

Abstract
The current understanding of epigenetic signaling assigns a central role to post-translational modifications that occur in the histone tails. In this context, it has been proposed that methylation of K9 and phosphorylation of S10 in the tail of histone H3 represent a binary switch that controls its reversible association to heterochromatin protein 1 (HP1). To test this hypothesis, we performed a comprehensive molecular dynamics study in which we analyzed a crystallographically defined complex that involves the HP1 chromodomain and an H3 tail peptide. Microsecond-long simulations show that the binding of the trimethylated K9 H3 peptide in the aromatic cage of HP1 is only slightly affected by S10 phosphorylation, because the modified K9 and S10 do not interact directly with one another. Instead, the phosphate group of S10 seems to form a persistent intramolecular salt bridge with R8, an interaction that can provoke a major structural change and alter the hydrogen-bonding regime in the H3-HP1 complex. These observations suggest that interactions between adjacent methyl-lysine and phosphoserine side chains do not by themselves provide a binary switch in the H3-HP1 system, but arginine-phosphoserine interactions, which occur in both histones and nonhistone proteins in the context of a conserved RKS motif, are likely to serve a key regulatory function.
Introduction
Histone proteins, the building blocks of the nucleosome core particle, are subject to a variety of posttranslational modifications (PTMs) (1). PTMs are thought to modulate the compaction state of chromatin, exposing or concealing different stretches of DNA and translating modification chemistry to active or repressive signaling marks (2–5). Because the combinatorial repertoire of histone modifications is theoretically very large, it has been proposed that PTMs serve a coding function. According to this histone code hypothesis, specific modifications or combinations thereof can affect distinct cellular events by altering the structure of chromatin (the cis mechanism) or by generating a binding platform for other effector proteins (the trans mechanism) (5,6). Such effector proteins contain evolutionarily conserved domains (e.g., the chromo-, bromo-, Tudor, MBT, PWWP, PHD, and 14-3-3 domains (7)) that are able to recognize specific PTMs and initiate a cascade of downstream events that determine the outcome of chromatin-dependent processes ranging from DNA repair, replication, and recombination to regulation of gene expression.
A key example of how modified histone molecules interact with their effectors is the recognition of the K9-methylated H3 tail by the chromo domain of heterochromatin protein 1 (HP1) (8–10). The tertiary structure of several complexes that form between the HP1 chromodomain and H3 tail peptides mono-, di-, and trimethylated at K9 (11,12) reveals that the modified side chain of K9 is caged by three conserved aromatic rings that are presented on the surface of the HP1 chromodomain. Binding affinity measurements indicate that the H3-HP1 interaction is strongest when K9 is fully modified, i.e., trimethylated (11).
Although H3K9 methylation and several other single-site histone modifications have been established as epigenetic marks, their potential cross talk has not been sufficiently investigated. An early speculation that accompanied the histone code hypothesis involved the existence of putative binary switches in which phosphorylated serine or threonine residues prevent or abrogate the binding of regulatory proteins to adjacent methylated lysines (13). In the framework of this model, it was initially suggested that the H3 tail cannot be simultaneously phosphorylated at S10 and methylated at K9 (14), or that a methyl-K9/phospho-S10 binary switch operating during mitosis abrogates the interaction of HP1 with the K9-methylated target site (15,16). Contrary to these reports, experimental in vitro and in vivo evidence showed that S10 phosphorylation is compatible with K9 methylation during mitosis and that this modification is not necessarily linked to HP1 dissociation (17,18). The discrepancies among the different studies may have originated from the experimental techniques used, the isoform-specific features of the different HP1 proteins employed (19), and/or the use of different antibodies, which often fail to recognize their epitopes when residues in their vicinity are modified. Be that as it may, and despite recent advances in the field, the role of binary switches operating in the H3 molecule remains controversial (20–22).
Notwithstanding the complexity of chromatin assemblies and the extended spatial and temporal scales involved, computational modeling is well suited to tackle the fundamental questions regarding chromatin organization and interactions with regulatory proteins. Several published macroscopic and CG-mesoscale models of chromatin (23–30) were able to capture the relevant physicochemical features of DNA and nucleosomes, but, due to inherent limitations, could only approximate histone tails. The interactions and dynamics of modified and unmodified histone tails were recently studied by means of atomistic molecular dynamics (MD) simulations (31,32). A 10 ns MD simulation of HP1 in complex with the K9-trimethylated and S10-phosphorylated H3 tail suggested that the two adjacent modifications increase the flexibility of the H3 tail and weaken HP1 binding to chromatin (33). Another recent study showed that cation-π− and charge-independent interactions play an essential role in binding (34). Molecular simulations of modified H3 tail peptides, as well as in vivo experiments, suggested that the K9-dimethylated/S10-phosphorylated H3 tail can adopt an ensemble of conformations, one of which is specifically recognized by HP1 between the early prophase and the early anaphase of mitosis and meiosis (35). This conformation involves the interaction between dimethylated K9 and phosphorylated S10. Questions about the isoform-specific behavior of HP1 proteins with regard to their mitotic release from H3 were also addressed by molecular simulations that indicated that all HP1 isoforms showed no tendency to dissociate from H3 upon S10 phosphorylation, at least within the timescale explored (up to 100 ns) (36).
