. Though the production rates of the 42-residue peptide are approximately eight times lower than the 40-residue form, it has greater propensity to oligomerize and aggregate and is associated with far greater neurotoxic effects (2,3). There is evidence that the peptide's self-assembly is influenced by mutations, changes in the solvent environment, and thermodynamics conditions (4–7). Interestingly, a number of emerging studies suggest that the soluble, oligomeric intermediates could be more toxic than the insoluble aggregates (2,8,9).
Structural and computational studies indicate that residues belonging to three contiguous regions within the Aβ peptide play prominent roles in its self-assembly: the central hydrophobic core spanning the residues L17VFFA21 (6,10,11); the turn region spanning V24GSN27 (12,13); and an additional C-terminal hydrophobic domain thought to span a subset of the residues G29AIIGLMVGGVVIA42 (6,11). The importance of these regions and the interplay of their dynamics has been demonstrated by several experimental and computational studies (6,10,14,15). Self-assembly of the peptide includes a number of key steps that involve the interplay of enthalpic (favoring association) and entropic (favoring dissociation) effects, and at optimal thermodynamic conditions result in the formation of insoluble fibrils, wherein the two strands of the peptide separated by the turn region form a collapsed, hairpin-like structure (10,14).
Interactions between the peptide's hydrophobic domains leading to its collapse is thus considered one of the key steps in Aβ self-assembly, whether into soluble oligomers or into insoluble aggregates (6,15). Efforts to interfere with or destabilize the self-assembly of the peptide have included approaches that impose external perturbations to the physico-chemical behavior of these domains. Because the central hydrophobic core is thought to play a key role in the adhesion of peptide units, it had been suggested that fibrillar extension could be prevented with the binding of small peptides or ligands to this patch (10). It has been shown that potential Alzheimer's disease drug candidates preferentially interact with and stabilize the central hydrophobic core of the peptide, and perhaps work by preventing conformational changes that could lead to amyloid formation (16). Interestingly, a very recent study points out the inverse correlation between the strength of ligand binding to this core and the aggregation rate (17).
The convergence of many unique properties in carbon nanotubes (CNTs) (such as ordered structure, high aspect ratio and surface area, ultralight weight, high mechanical strength, etc.) has opened up several avenues of their applicability (18–20). Particularly in biology, CNTs are, as of this writing, being considered for usage in biomolecular sensors and in biomedical devices (21,22), as well as in therapeutics, either for direct interaction with disease-causing biomolecules (23) or for site-specific drug delivery (24). Such potential applications have necessitated detailed studies of CNT interactions with peptides, proteins, sugars, membrane lipids, and nucleic acids, and in fact, a number of recent studies demonstrate interesting structural, conformational, and functional changes in biological molecules adsorbed on CNT surfaces (25–29). Molecular dynamics (MD) simulations have demonstrated how single-walled carbon nanotubes (SWCNTs) induce curvature increases in biomolecules around them, leading to the observation of biomolecular wrapping around nanotubes (25–28). SWCNTs have also been shown to interfere with backbone hydrogen bonding of helical peptides, and the dimensions of the SWCNT has been shown to have an effect on the interaction propensities (25,26,30–32).
Experimentally, it has been shown that CNTs (and related nanoparticles such as fullerenes) affect protein aggregation kinetics (33). This is noteworthy in light of a number of earlier studies that have implicated π-π stacking of aromatic residues in peptide self-assembly (34–36), and as inhibition of amyloid formation has oftentimes been attributed to the interference by drug molecules to the interactions between aromatic side chains of nearby peptide units (37,38). Conversely, very recent experimental work involving mutations of Phe19 and Phe20 of Aβ1-42 with aliphatic side chains such as Leu and Ile show that aggregation propensities remain unchanged (or even enhanced) (39,40). Interestingly, substitution of the aromatic residues with Valine causes a decreased stability of the aggregates (40). Detailed simulation studies have already shown that oligomeric Aβ fragments belonging to the second hydrophobic domain (G25SNKGAIIGLM35) form barrel-like structures on SWCNTs surfaces and can hinder further self-assembly (41). However, as noted in that study, details of SWCNT interactions with other important regions of the peptide (such as the central hydrophobic core and the C-terminal hydrophobic tail) require further investigations.
The body of research discussed above suggests that CNTs could significantly affect the intrinsic behavior of disease causing peptides. However, barring a few insightful studies that describe the interaction mechanisms between CNTs and such peptide candidates (29,33,41), this area of research remains largely unexplored. As mentioned earlier, CNTs can be used either directly as site-specific drug-delivery agents, or as entities that modulate the conformational landscape of amyloidogenic proteins, thus interfering with self-assembly and amyloid formation. In this article, we report results obtained with fully atomistic MD simulations of the full-length, Aβ1-42 peptide (PDB:1Z0Q) in proximity to SWCNTs in aqueous environment. We have carried out a total of nine simulations of 80 ns each, with five different relative orientations of the peptide's central hydrophobic core and the second, C-terminal hydrophobic domain with the SWCNT. The five configurations differ in their initial peptide-SWCNT contact areas, as well as distances of the important hydrophobic regions from the SWCNT; the contact area was initially the least (greatest) and the distance from the central hydrophobic core was the greatest (least) when the second (first) helical region was placed parallel to the SWCNT. Further, a control simulation of the same trajectory length has been generated for the pure peptide in absence of SWCNT.