In short, experimental and theoretical approaches so far have not succeeded in resolving the molecular mechanism and the synergistic interactions that can support the association/dissociation of HP1 (and possibly of other proteins as well) to methylated histones and chromatin during the formation of hypercondensed, mitotic chromosomes. Structural analysis of HP1/H3 complexes has provided valuable information, but is limited to only a few known structures of HP1 in complex with methylated histone tails and none with the phosphorylated form. It is now widely recognized that additional experimental and computational work is required before we can gain a better understanding of the histone modification cross talk and its effect on chromatin interactions (37). To that end, we studied the interaction between HP1 and an assortment of differentially modified H3 tails by means of MD simulations.
The work reported here focuses on the S10 phosphorylation of histone H3 methylated at K9 and on the effect this modification has on the reversible association of the nucleosome with HP1. We performed full-atomistic MD simulations for sufficiently long times (1 μs) of the complex formed between the chromodomain of HP1 and a hexapeptide from the H3 tail, mono-, di-, and trimethylated at K9, in the presence or absence of Ser-10 phosphorylation. We also examined the contribution of H3 sites that are neighbors to the methyl/phos switch in the H3-HP1 interaction, by mutating these residues. Our simulations reveal novel (to our knowledge) structural determinants that are involved in H3-HP1 interactions, and highlight the crucial role of H3 residues other than K9 and S10 in the H3/HP1 association-dissociation mechanism.
Materials and Methods
Selection of structural models
The MD simulations were based on three different structures from the PDB database, namely, 1Q3L, 1KNA, and 1KNE. All three structures comprise HP1 residues 23–74 bound to H3 tail residues 5–10 with K9 monomethylated (1Q3L), dimethylated (1KNA), and trimethylated (1KNE). To investigate the effect of S10 phosphorylation, we added a phosphate group to residue S10 of the H3 tail. Simulations were also performed for a HP1-H3 complex in which we replaced residue R8 of the doubly modified K9-methylated/S10 phosphorylated H3 peptide with glycine to further investigate the specific effect of phosphorylation on intramolecular bonding.
Force field and derivation of partial charges
All simulations were based on the Amber parm99 (38,39) force field as modified by Hornak et al. (40) (ff99SB). Parameters derived by Craft and Legge (41) were used for phosphoserine. The partial charge parameters for the mono-, di-, and trimethylated K9 (see Table S1 in the Supporting Material) were derived via the Amber charge fitting procedure (42). We constructed small model systems that consisted of a modified amino acid and were blocked by the acetyl (Ac) and N-methyl (NME) groups. For each one of the model systems, we considered two different initial conformations (α-helical (φ,ψ) = (−60°, −40°) and β-sheet (φ,ψ) = (−120°, 140°)) with side-chain χ-values chosen as in the original charge calculation of the common amino acids (43). In the case of K9 monomethylation, there is no clear conformational preference for the orientation of the methyl group; hence, we considered all three possible orientations for both the α-helical and β-sheet conformers, leading to a total of six different structures. The same is true in the case of dimethylated K9, again leading to a total of six different structures. The geometry of all model systems was optimized at the HF/6-31G∗ level of theory and the electrostatic potential was calculated for the optimized structures. The addition of the methyl and acetyl groups affects the values of the φ, ψ, and χ angles of the corresponding wild types only marginally. Subsequently, we carried out a multiconformational, two-stage RESP fit (39,44). During the RESP fit, the charges of the Ac and NME groups along with the N, CA, C, and O atoms of the peptide backbone were fixed to their values in the ff99SB parameter set. We employed Gaussian98 (45) for all ab initio calculations. Partial charges were derived using the AmberTools software package (46).
MD simulations
The simulation of the eight systems started from the original PDB structures. The HP1 and H3 chains were terminated with the NME and Ac groups, and the water molecules contained in the PDB files were removed and hydrogen atoms were added when appropriate. Subsequently, each one of the systems was placed in the center of a periodic rectangular simulation box filled with ∼4000 water molecules modeled by the TIP3P approach (47). The distance between the solute and the box sides was kept at >10 Å. Charged systems were rendered neutral by addition of Na+ and Cl− counterions as required. An additional concentration of 200 mM NaCl was imposed to mimic physiological conditions. The nonbonded interaction cutoff was set to 10 Å. The particle mesh Ewald method was used to calculate electrostatic interactions in the periodic box.
Initially, the energy of the system was minimized in two stages: First, the heavy solute atoms were kept fixed while the water molecules and the solute hydrogen atoms were allowed to relax during 5 × 105 conjugate gradient minimization steps. Second, the constraints were removed and the energy of the entire solute-solvent system was minimized until the root mean-square gradient reached the value of 10−2 Kcal/mol/Å. The system was subsequently slowly heated at 310 K for a time period of 240 ps. The temperature was kept fixed by means of a Langevin thermostat for a 200 ps equilibration period, followed by a 200 ps equilibration under constant pressure of 1 atm by application of the Langevin piston (48). The system was then propagated in time for 1000 ns using the isothermal-isobaric ensemble. In all of the MD simulations, SHAKE constraints were applied, the time step was set to 2 fs, and trajectory information was recorded at intervals of 2.5 ps. More details about the method used, as well as evidence on the convergence of the simulations, are provided in Table S2 and Fig. S1.
All energy minimizations and MD simulations were carried out with the use of NAMD software (49), and trajectories and pictorial representation of molecules were visualized with VMD (50). Additional in-house code, necessary for computational analysis, was developed.