We find that the central hydrophobic region and the first 16 residues at the N-terminus play an important role in influencing the interactions with the SWCNT. Though these regions are distal to the nanotube in two of the setups, the interaction strength is such that there is rapid convergence in the peptide-SWCNT distance and contact areas with the values obtained with the other setups. For all simulations, there is a manifold increase in the peptide-SWCNT contact area within tens of nanoseconds, resulting in similar wrapping propensities around the SWCNT. Our adaptive biasing force (ABF)-based calculations of the free energies as a function of the distance between centers of mass of the Aβ strands (i.e., the collapse distance), performed with the setups where the SWCNT is parallel to either of the hydrophobic domains, demonstrates that the presence of SWCNTs hinders the propensity of peptide collapse. This is corroborated by the converged, high distance of collapse obtained with all the simulation trajectories. We discuss the implications of peptide localization and adsorption, and the thermodynamic favorability of the open relative to the closed states in the presence of SWCNT, in the development of methods to prevent self-assembly of Aβ peptides.
Materials and Methods
Setup of peptide-SWCNT complexes
We have considered an armchair (6,6), single-walled carbon nanotube (SWCNTs) of length 99.5 Å, radius 4.1 Å, containing 984 carbon atoms. The SWCNT coordinates were obtained using the VMD package (42). To avoid artifacts that may arise due to edge effects, periodic boundary conditions were used to create infinitely long SWCNTs parallel to the x axis; this approach has been adopted in relatively recent simulation studies (25). The coordinates for the Aβ1-42 peptide were obtained from the PDB database (PDB:1Z0Q) (7). These coordinates, obtained with solution NMR studies in a 3:7 mixture of hexafluoro-2-propanol and water, correspond to a largely helical form of the monomeric peptide. The structure consists of a long N-terminal helix (S8GYEVHHQKLVFFAEDVG25) and a shorter, C-terminal helix (K28GAIIGLMVGG38), separated by a two-residue turn (S26N27). In our first system (referred to henceforth as NT1), the SWCNT axis is made parallel to the vector joining the Cα atoms of the 8th and the 25th residues. In our second system, (referred to henceforth as NT2), the SWCNT axis is made parallel to the vector joining the Cα atoms of the 28th and the 38th residues. Three more systems (referred to henceforth as NT3, NT4, and NT5) were created with either helical domain oriented at 30° or 45° relative to the SWCNT.
In all the setups, the SWCNT axis is set at exactly 12.6 Å away from the center of mass of the closest helix; thus, the SWCNT surface is, in all cases, 8.5 Å away from the respective center of mass. The initial setups are depicted in Fig. 1. The relative orientations of the two helices with the SWCNT in each setup, the distances and angles of the two helical domains relative to the SWCNT, are provided in Table S2 in the Supporting Material. The distances of residues 8–38 from the SWCNT in the setups are provided in Table S3 and Table S4.
Figure 1.
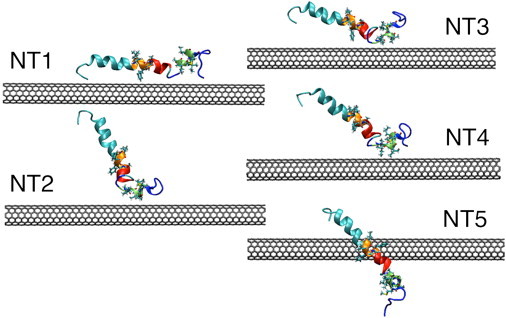
(Color online) Setups of the Aβ1-42 peptide-SWCNT complex. Segments L17VFFAEDVGS26 (red-orange) and K28GAIIGLMVGGVVIA42 (blue-green). L17VFFA21 (the central hydrophobic core, HP1) lies at one end to the first patch (highlighted in orange), and A30IIGLM35 (HP2) lies within the second patch (highlighted in green). The side chains of these segments are shown in line representation.
Throughout the simulations and the free energy calculations, the SWCNTs were held fixed at its setup position with a constant harmonic force with a force constant of 2 kcal mol−1. The Aβ1-42 peptide is characterized by the presence of two important hydrophobic regions that have been referred to throughout this article as HP1 and HP2. The central hydrophobic core, comprised of residues 17–21 (LVFFA), has been designated “HP1”. Of the C-terminal residues, the importance of residues 30–35 (A30IIGLM35) has often been discussed (2,16,43). We refer to the residue stretch of 30–35 as “HP2”. A control simulation for the peptide was also performed. The NH3+ and COO− groups were added to the N- and C-termini, respectively, of the peptide in all six setups, and each system was solvated with the TIP3P water model (44). Three sodium counterions were added to neutralize each system. The dimensions of the periodic box and the number of water molecules added to each system are provided in Table S1.
MD simulations and free energy calculations
Molecular dynamics (MD) simulations were performed with NAMD2.7 simulation package (45) and visualizations were made using the VMD tool (42). The CHARMM22 all-atom force field with the CMAP correction was used (46,47). Bond lengths involving hydrogen atoms were held fixed using the SHAKE algorithm (48). Energy minimization, using the conjugate gradient technique, was first performed for 10,000 steps on all the systems. After this, simulations were carried out in the isothermal-isobaric (NPT) ensemble. Three independent trajectories of 80-ns duration were generated for the NT1 and NT2 simulations, and one each for NT3, NT4, NT5, and the free peptide. Thus, the total simulation data for the peptide-nanotube complex were for 720 ns. A constant temperature of 310 K was maintained using Langevin dynamics with a collision frequency of 1 ps−1, and a 1 atmosphere pressure was maintained using the Langevin piston Nosé-Hoover method (49,50). A timestep of 2 fs was used. Three-dimensional orthorhombic periodic boundary conditions were employed. Electrostatic interactions were calculated with the particle-mesh Ewald method (51). The cutoff distance for nonbonded interactions were set to 12 Å with a smoothing function employed from 10.5 Å.