Results
Effect of S10 phosphorylation on the structure of H3 tail
In the MD simulations, we examined in detail the complex formed between the chromodomain of HP1 and a K9-methylated H3 tail hexapeptide in the presence and absence of S10 phosphorylation.
The distance d1 between the Nζ atom of K9 and the Oγ atom of S10 was initially used as a measure of the proximity between the two residues in the putative methyl/phos switch (see Fig. 1 A). As shown in Fig. 1 B, panel a, the probability density functions (PDFs) of d1 in the presence and absence of S10 phosphorylation are very similar, with PDFs peaking in both cases between 9–11 Å when K9 is trimethylated. Upon S10 phosphorylation of the mono- and dimethylated K9 peptide, the PDF curve shifts slightly toward longer values (Fig. S2, A and C), but this shift is marginal, suggesting a local structural change rather than a specific interaction between methylated K9 and S10.
Figure 1.
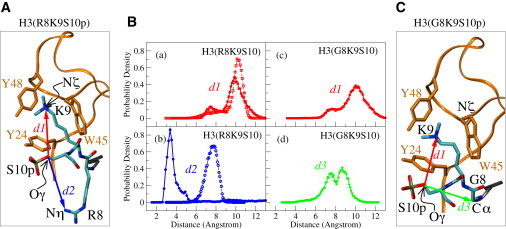
Effect of S10 phosphorylation on the local geometry of the H3 8RKS10 stretch. (A) The variables d1 (red arrow) and d2 (blue arrow) are defined as the distance between atoms labeled Oγ on S10 and Nζ on K9, and the distance between Oγ on S10 and Nη on R8, respectively. (B) PDFs of distance variables d1, d2, d3 in the phosphorylated (solid lines and solid symbols) and unphosphorylated (dashed lines and open symbols) forms of K9-trimethylated H3 tail. (C) The variables in the mutated form of H3 peptide: d1 (red arrow), which is the same as in A, and d3 (green arrow), which is defined as the distance between atoms Oγ on S10 and Cα on G8.
A closer inspection of the trajectories followed during the simulations confirms the absence of interaction between modified K9 and S10, but reveals a distinct salt bridge forming between phosphorylated S10 and the residue preceding K9, i.e., R8. As shown in Fig. 2 B, panel b, the PDF of d2 (the distance between the Nη atom of R8 and the hydroxyl Oγ atom of S10) is significantly altered when S10 is modified and its peak moves from 6–9 Å in the nonphosphorylated peptide to 3–5 Å in the phosphorylated one. This change is also observed in the mono- and dimethylated versions of the H3 tail peptide (Fig. S2, B and D).
Figure 2.

Clustering of conformations adopted by the 8RKS10 stretch of H3 when S10 is phosphorylated.
Because both R8 and methylated K9 possess a net positive charge, it is plausible that the two residues might compete with one another for salt-bridging with the negatively charged phosphorylated S10. To explore this scenario, we replaced R8 with a glycine residue (G/R8) and repeated the simulations in the standard fashion. We chose to perform the MD simulations of the mutant R8G with the two extreme states of K9 modification (i.e., the mono- and trimethylated forms) because the simulations of the HP1/H3 complex clearly showed that there are no significant differences between the mono-, di-, and trimethylated histone tails. As illustrated in Fig. 1 B, panels a and c, the PDF curves of d1 are not affected when the mutant peptide is phosphorylated in S10. Moreover, the PDF of d3 (the distance between the Cα atom of G8 and the Oγ atom of phosphorylated S10) peaks at >7 Å, which precludes an interaction between G8 and S10. Similar observations can be made for the monomethylated H3 tail (Fig. S2, E and F), suggesting the absence of a direct interaction between S10 and K9, even if a competing group is no longer there. The lack of interaction with S10 can be attributed to the fact that the side chain of K9 is tightly confined in the aromatic cage of HP1 throughout the simulation time. From these results we also infer that the G/R8 mutation results in a wider distribution of distances d1 and d3, most likely because of the flexibility of the S10 phosphate group, which is no longer engaged in a strong interaction with neighboring residues. The relative spatial disposition of residues R8, K9, and S10 during the simulations can be clustered in three dominant conformations, as shown in Fig. 2. The atoms selected for structural analysis and the dihedral angles in the respective structures are shown in Table S3.
Interactions of K9 and S10 with the HP1 aromatic cage
Having established that H3 phosphorylation at S10 does not promote a strong intramolecular interaction with methylated K9, we attempted to evaluate the effect of the same modification in the tail peptide on H3 binding to the aromatic cage of HP1. To that end, we analyzed the trajectories of the various complexes during MD simulations and calculated the distribution of distances between the key H3 residues K9 and S10 and the aromatic rings that form the binding pocket. The average distances between the Nζ atom of K9 and the calculated centroid in each of the three aromatic rings (Table S4) are consistent with previous studies (11) and indicate an increase of HP1-H3 affinity with increasing K9 methylation. Overall, the net result of S10 phosphorylation on the system under study is apparently not sufficient to force the H3 tail out of the aromatic cage.