The adaptive biasing force (ABF) method, as implemented in the NAMD2.7 package, was used to calculate the free energy profiles of the NT1 and NT2 systems, as well as for the pure peptide (52,53). Unlike the umbrella sampling technique, this method for calculating the potential of mean force (PMF) requires no prior knowledge of the free energy surface, and has been used in a number of recent studies to probe conformational changes in biomolecular systems, using a variety of spatial reaction coordinates (54–57). In our calculations, the center-of-mass distance of the residue segments L17VFFAEDVGS26 and K28GAIIGLMVGGVVIA42 was taken as the reaction coordinate to characterize the peptide's collapse. This distance, dcollapse, spanned a separation of 8–21 Å, which was divided into 52 windows, each 0.25 Å wide. The free energy profiles used for comparing barrier heights converged over a period of at least 18 ns. The total sampling time for the NT1 and NT2 systems was 180 ns and 198 ns, respectively, while it was 130 ns for the pure peptide.
Results and Discussion
Peptide localization and adsorption on the SWCNT surface
We began by comparing the center-of-mass motion of the peptide relative to its position at the setup configuration (time = 0 ns), when in the free state, and when in complex with the SWCNT. We find that irrespective of the initial configuration, diffusion of the peptide is markedly restricted in the presence of the SWCNT. Further, the contact angle between HP1 and the SWCNT is restricted to relatively low values (∼15°) within few tens of nanoseconds. See Supporting Material for greater details.
It has been observed that amphiphilic peptides have a distinctive tendency to adsorb around the rigid hydrophobic surface of high curvature provided by CNTs (25). For the 42-residue Aβ peptide with relatively long, contiguous hydrophobic regions, the contact area with the SWCNT can be used as a quantitative measure for characterizing the extent of adsorption on to the nanotube. For our simulation trajectories, we have estimated this value by the method specified in recent reports (25,26), as half the difference between the sum of the solvent-accessible surface areas of the peptide and the SWCNT, and that of the peptide-SWCNT complex. A probe of 1.8 Å diameter is used to calculate the individual solvent accessible surface areas of the peptide, SWCNT, and the complexes.
In Fig. 2, we show temporal evolution of the full peptide-SWCNT contact area for the nine peptide-SWCNT simulations. Further, in Table S5 and Table S6, we report the values of the contact area with SWCNT for the full peptide, the N-terminal segment (residues 1–16), and regions HP1 and HP2, averaged over different time segments for all nine trajectories. Despite initial differences in dynamics in the early stages, all the peptide-SWCNT simulations show marked increase in the contact area; the final values attained are in the vicinity of 900 Å2. In Fig. S3 in the Supporting Material, we show the peptide configuration around the nanotube cross section at selected points along the trajectories. For the NT1, NT3, NT4, and NT5 simulations, the contact areas rise rapidly and attain saturation values within a few tens of nanoseconds. For NT2, the setup with the least initial contact area, the snapshots describe the role of the HP1 region in the peptide's wrapping around the nanotube.
Figure 2.
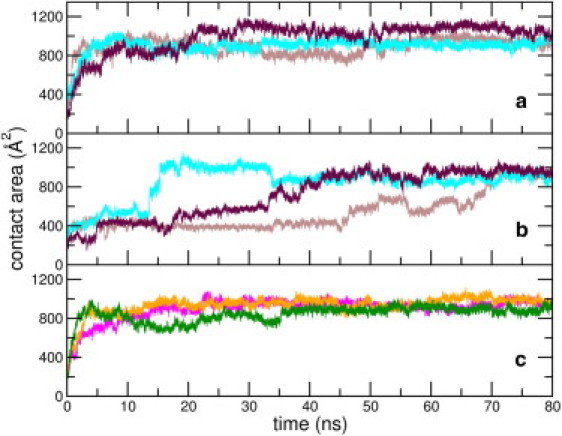
(Color online) Evolution of peptide-SWCNT contact area over (a) three NT1 trajectories, (b) three NT2 trajectories, and (c) NT3 (in green), NT4 (in magenta), and NT5 (in orange) trajectories. Snapshots of NT1 and NT2 shown in the Supporting Material correspond to the plotted data (in brown).
In Fig. 3, we have plotted the evolution of the mean radial distance of the centers of mass of HP1 and HP2 from the SWCNT surface for the NT1 and NT2 trajectories (see Fig. S4 for plots of the other systems). The radial distance between HP1 and the nanotube converges for all systems by the end of the simulations. For the HP2 patch, the corresponding changes are minor, and the radial distance from the nanotube remains largely constant for all trajectories. We note that for systems with low initial contact areas (NT2 and NT4), the decrease in the approach distance of HP1 to the nanotube precedes the increase in the peptide-SWCNT contact area. Thus, it appears that the adsorption mechanism initially involves an approaching closeness of the SWCNT with the central hydrophobic core. The eventual convergence of the radial distances and the contact areas suggest that the initial relative configuration between the peptide and the SWCNT is of minor importance in the adsorption mechanism.
Figure 3.
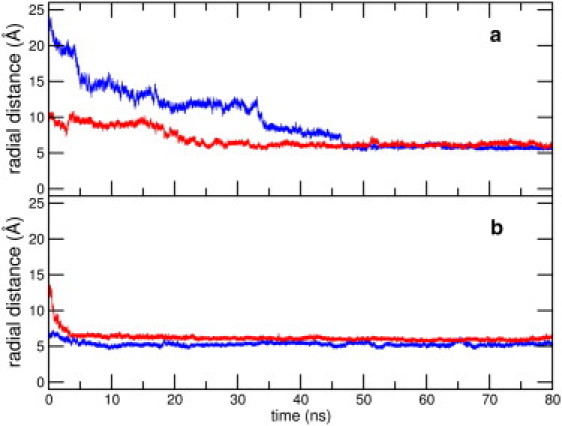
(Color online) Time evolution of the radial distance of SWCNT and with (a) HP1 and (b) HP2 segments of Aβ1-42 for the NT1 (in red) and NT2 (in blue) setups, averaged over three trajectories each.