We also explored the possibility of repulsive interactions between the phosphorylated S10 and the aromatic residues of the HP1 binding pocket. Such interactions between a phosphorylated side chain and an aromatic residue have been reported to disrupt the structure and control the folding/unfolding of short β-hairpin peptides mimicking the H3/HP1 complex (51–53). The distances d4, d5, and d6 between the hydroxyl oxygen of S10 and the centroids in the aromatic rings of Y24, W45, and Y48, respectively, were monitored during the simulation of the complex (Fig. 3 A). The PDF plots for these distances when the complex included the trimethylated K9 H3 tail in its phosphorylated and nonphosphorylated forms indicate a broadening of the corresponding peaks upon phosphorylation of S10 (Fig. 3 B, panels a–c). As suggested by the shift in d4 and d6 toward smaller values, upon phosphorylation, S10 seems to come closer to two of the three aromatic residues, Y24 and Y48, which form the HP1 cage. However, S10 has a tendency to move away from the third aromatic residue, W45, when it is phosphorylated. Of interest, the relative disposition of S10 and W45 is similar to that observed in the β-hairpin model peptides used to study the disruptive interaction between a phosphorylated side chain and an aromatic residue (51). Therefore, such an interaction cannot be excluded, but seems to be marginal in the system we have been studying. An analysis of the results obtained for the mono- and dimethylated K9 tail peptide (Fig. S3, A–F) leads to similar conclusions. On the other hand, the G/R8 mutation results in an even broader distribution of the PDF peaks when S10 is phosphorylated (Fig. S3, G–I), indicating an increased mobility upon elimination of the R8 side chain, which is presumably involved in a tight salt-bridge formation. Taken together, these observations strongly suggest that S10 phosphorylation weakens the interaction between methylated K9 and the binding pocket on the surface of HP1, but, for the simplified model system studied and the timescale investigated here, this does not seem to be sufficient to disrupt the H3-HP1 complex.
Figure 3.
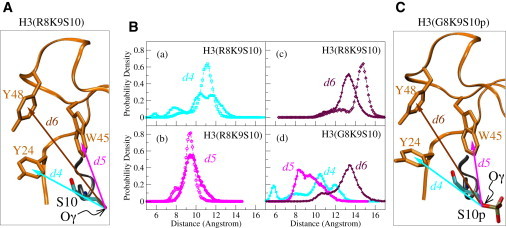
Effect of S10 phosphorylation on the interaction of S10 with the HP1 aromatic cage. (A) The definition of variables d4, d5, d6, shown as arrows in the structural representation of K9-trimethylated H3(R8K9S10): d4 (cyan) is the distance between the atom labeled Oγ on S10 and the centroid of the aromatic ring in Y24; d5 (pink) is the distance between Oγ on S10 and the centroid of the aromatic ring in W45; d6 (brown), is the distance between Oγ on S10 and the centroid of the aromatic ring in Y48. (B) PDFs of d4, d5, d6 in the phosphorylated (solid lines and solid symbols) and unphosphorylated (dashed lines and open symbols) forms of H3. (C) Same as in A for the mutant H3 peptide (R8 replaced by G).
Hydrogen bonding between HP1 and H3
It has been shown that in addition to the conserved aromatic pocket, the recognition of methylated H3 by HP1 involves a series of backbone hydrogen bonds and side-chain interactions between the H3 tail and the HP1 chromodomain (54). In particular, residues Q5, T6, A7, R8, and S10 of the H3 tail form a network of hydrogen bonds with the HP1 chromodomain, as shown in Fig. 4 A. Investigators assessed the contribution of these additional, but no less critical, interactions to the stability of the H3/HP1 complex experimentally by mutating specific residues and measuring the resulting binding affinities (11,54). To evaluate the effect of S10 phosphorylation on these interactions with our methods, we analyzed in detail the backbone and side-chain hydrogen-bonding patterns between H3 and HP1 during the simulations. We identified hydrogen bonds on the basis of their geometry, using as cutoff criteria the distance between bonded atoms (which must be <3.2 Å), as well as their angle (which should be ≥120° in a hydrogen bond). Results obtained with the mono-, di-, and trimethylated K9 H3-HP1 complexes show that upon S10 phosphorylation, the hydrogen-bonding pattern between HP1 and H3 is considerably disturbed, suggesting a strong destabilizing effect. More specifically, upon S10 phosphorylation, almost all backbone hydrogen bonds are weakened, as established by a comparison of the blue and red bars in Fig. 4 B (panels a and b) and Fig. S4, with the only exception being the one between O/T6 and N/D62. A more pronounced effect, though, is the dramatic rearrangement of the hydrogen bonds formed by the side chains of S10 (S10-E56) and R8 (R8-E23 and R8-E56), shown in Fig. 4 B (panels c and d) and Fig. S4. Phosphorylation removes the hydroxyl moiety from S10 and results in the replacement of the strong hydrogen bond between S10 and E56 by their enhanced hydrogen bonding with water molecules (1.4–2.6 times more for E56, and 8.7 times more for S10) and by the R8-E56 interaction, which is hardly present in the initial crystal structures and in the respective trajectories produced by the MD simulations. As a result, the initially present R8-E23 hydrogen bond persists in only a minor proportion (∼15%) of the simulated structures in the case of the phosphorylated complex. The newly formed hydrogen-bonding pattern between HP1 and H3, along with the intramolecular R8-S10p interaction described above, is arranged in a triangular pattern of alternating charges (negative-positive-negative), as depicted in Fig. 4 C.
Figure 4.