Peptide-nanotube interaction energies
It is known that hydrophobic interactions are the dominant forces behind peptide and protein adsorption on fullerene balls, pristine graphene sheets, and CNTs (25,32,33). The different initial geometries used in our study are meant to delineate interactions of the important parts of the Aβ1-42 peptide with the SWCNT; however, we observe strong convergence in the peptide behavior in these systems within a few tens of nanoseconds. For understanding the differential contributions of the principle hydrophobic regions in the Aβ1-42 peptide in the observed adsorption mechanism, we have calculated for all the trajectories, the self, nonbonded energy of the peptide; the peptide's interaction energies with the SWCNT; and interaction energies of important hydrophobic regions of the peptide with the SWCNT.
We show, in Fig. 4 a, the total nonbonded energy of the peptide over the peptide-nanotube complexes, as well as for the control simulation of the pure peptide; data for NT1 and NT2 are averaged over three trajectories each. This energy is higher overall when in proximity to the SWCNT. Toward the end of the simulations, the total nonbonded energy of the peptide, averaged over all nine simulations with the SWCNT, is lesser than that of the pure peptide by ∼105 kcal mol−1. Not surprisingly, the peptide's bonded self-energy (the sum total of bond, angle, dihedral, and improper components) displays no significant differences among any of the simulated trajectories (data not shown). It is thus obvious that interactions with the SWCNT are responsible for the decreased stability of the peptide. In Fig. 4 b, we show evolutions of interaction energy of the nanotube with the full peptide. This interaction is found to stabilize rapidly for all, except the NT2 trajectories. Toward the end of the simulations, however, we find clear convergence in the interactions, irrespective of the initial configurations. The mean peptide-SWCNT interaction in the last 10 ns is ×128.8 (±11.2) kcal mol−1.
Figure 4.
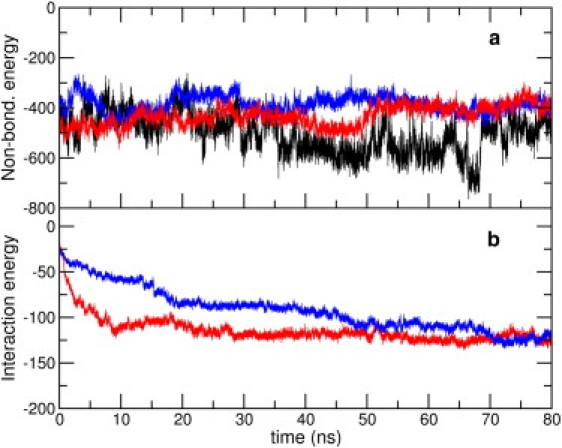
(a) (Color online) Total nonbonded energy of the full peptide along the control, NT1 and NT2 simulation trajectories. (b) Total interaction energy of the full peptide and the SWCNT along the NT1 and NT2 simulation trajectories. Energy units used are kcal mol−1. The data for NT1 and NT2 have been averaged over the three independent trajectories each.
To understand the role of key regions in the peptide's adsorption on the SWCNT, we have calculated the interaction energies of the N-terminal segment (residues 1–16), HP1, and HP2 with the SWCNT, for all the nine peptide-SWCNT trajectories. As seen from plots of these interactions in Figs. 5 and 6, corresponding interactions obtained from different trajectories converge toward the end of the simulations. Averaged over all trajectories, between 70 and 80 ns, the interaction with the N-terminal residues is −55.0 (±7.8) kcal mol−1; with the HP1 segment is −17.1 (±3.1) kcal mol−1; and with the HP2 segment is −16.2 (±2.6) kcal mol−1. For the NT2 simulations, we observe an interesting correlation between the interaction strengths and the peptide's adsorption on the SWCNT surface. Initially, we observe an onset in the interaction strength with the HP1 domain and a concurrent sharp drop in the distance from the SWCNT. This is followed by a more substantial increase in the interaction strength with the N-terminal domain, and the attainment of saturation values of the peptide-nanotube contact area. This reiterates the role of HP1 in initiating the adsorption process, whose interactions with the SWCNT appear to initiate the adsorption process by bringing the N-terminal domain closer to the nanotube.
Figure 5.
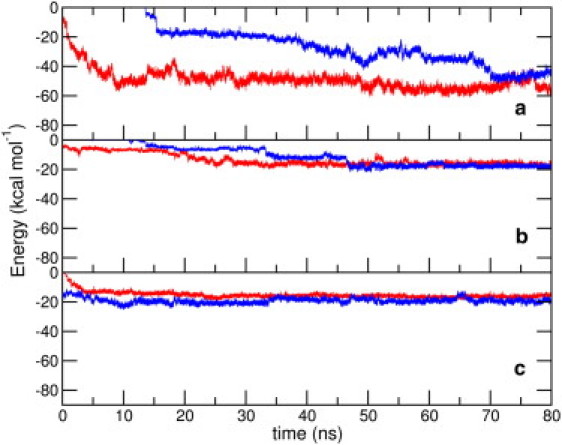
(Color online) Time evolution of interaction energies of the SWCNT with (a) residues 1–16, (b) HP1, and (c) HP2 segments of the peptide in the NT1 (in red) and NT2 (in blue) setups, averaged over three trajectories each.
Figure 6.