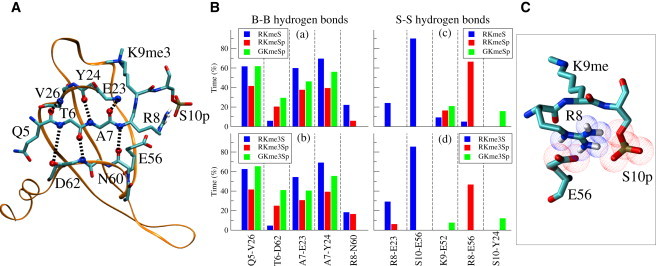
Hydrogen bonding between the H3 oligopeptide and HP1. (A) Hydrogen-bonding pattern in the complex between the H3 oligopeptide and HP1. (B) Vertical bars represent the percentage of time during which hydrogen bonds between backbone atoms (B-B) persist for the various amino acid pairs in (a) the RKmeS (blue), RKmeSp (red), and GKmeSp (green) structures, and (b) the RKme3S (blue), RKme3Sp (red) and GKme3Sp (green) structures. The same notation is used in c and d for the hydrogen bonds between side-chain atoms (S-S). (C) Part of the structure of the complex with the O atoms of phosphorylated S10 and E56 (red) and the N atoms on R8 (blue) shown as van der Waals spheres.
Simulations with the phosphorylated and G/R8 mutated complex reveal that elimination of the positively charged side chain of R8 restores almost completely the hydrogen-bonding pattern between HP1 and H3 to that of the nonphosphorylated complex, as seen from the green bars in Fig. 4 B, panels a and b. In addition, in the absence of a strong intramolecular interaction with R8, S10 is free to form a hydrogen bond with Y24 (Fig. 4 B, panels c and d), which may also account for the variation in the distance d4 between phosphorylated S10 and Y24 in the mutated complex (see Fig. 3 B, panel d, and Fig. S3 G). These observations support the importance of the R8-S10p interaction in the overall destabilizing effect brought about by S10 phosphorylation.
Discussion
Several recent reports have provided evidence for the ability of lysine residues to interact strongly with phosphoserine in the context of isolated oligopeptides (35,52,55). However, on the timescale of 1 μs used in our MD simulations (the longest reported for these systems, to our knowledge) and in the context of the H3/HP1 model complex studied, we find no indication of a direct interaction between methylated K9 and phosphorylated S10 that is sufficiently strong to destabilize the complex and drive the dissociation of HP1 from chromatin. On the other hand, phosphorylation of S10 seems to trigger the formation of a strong intramolecular bond between the corresponding phosphate group and the positively charged guanidinium group of R8. Also noticeable under these conditions is the simultaneous rearrangement of some of the intermolecular hydrogen bonds between H3 and HP1, which has not been previously reported for the HP1-H3 complex.
According to our results, the network of intermolecular hydrogen bonds between H3 and HP1 seems to be the most vulnerable part of the complex and possibly the first to be disturbed upon S10 phosphorylation in mitosis, mainly because of the competing strong intramolecular interaction between phosphorylated S10 and R8. It is likely that this interaction can be competed against by positively charged moieties that might shield the S10 phosphate group, such as the side chain of K14, which, according to a recent report (18), has to be acetylated before HP1 is released from the K9-methylated and S10-phosphorylated H3 during mitosis. These pieces of evidence indicate that these effects may represent only one of the stages in the molecular mechanism underlying the dissociation of HP1 from H3.
We have to note at this point that an important issue to be addressed in computational studies, such as the one presented here, is the suitability of the force field employed for the simulated system (56–59). In our work we used a well-established force field, AMBER. To our knowledge, there are no widely accepted gold standards for the comparison of force fields and the respective parameters used (60, 61). In this respect, it is very useful to compare the results of simulations, if available, that have employed different force fields to study the same system. In similarity to our work, MD simulations of isolated native and variably modified K9 and S10 H3 histone tails were recently reported by Eberlin et al. (35). The main focus of the analysis was the K9-S10p interaction, the force field employed was CHARMM, and the conditions for the MD runs were identical to ours (in explicit water (TIP3P) with periodic boundary conditions, in a cubic box of 36 Å and at an ionic strength of 0.2 M). Inspection of those results reveals that the R8-S10p interaction clearly dominates the dimethylated or trimethylated K9-S10p interaction. This is reasonable because arginine residues are known to be more basic than lysine residues. In the complex we studied, the methylated K9 is additionally protected by the aromatic cage. Therefore, our results show a very good agreement with those derived with the use of a different force field.
The interaction between guanidinium and phosph(on)ate groups has been the subject of several other experimental and theoretical studies (62). For example, binding of arginine to phosphorylated serine or threonine residues was experimentally shown to be involved in receptor heteromerization, and its stability was described as being surprisingly high (comparable to that of a covalent bond) on the basis of data derived from mass spectrometry and plasmon resonance experiments (63). Of interest, the interdigitation of the guanidinium group of R8 between the phosphate oxyanion of S10 and the carboxyl group of a glutamic acid residue on the surface of the binding partner was recently reported for the complex of a 14-3-3 protein with the H3 tail. In this complex, the R-S interaction plays a different role, mediating the binding of 14-3-3 protein to H3 (64). Moreover, it was shown that other complexes of 14-3-3 with various phosphopeptides are stabilized by the intramolecular R-S interaction (65,66). Taking this into account, our computational analysis underscores the importance of the RKS motif in the H3 tail for the regulation of epigenetic events and provides the theoretical framework for the experimental observations described above.