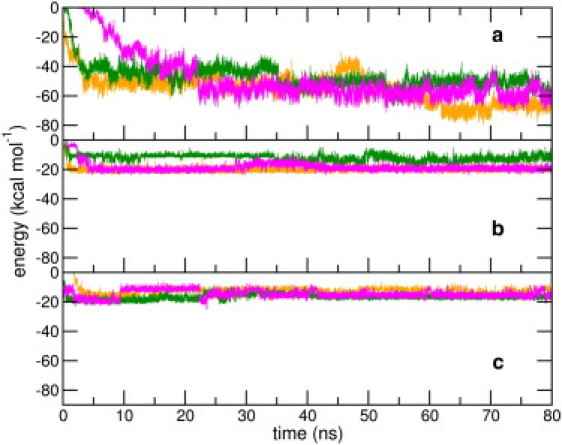
(Color online) Time evolution of interaction energies of the SWCNT with (a) residues 1–16, (b) HP1, and (c) HP2 segments of the peptide in the NT3 (in green), NT4 (in magenta), and NT5 (in orange) setups.
Propensity of peptide collapse
A key step in the self-assembly of the Aβ peptide in a primarily aqueous environment either into oligomers or into insoluble aggregates, is the formation of collapsed units of the monomeric form. This collapse is thought to be largely due to the strong hydrophobic interactions of the central hydrophobic core, and the hydrophobic patches near the peptide's C-terminus (11,12,15). Therefore, devising methods in which the propensity for peptide collapse is decreased can lead to effective ways of preventing Aβ self-assembly and amyloidogenesis. As mentioned earlier, we have chosen the center-of-mass distance dcollapse between the contiguous sequences L17VFFAEDVGS26 and K28GAIIGLMVGGVVIA42 of the Aβ1-42 for comparing the free energy changes due to peptide collapse; this has been done for the peptide-SWCNT systems with the largest and the smallest initial contact areas (i.e., NT1 and NT2), and the pure peptide. The first sequence contains the central hydrophobic core and the greater part of the first helix of the PDB:1Z0Q structure, and the second sequence contains the main C-terminal hydrophobic sequence. It is to be noted that the collapse (opening) of the peptide will effectively bring these patches closer (farther away), and therefore the separation between their centers of mass can be considered as an effective metric to probe loop closure propensity of the monomeric Aβ.
In Fig. 7, we show the PMF profiles obtained with the three setups mentioned above. (Details pertaining to the PMF calculation and the order parameter involved are provided in the Materials and Methods.) For the peptide monomer in the purely aqueous environment, there is a clear propensity for the patches to come together, or to collapse, and the energetic cost of maintaining the open state is >∼15 kcal mol−1. In the presence of the SWCNT, there is a striking decrease in the energetic cost of maintaining the open states, which are energetically more favorable compared to the closed states. The PMF profiles indicate the appearance of a barrier between the two states, and considering the initial dcollapse value of 16.7 Å, a marked propensity for the open states. From estimates of the peptide-nanotube contact area, we ascertained that the peptide remains in the vicinity of the SWCNT after a transient time during the course of the ABF calculation.
Figure 7.
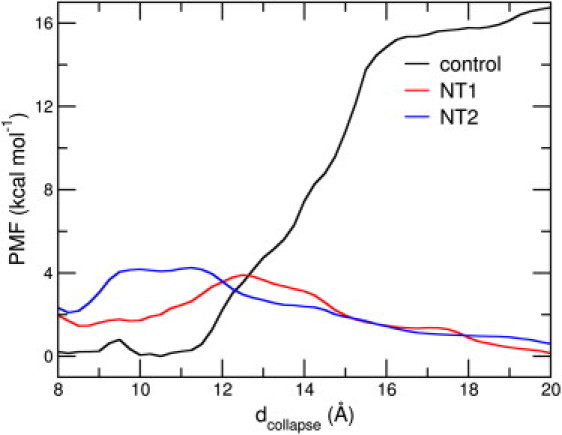
(Color online) Free energy profiles along dcollapse for the control, NT1, and NT2 setups obtained with ABF calculations.
Shown in Fig. 8 are plots of dcollapse obtained from the canonical MD simulation trajectories of the free peptide, and the NT1 and NT2 systems; corresponding data for NT3, NT4, and NT5 are shown in Fig. S6. For the peptide in NT1–NT5, dcollapse, for the last 40 ns is 17.8 (±1.4) Å. On the other hand, the average value over the last 40 ns of the free peptide trajectory is 6.9 (±0.8) Å, representing a decrease by 58.7% over the initial value. We mention here that three other independent 50-ns simulations of the same peptide coordinates in a purely aqueous environment, performed at 300 K, show a consistent decrease in dcollapse; the average value of 8.2 Å (or a decrease of ∼51%) at the end of the 50-ns simulations reiterates the spontaneity of collapse of the peptide when placed in a purely aqueous environment (data not shown). The presence of the SWCNT thus acts to disrupt the spontaneity of the collapse process.
Figure 8.
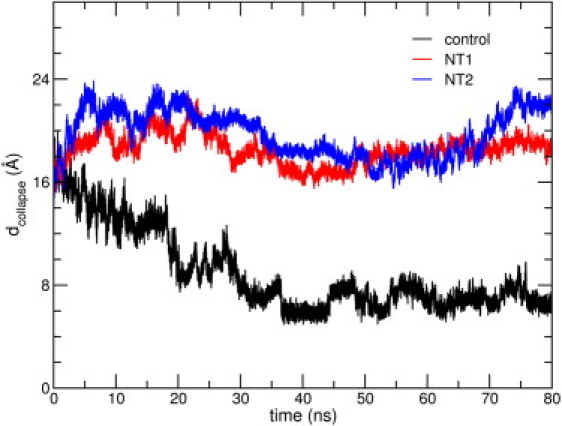
(Color online) Time evolution of dcollapse for the control, NT1, and NT2 trajectories. Data for NT1 and NT2 have been averaged over three independent trajectories.