The significance of the RKS motif has already been reported in the literature in the context of the ARKS/T motifs (67). An ARKS motif centered on K27 was found to bind in its methylated form to several chromodomains with varying affinity and specificity. In addition, similar motifs present in other chromatin-associated proteins, such as linker histone H1.4 and lysine methyltransferase G9a, are thought to function as peripheral docking sites for chromodomains (68,69). Finally, an ARKS motif was identified within the ADNP homeodomain involved in HP1-dependent association and localization to pericentromeric heterochromatin (70). The low hydropathy, high charge content, and ability of the RKS motif to undergo multiple modifications (all three residues are modifiable, and two can be modified in several possible ways) are all unique structural features associated with a special class of proteins called intrinsically disordered proteins (IDPs). IDPs lack stable tertiary and/or secondary structure but are abundant in eukaryotic organisms and are known to undergo multiple post-translational modifications (71–74). A survey of the DisProt (75) database (DisProt Release 5.7, 2011-02-28) reveals that the RKS/T sequence is very common among IDPs (128 such motifs (58 RKS and 70 RKT) were identified in 101 out of a total of 710 IDPs that were searched). Based on the above considerations, we suggest that the electrostatic interaction between arginine and phosphorylated serine or threonine in the context of an RKS/T motif may be essential for the regulation of a wide range of protein-protein interactions, especially those involving IDPs.
Conclusion
MD simulations show that the K9-S10 interaction alone is not sufficient for the dissociation of H3 from HP1. Instead, the H3-HP1 binding can be considerably affected by the formation of a persistent intramolecular salt bridge between phosphorylated S10 and R8 that can provoke a major structural change in the H3 tail and rearrange the intermolecular hydrogen-bonding network in the H3-HP1 complex.
Our data further suggest that the electrostatic interaction between arginine and phosphorylated serine or threonine in the context of an RKS/T motif occurring in both histones and nonhistone proteins may be essential for the regulation of a wide range of protein-protein interactions, especially those involving IDPs.
Acknowledgments
This work was supported in part by funds from the European Union and the Hellenic Ministry of Education, in the framework of the programs Pythagoras II and PENED. Computational resources were provided by the Research Center for Scientific Calculations at the University of Ioannina, and CADMOS at École Polytechnique Fédérale de Lausanne (funded by the cantons of Geneva and Vaud, H. Wilsdorf Foundation, L. Jeantet Foundation, University of Geneva, University of Lausanne, and École Polytechnique Fédérale de Lausanne) on the IBM-BG/P system.
Contributor Information
Anastasia S. Politou, Email: apolitou@cc.uoi.gr.
Efthimios Kaxiras, Email: kaxiras@physics.harvard.edu.
Supporting Material
References
- 1.Wang Y., Wysocka J., Coonrod S.A. Linking covalent histone modifications to epigenetics: the rigidity and plasticity of the marks. Cold Spring Harb. Symp. Quant. Biol. 2004;69:161–169. doi: 10.1101/sqb.2004.69.161. [DOI] [PubMed] [Google Scholar]
- 2.Strahl B.D., Allis C.D. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 3.Nightingale K.P., O'Neill L.P., Turner B.M. Histone modifications: signalling receptors and potential elements of a heritable epigenetic code. Curr. Opin. Genet. Dev. 2006;16:125–136. doi: 10.1016/j.gde.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 4.Henikoff S. Histone modifications: combinatorial complexity or cumulative simplicity? Proc. Natl. Acad. Sci. USA. 2005;102:5308–5309. doi: 10.1073/pnas.0501853102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenuwein T., Allis C.D. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 6.Hake S.B., Allis C.D. Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proc. Natl. Acad. Sci. USA. 2006;103:6428–6435. doi: 10.1073/pnas.0600803103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taverna S.D., Li H., Patel D.J. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bannister A.J., Zegerman P., Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 9.Lachner M., O'Carroll D., Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 10.Nakayama J., Rice J.C., Grewal S.I. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 11.Jacobs S.A., Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen P.R., Nietlispach D., Laue E.D. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- 13.Fischle W., Wang Y., Allis C.D. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–479. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 14.Rea S., Eisenhaber F., Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 15.Fischle W., Tseng B.S., Allis C.D. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 16.Hirota T., Lipp J.J., Peters J.M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- 17.Terada Y. Aurora-B/AIM-1 regulates the dynamic behavior of HP1alpha at the G2-M transition. Mol. Biol. Cell. 2006;17:3232–3241. doi: 10.1091/mbc.E05-09-0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mateescu B., England P., Muchardt C. Tethering of HP1 proteins to chromatin is relieved by phosphoacetylation of histone H3. EMBO Rep. 2004;5:490–496. doi: 10.1038/sj.embor.7400139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dormann H.L., Tseng B.S., Fischle W. Dynamic regulation of effector protein binding to histone modifications: the biology of HP1 switching. Cell Cycle. 2006;5:2842–2851. doi: 10.4161/cc.5.24.3540. [DOI] [PubMed] [Google Scholar]