Conclusion
In this article, we have presented the results of our investigations on the effects of a SWCNT on the full-length, monomeric Aβ1-42 peptide. Five different initial configurations of the peptide and the SWCNT, with differences in the distances of and the angles made by the nanotube with key domains, as well as in the initial peptide-nanotube contact area, have been used. Compared with the pure monomeric peptide, we find significant changes in the behavior of the peptide when in proximity to the nanotube. We observe that peptide diffusion is largely arrested in the presence of the SWCNT, resulting in an overall localization of the peptide in its vicinity. The structural stability of the peptide is decreased; and the change in the peptide's nonbonded energy is approximately equivalent to the interaction strength of the peptide with the SWCNT. The adsorption is independent of the initial peptide-nanotube configuration, as indicated by the converged values of the contact area obtained from all trajectories.
Our studies show that the peptide's collapse propensity is markedly reduced in the presence of the SWCNT. ABF-based free energy calculations using two representative peptide-SWCNT setups indicate that the open state should be largely populated at physiological temperatures. Preliminary analyses show that interactions of the HP1 segment with HP2 are hindered due to the onset of interactions with the nanotube. Further studies will be required to unravel, in detail, the competition between interactions that lead to spontaneous collapse of the free peptide, with interactions arising due to the nanotube that act to prevent the collapse.
We propose that some of the observations from this study, and other computational studies of SWCNT interactions with the Aβ peptide fragments (41), may be leveraged as means to disrupt Aβ aggregation in aqueous environment. As mentioned previously, in recent years, CNTs are being considered for applications such as site-directed drug delivery and gene therapy. In the case of the Aβ peptide, the observance of peptide localization near the SWCNT could thus be advantageous as it could increase the specificity of drug delivery mechanisms. Importantly, the inhibition of peptide collapse suggests that SWCNTs could be a powerful agent for prevention of the peptide's self-assembly.
It is necessary, however, to keep in mind some of the extant limitations and challenges that need to be met. To begin with, CNTs of very large dimensions can be highly cytotoxic (as they are capable of deforming the walls of living cells) (58). Thus, only CNTs of limited sizes may be used in actual, cell-based studies. Further, we know that pristine CNTs are insoluble in water (21). However, recent experimental work shows that suitably functionalized CNTs have vastly increased solubilities (59,60), and several approaches for increasing the dispersion of CNTs in soluble media with noncovalent functionalization also exist (27,61).
Further studies of the Aβ peptide with such suitably functionalized SWCNTs that retain sufficient hydrophobic surface area for the peptide's adsorption will be required; this may require finding optimal aspect ratios of the nanotubes. Lastly, this study has been performed with the monomeric form of the Aβ1-42 peptide, and this approach may only work before the formation of insoluble aggregates, or even soluble oligomers and protofibrils. Detailed studies probing the competition between adsorption of a free monomeric peptide onto a SWCNT surface and its propensity for deposition on the edge of a protofibril would be required; it needs to be determined whether or not the strength of the interactions with the SWCNT is sufficient to overcome the enthalpy of protofibrillar elongation.
Acknowledgments
We thank Ms. Jaya Jose for help in setting up the simulations and data from simulations at 300 K, and Dr. Chetan Gadgil for discussions.
This work was supported by funds, and an initial fellowship to A.K.J., provided by the Department of Science and Technology (project code No. GAP280526). A.K.J. thanks the University Grants Commission for his current Junior Research Fellowship. N.S. thanks the National Chemical Laboratory, India, for startup grants and facilities provided through the Centre of Excellence in Scientific Computing. The Center for Development of Advanced Computing, Pune is thanked for generously providing additional computational resources.
Supporting Material
References
- 1.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Liao M.Q., Tzeng Y.J., Chen Y.C. The correlation between neurotoxicity, aggregative ability and secondary structure studied by sequence truncated Aβ peptides. FEBS Lett. 2007;581:1161–1165. doi: 10.1016/j.febslet.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 3.Bugiani O. Alzheimer's disease: ageing-related or age-related? New hypotheses from an old debate. Neurol. Sci. 2011;32:1241–1247. doi: 10.1007/s10072-011-0614-4. [DOI] [PubMed] [Google Scholar]
- 4.Demeester N., Mertens C., Labeur C. Comparison of the aggregation properties, secondary structure and apoptotic effects of wild-type, Flemish and Dutch N-terminally truncated amyloid β peptides. Eur. J. Neurosci. 2001;13:2015–2024. doi: 10.1046/j.0953-816x.2001.01579.x. [DOI] [PubMed] [Google Scholar]
- 5.Crescenzi O., Tomaselli S., Picone D. Solution structure of the Alzheimer amyloid β-peptide (1-42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 2002;269:5642–5648. doi: 10.1046/j.1432-1033.2002.03271.x. [DOI] [PubMed] [Google Scholar]