- 20.Lee J.S., Smith E., Shilatifard A. The language of histone crosstalk. Cell. 2010;142:682–685. doi: 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gardner K.E., Allis C.D., Strahl B.D. Operating on chromatin, a colorful language where context matters. J. Mol. Biol. 2011;409:36–46. doi: 10.1016/j.jmb.2011.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henikoff S., Shilatifard A. Histone modification: cause or cog? Trends Genet. 2011;27:389–396. doi: 10.1016/j.tig.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Langowski J., Schiessel H. Theory and computational modeling of the 30 nm chromatin fiber. In: Zlatanova J., Leuba S.H., editors. Chromatin Structure and Dynamics: State-of-the-Art. Elsevier B.V.; Amsterdam: 2004. pp. 397–420. [Google Scholar]
- 24.Arya G., Zhang Q., Schlick T. Flexible histone tails in a new mesoscopic oligonucleosome model. Biophys. J. 2006;91:133–150. doi: 10.1529/biophysj.106.083006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arya G., Schlick T. Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc. Natl. Acad. Sci. USA. 2006;103:16236–16241. doi: 10.1073/pnas.0604817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arya G., Schlick T. A tale of tails: how histone tails mediate chromatin compaction in different salt and linker histone environments. J. Phys. Chem. A. 2009;113:4045–4059. doi: 10.1021/jp810375d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma S., Ding F., Dokholyan N.V. Multiscale modeling of nucleosome dynamics. Biophys. J. 2007;92:1457–1470. doi: 10.1529/biophysj.106.094805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kepper N., Foethke D., Rippe K. Nucleosome geometry and internucleosomal interactions control the chromatin fiber conformation. Biophys. J. 2008;95:3692–3705. doi: 10.1529/biophysj.107.121079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Y., Lyubartsev A.P., Nordenskiöld L. Computer modeling reveals that modifications of the histone tail charges define salt-dependent interaction of the nucleosome core particles. Biophys. J. 2009;96:2082–2094. doi: 10.1016/j.bpj.2008.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korolev N., Lyubartsev A.P., Nordenskiöld L. Computer modeling demonstrates that electrostatic attraction of nucleosomal DNA is mediated by histone tails. Biophys. J. 2006;90:4305–4316. doi: 10.1529/biophysj.105.080226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu H., Duan Y. Effects of posttranslational modifications on the structure and dynamics of histone H3 N-terminal peptide. Biophys. J. 2008;94:4579–4585. doi: 10.1529/biophysj.107.115824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Y.-K., Zou J.-W., Yu Q.-S. Dynamics simulation on the flexibility and backbone motions of HP1 chromodomain bound to free and lysine 9-methylated histone H3 tails. Int. J. Quantum Chem. 2009;109:1135–1147. [Google Scholar]
- 33.Jiang Y.-K., Zou J.-W., Jiang Y.-J. Molecular dynamics simulation on HP1 protein binding by histone H3 tail methylation and phosphorylation. Int. J. Quantum Chem. 2009;109:746–755. [Google Scholar]
- 34.Lu Z., Lai J., Zhang Y. Importance of charge independent effects in readout of the trimethyllysine mark by HP1 chromodomain. J. Am. Chem. Soc. 2009;131:14928–14931. doi: 10.1021/ja904951t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eberlin A., Grauffel C., Tora L. Histone H3 tails containing dimethylated lysine and adjacent phosphorylated serine modifications adopt a specific conformation during mitosis and meiosis. Mol. Cell. Biol. 2008;28:1739–1754. doi: 10.1128/MCB.01180-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Machado M.R., Dans P.D., Pantano S. Isoform-specific determinants in the HP1 binding to histone 3: insights from molecular simulations. Amino Acids. 2010;38:1571–1581. doi: 10.1007/s00726-009-0371-3. [DOI] [PubMed] [Google Scholar]
- 37.Allis C.D., Muir T.W. Spreading chromatin into chemical biology. Chem. Bio. Chem. 2011;12:264–279. doi: 10.1002/cbic.201000761. [DOI] [PubMed] [Google Scholar]
- 38.Cheatham T.E., 3rd, Cieplak P., Kollman P.A. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 1999;16:845–862. doi: 10.1080/07391102.1999.10508297. [DOI] [PubMed] [Google Scholar]
- 39.Cornell W.D., Cieplak P., Kollman P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 40.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Craft J.W., Jr., Legge G.B. An AMBER/DYANA/MOLMOL phosphorylated amino acid library set and incorporation into NMR structure calculations. J. Biomol. NMR. 2005;33:15–24. doi: 10.1007/s10858-005-1199-0. [DOI] [PubMed] [Google Scholar]
- 42.Wang J., Cieplak P., Kollman P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 43.Cieplak P., Cornell W.D., Kollman P.A. Application of the multimolecule and multiconformational RESP methodology to biopolymers: charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995;16:1357–1377. [Google Scholar]
- 44.Bayly C.I., Cieplak P., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
- 45.Frisch M.J., Trucks G.W., Pople J.A. Gaussian, Inc.; Pittsburgh, PA: 1998. Gaussian 98 (Revision A.9) [Google Scholar]
- 46.Amber home page. 2009. http://ambermd.org/#AmberTools.