- 6.Khandogin J., Brooks C.L., 3rd Linking folding with aggregation in Alzheimer's β-amyloid peptides. Proc. Natl. Acad. Sci. USA. 2007;104:16880–16885. doi: 10.1073/pnas.0703832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomaselli S., Esposito V., Picone D. The α-to-β conformational transition of Alzheimer's Aβ-(1–42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of β -conformation seeding. ChemBioChem. 2006;7:257–267. doi: 10.1002/cbic.200500223. [DOI] [PubMed] [Google Scholar]
- 8.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 9.Cappai R., Barnham K.J. Delineating the mechanism of Alzheimer's disease Aβ peptide neurotoxicity. Neurochem. Res. 2008;33:526–532. doi: 10.1007/s11064-007-9469-8. [DOI] [PubMed] [Google Scholar]
- 10.Lührs T., Ritter C., Riek R. 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernstein S.L., Wyttenbach T., Bowers M.T. Amyloid β-protein: monomer structure and early aggregation states of Aβ42 and its Pro19 alloform. J. Am. Chem. Soc. 2005;127:2075–2084. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]
- 12.Baumketner A., Shea J.E. The structure of the Alzheimer amyloid β 10-35 peptide probed through replica-exchange molecular dynamics simulations in explicit solvent. J. Mol. Biol. 2007;366:275–285. doi: 10.1016/j.jmb.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 13.Triguero L., Singh R., Prabhakar R. Comparative molecular dynamics studies of wild-type and oxidized forms of full-length Alzheimer amyloid β-peptides Aβ (1-40) and Aβ (1-42) J. Phys. Chem. B. 2008;112:7123–7131. doi: 10.1021/jp801168v. [DOI] [PubMed] [Google Scholar]
- 14.Petkova A.T., Ishii Y., Tycko R. A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee C., Ham S. Characterizing amyloid-β protein misfolding from molecular dynamics simulations with explicit water. J. Comput. Chem. 2011;32:349–355. doi: 10.1002/jcc.21628. [DOI] [PubMed] [Google Scholar]
- 16.Li J., Liu R., Duan Y. Alzheimer's disease drug candidates stabilize A-β protein native structure by interacting with the hydrophobic core. Biophys. J. 2011;100:1076–1082. doi: 10.1016/j.bpj.2010.12.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Viet M.H., Ngo S.T., Li M.S. Inhibition of aggregation of amyloid peptides by β-sheet breaker peptides and their binding affinity. J. Phys. Chem. B. 2011;115:7433–7446. doi: 10.1021/jp1116728. [DOI] [PubMed] [Google Scholar]
- 18.Mattson M.P., Haddon R.C., Rao A.M. Molecular functionalization of carbon nanotubes and use as substrates for neuronal growth. J. Mol. Neurosci. 2000;14:175–182. doi: 10.1385/JMN:14:3:175. [DOI] [PubMed] [Google Scholar]
- 19.Zanello L.P., Zhao B., Haddon R.C. Bone cell proliferation on carbon nanotubes. Nano Lett. 2006;6:562–567. doi: 10.1021/nl051861e. [DOI] [PubMed] [Google Scholar]
- 20.Shi X., Sitharaman B., Mikos A.G. Fabrication of porous ultra-short single-walled carbon nanotube nanocomposite scaffolds for bone tissue engineering. Biomaterials. 2007;28:4078–4090. doi: 10.1016/j.biomaterials.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lacerda L., Bianco A., Kostarelos K. Carbon nanotubes as nanomedicines: from toxicology to pharmacology. Adv. Drug Deliv. Rev. 2006;58:1460–1470. doi: 10.1016/j.addr.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 22.Bianco A., Hoebeke J., Partidos C.D. Cationic carbon nanotubes bind to CpG oligodeoxynucleotides and enhance their immunostimulatory properties. J. Am. Chem. Soc. 2005;127:58–59. doi: 10.1021/ja044293y. [DOI] [PubMed] [Google Scholar]
- 23.Wenrong Y., Thordarson P., Braet F. Carbon nanotubes for biological and biomedical applications. Nanotechnology. 2007;18:412001. [Google Scholar]
- 24.Chaban V.V., Savchenko T.I., Prezhdo O.V. Heat-driven release of a drug molecule from carbon nanotubes: a molecular dynamics study. J. Phys. Chem. B. 2010;114:13481–13486. doi: 10.1021/jp104507g. [DOI] [PubMed] [Google Scholar]
- 25.Chiu C.C., Dieckmann G.R., Nielsen S.O. Molecular dynamics study of a nanotube-binding amphiphilic helical peptide at different water/hydrophobic interfaces. J. Phys. Chem. B. 2008;112:16326–16333. doi: 10.1021/jp805313p. [DOI] [PubMed] [Google Scholar]
- 26.Balamurugan K., Gopalakrishnan R., Subramanian V. Exploring the changes in the structure of α-helical peptides adsorbed onto a single walled carbon nanotube using classical molecular dynamics simulation. J. Phys. Chem. B. 2010;114:14048–14058. doi: 10.1021/jp106177n. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y., Chipot C., Cai W. Free-energy landscape of the helical wrapping of a carbon nanotube by a polysaccharide. J. Phys. Chem. C. 2011;115:1851–1856. [Google Scholar]
- 28.Johnson R.R., Rego B.J., Klein M.L. Computational study of a nanobiosensor: a single-walled carbon nanotube functionalized with the Coxsackie-adenovirus receptor. J. Phys. Chem. B. 2009;113:11589–11593. doi: 10.1021/jp901999a. [DOI] [PubMed] [Google Scholar]
- 29.Hirano A., Uda K., Shiraki K. One-dimensional protein-based nanoparticles induce lipid bilayer disruption: carbon nanotube conjugates and amyloid fibrils. Langmuir. 2010;26:17256–17259. doi: 10.1021/la103615b. [DOI] [PubMed] [Google Scholar]
- 30.Sorin E.J., Pande V.S. Nanotube confinement denatures protein helices. J. Am. Chem. Soc. 2006;128:6316–6317. doi: 10.1021/ja060917j. [DOI] [PubMed] [Google Scholar]