- 47.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 48.Feller S.E., Zhang Y., Pastor R.W. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 49.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 51.Riemen A.J., Waters M.L. Controlling peptide folding with repulsive interactions between phosphorylated amino acids and tryptophan. J. Am. Chem. Soc. 2009;131:14081–14087. doi: 10.1021/ja9047575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riemen A.J., Waters M.L. Dueling post-translational modifications trigger folding and unfolding of a β-hairpin peptide. J. Am. Chem. Soc. 2010;132:9007–9013. doi: 10.1021/ja101079z. [DOI] [PubMed] [Google Scholar]
- 53.Riemen A.J., Waters M.L. Positional effects of phosphoserine on β-hairpin stability. Org. Biomol. Chem. 2010;8:5411–5417. doi: 10.1039/c0ob00202j. [DOI] [PubMed] [Google Scholar]
- 54.Jacobs S.A.S.D., Taverna S.D., Khorasanizadeh S. Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 2001;20:5232–5241. doi: 10.1093/emboj/20.18.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Errington N., Doig A.J. A phosphoserine-lysine salt bridge within an α-helical peptide, the strongest α-helix side-chain interaction measured to date. Biochemistry. 2005;44:7553–7558. doi: 10.1021/bi050297j. [DOI] [PubMed] [Google Scholar]
- 56.Salonen L.M., Ellermann M., Diederich F. Aromatic rings in chemical and biological recognition: energetics and structures. Angew. Chem. Int. Ed. Engl. 2011;50:4808–4842. doi: 10.1002/anie.201007560. [DOI] [PubMed] [Google Scholar]
- 57.The CH/π Interaction. http://www.tim.hi-ho.ne.jp/dionisio/. Accessed December 20, 2011.
- 58.Takahashi O., Kohno Y., Nishio M. Relevance of weak hydrogen bonds in the conformation of organic compounds and bioconjugates: evidence from recent experimental data and high-level ab initio MO calculations. Chem. Rev. 2010;110:6049–6076. doi: 10.1021/cr100072x. [DOI] [PubMed] [Google Scholar]
- 59.Ma J.C., Dougherty D.A. The cation-π interaction. Chem. Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 60.Ponder J.W., Case D.A. Force fields for protein simulations. Adv. Protein Chem. 2003;66:27–85. doi: 10.1016/s0065-3233(03)66002-x. [DOI] [PubMed] [Google Scholar]
- 61.Lopes P.E.M., Roux B., Mackerell A.D., Jr. Molecular modeling and dynamics studies with explicit inclusion of electronic polarizability. Theory and applications. Theor. Chem. Acc. 2009;124:11–28. doi: 10.1007/s00214-009-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schug K.A., Lindner W. Noncovalent binding between guanidinium and anionic groups: focus on biological- and synthetic-based arginine/guanidinium interactions with phosph[on]ate and sulf[on]ate residues. Chem. Rev. 2005;105:67–114. doi: 10.1021/cr040603j. [DOI] [PubMed] [Google Scholar]
- 63.Woods A.S., Ferré S. Amazing stability of the arginine-phosphate electrostatic interaction. J. Proteome Res. 2005;4:1397–1402. doi: 10.1021/pr050077s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Macdonald N.J., Welburn J.P., Mahadevan L.C. Molecular basis for the recognition of phosphorylated and phosphoacetylated histone h3 by 14-3-3. Mol. Cell. 2005;20:199–211. doi: 10.1016/j.molcel.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 65.Obsil T., Ghirlando R., Dyda F. Crystal structure of the 14-3-3ζ:serotonin N-acetyltransferase complex a role for scaffolding in enzyme regulation. Cell. 2001;105:257–267. doi: 10.1016/s0092-8674(01)00316-6. [DOI] [PubMed] [Google Scholar]
- 66.Rittinger K., Budman J., Yaffe M.B. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol. Cell. 1999;4:153–166. doi: 10.1016/s1097-2765(00)80363-9. [DOI] [PubMed] [Google Scholar]
- 67.Fischle W., Franz H., Khorasanizadeh S. Specificity of the chromodomain Y chromosome family of chromodomains for lysine-methylated ARK(S/T) motifs. J. Biol. Chem. 2008;283:19626–19635. doi: 10.1074/jbc.M802655200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garcia B.A., Busby S.A., Hunt D.F. Characterization of phosphorylation sites on histone H1 isoforms by tandem mass spectrometry. J. Proteome Res. 2004;3:1219–1227. doi: 10.1021/pr0498887. [DOI] [PubMed] [Google Scholar]
- 69.Sampath S.C., Marazzi I., Tarakhovsky A. Methylation of a histone mimic within the histone methyltransferase G9a regulates protein complex assembly. Mol. Cell. 2007;27:596–608. doi: 10.1016/j.molcel.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 70.Mosch K., Franz H., Fischle W. HP1 recruits activity-dependent neuroprotective protein to H3K9me3 marked pericentromeric heterochromatin for silencing of major satellite repeats. PLoS ONE. 2011;6:e15894. doi: 10.1371/journal.pone.0015894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uversky V.N., Dunker A.K. Biochemistry. Controlled chaos. Science. 2008;322:1340–1341. doi: 10.1126/science.1167453. [DOI] [PubMed] [Google Scholar]
- 72.Gsponer J., Futschik M.E., Babu M.M. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science. 2008;322:1365–1368. doi: 10.1126/science.1163581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dunker A.K. Encyclopedia of Life Sciences. John Wiley & Sons Ltd.; Chichester: 2007. Disordered proteins; pp. 1–9. [Google Scholar]
- 74.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 75.Sickmeier M., Hamilton J.A., Dunker A.K. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007;35(Database issue):D786–D793. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.