- 31.Kang Y., Wang Q., Wu T. Diameter selectivity of protein encapsulation in carbon nanotubes. J. Phys. Chem. B. 2010;114:2869–2875. doi: 10.1021/jp905995s. [DOI] [PubMed] [Google Scholar]
- 32.Balamurugan K., Singam E.R.A., Subramanian V. Effect of curvature on the α-Helix breaking tendency of carbon based nanomaterials. J. Phys. Chem. C. 2011;115:8886–8892. [Google Scholar]
- 33.Kim J.E., Lee M. Fullerene inhibits β-amyloid peptide aggregation. Biochem. Biophys. Res. Commun. 2003;303:576–579. doi: 10.1016/s0006-291x(03)00393-0. [DOI] [PubMed] [Google Scholar]
- 34.Tatko C.D., Waters M.L. Selective aromatic interactions in β-hairpin peptides. J. Am. Chem. Soc. 2002;124:9372–9373. doi: 10.1021/ja0262481. [DOI] [PubMed] [Google Scholar]
- 35.Tracz S.M., Abedini A., Raleigh D.P. Role of aromatic interactions in amyloid formation by peptides derived from human Amylin. Biochemistry. 2004;43:15901–15908. doi: 10.1021/bi048812l. [DOI] [PubMed] [Google Scholar]
- 36.Platt G.W., Routledge K.E., Radford S.E. Fibril growth kinetics reveal a region of β2-microglobulin important for nucleation and elongation of aggregation. J. Mol. Biol. 2008;378:251–263. doi: 10.1016/j.jmb.2008.01.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porat Y., Abramowitz A., Gazit E. Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug Des. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 38.Scherzer-Attali R., Pellarin R., Segal D. Complete phenotypic recovery of an Alzheimer's disease model by a quinone-tryptophan hybrid aggregation inhibitor. PLoS ONE. 2010;5:e11101. doi: 10.1371/journal.pone.0011101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Armstrong A.H., Chen J., Hecht M.H. Mutations that replace aromatic side chains promote aggregation of the Alzheimer's Aβ peptide. Biochemistry. 2011;50:4058–4067. doi: 10.1021/bi200268w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Senguen F.T., Doran T.M., Nilsson B.L. Clarifying the influence of core amino acid hydrophobicity, secondary structure propensity, and molecular volume on amyloid-β 16-22 self-assembly. Mol. Biosyst. 2011;7:497–510. doi: 10.1039/c0mb00210k. [DOI] [PubMed] [Google Scholar]
- 41.Fu Z., Luo Y., Wei G. Induced β-barrel formation of the Alzheimer's Aβ 25-35 oligomers on carbon nanotube surfaces: implication for amyloid fibril inhibition. Biophys. J. 2009;97:1795–1803. doi: 10.1016/j.bpj.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 43.Hamodrakas S.J., Liappa C., Iconomidou V.A. Consensus prediction of amyloidogenic determinants in amyloid fibril-forming proteins. Int. J. Biol. Macromol. 2007;41:295–300. doi: 10.1016/j.ijbiomac.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 44.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 45.Kale L., Skeel R., Schulten K. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999;151:283–312. [Google Scholar]
- 46.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 47.MacKerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 48.Ryckaert J.P., Ciccotti G., Berendsen H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 49.Feller S.E., Zhang Y., Brooks B.R. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 50.Martyna G.J., Tobias D.J., Klein M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 51.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 52.Darve E., Rodríguez-Gómez D., Pohorille A. Adaptive biasing force method for scalar and vector free energy calculations. J. Chem. Phys. 2008;128:144120. doi: 10.1063/1.2829861. [DOI] [PubMed] [Google Scholar]
- 53.Hénin J., Fiorin G., Klein M.L. Exploring multidimensional free energy landscapes using time-dependent biases on collective variables. J. Chem. Theory Comput. 2009;6:35–47. doi: 10.1021/ct9004432. [DOI] [PubMed] [Google Scholar]
- 54.Tainer J.A., McCammon J.A., Ivanov I. Recognition of the ring-opened state of proliferating cell nuclear antigen by replication factor C promotes eukaryotic clamp-loading. J. Am. Chem. Soc. 2010;132:7372–7378. doi: 10.1021/ja100365x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gumbart J., Chipot C., Schulten K. Free energy of nascent-chain folding in the translocon. J. Am. Chem. Soc. 2011;133:7602–7607. doi: 10.1021/ja2019299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei C., Pohorille A. Permeation of nucleosides through lipid bilayers. J. Phys. Chem. B. 2011;115:3681–3688. doi: 10.1021/jp112104r. [DOI] [PubMed] [Google Scholar]
- 57.Fogolari F., Corazza A., Esposito G. Molecular dynamics simulation of β2-microglobulin in denaturing and stabilizing conditions. Proteins. 2011;79:986–1001. doi: 10.1002/prot.22940. [DOI] [PubMed] [Google Scholar]
- 58.Kostarelos K. The long and short of carbon nanotube toxicity. Nat. Biotechnol. 2008;26:774–776. doi: 10.1038/nbt0708-774. [DOI] [PubMed] [Google Scholar]
- 59.Dyke C.A., Tour J.M. Covalent functionalization of single-walled carbon nanotubes for materials applications. J. Phys. Chem. A. 2004;108:11151–11159. [Google Scholar]
- 60.Dyke C.A., Tour J.M. Overcoming the insolubility of carbon nanotubes through high degrees of sidewall functionalization. Chemistry. 2004;10:812–817. doi: 10.1002/chem.200305534. [DOI] [PubMed] [Google Scholar]
- 61.Vaisman L., Wagner H.D., Marom G. The role of surfactants in dispersion of carbon nanotubes. Adv. Colloid Interface Sci. 2006;128-130:37–46. doi: 10.1016/j.cis.2006.11.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.