

Abstract
Several high profile failures of lipid-related therapeutics in clinical trials have led to intense interest in improved discovery and preclinical prioritization of potential targets. The careful study of patients with rare monogenic disorders has played a key role in establishing the causal role of cholesterol in atherosclerosis and highlighting viable drug targets. Systematic efforts to extend the association of common variants linked with lipid levels to coronary disease allow assessment of the vascular consequences of lifelong differences in lipids due to variation in specific molecules. This application of genetic epidemiology, termed Mendelian randomization, may prove useful in informing ongoing drug development efforts.
Currently available therapies have proven effective in reducing the coronary heart disease (CHD) burden, particularly in high-income countries. The age-adjusted mortality from CHD in the United States was cut roughly in half between 1980 and 2000 – a recent analysis attributed 24% of this reduction to improvement in lipid profiles (Ford et al. 2007). This success is largely a reflection of systematic implementation of guidelines put forth by the National Cholesterol Education Program. Conclusive evidence of risk reduction, primarily derived from statin-based trials, has led to progressively lower low density lipoprotein-cholesterol (LDL-C) targets in patients at risk for CHD (Grundy et al. 2004). However, substantial residual risk remains, even in patients on intensive statin therapy.
Targeting of lipid metabolism is likely to remain a mainstay of future prevention strategies. For example, results from the JUPITER trial suggest that measurement of C-reactive protein may identify patients without elevated LDL-C who nevertheless derive benefit from statin therapy (Ridker et al. 2008). Furthermore, on-treatment LDL-C, high density lipoprotein cholesterol (HDL-C), and triglyceride (TG) levels have each been independently associated with clinical outcome in recent statin trials, stimulating intense interest in efforts to target these lipid subfractions (Ridker et al. 2009, Barter et al. 2007a, Miller et al. 2008). Finally, lipoprotein(a) [Lp(a)] levels have been robustly linked with CHD and may emerge as a bona fide drug target (Emerging Risk Factors Collaboration et al. 2009).
The private sector has continued to invest substantial resources in the research and development of novel treatments for dyslipidemia. According to a 2009 report from PhRMA, 26 drugs for lipid disorders are currently in various stages of clinical development (PhrMA, 2009). However, the majority of these are either in early Phase I trials or represent combinations of existing medications. Furthermore, several recent high-profile failures have dampened industry enthusiasm. Ezetimibe, a marketed inhibitor of cholesterol absorption that reduces LDL-C levels, offered no benefit with regard to progression of carotid intimal medial thickness when combined with (Taylor et al. 2009, Kastelein et al. 2008). This result led some to question whether the mechanism, in addition to the magnitude, of LDL-C lowering may be important. Fenofibrate, a marketed PPARα agonist that reduces TG levels, failed to meet the primary endpoint in reducing coronary events in patients with type 2 diabetes (Keech et al. 2005). Finally, torcetrapib, an inhibitor of cholesteryl ester transfer protein (CETP), was associated with increased mortality despite substantially increasing HDL-C levels (Barter et al. 2007b). This outcome, which came after a nearly $1 billion investment by Pfizer in the drug’s clinical development, may have played a role in the company’s decision to make major cutbacks in its cardiovascular drug development program (Garber 2009). Each of these examples has complicating features and current evidence limits definitive conclusions about these therapeutic strategies. However, the cumulative experience with lipid-targeting therapy in recent years has created a critical need for new lipid target discovery and improved preclinical prioritization of novel targets based on ability to influence clinical endpoints.
The study of genetic variants proven to effect lipid levels, and the systematic association of these same variants with CHD, may offer a way forward, particularly in light of recent genome-wide association study (GWAS) findings (Katan 1986). Technical advances, in conjunction with completion of the Human Genome and HapMap projects, have enabled high-throughput unbiased interrogation of relationships between common genetic variants and plasma lipid levels. Long known to be highly heritable, the robust association of single nucleotide polymorphisms (SNPs) at more than 30 loci with plasma lipid concentrations levels has helped validate the hypothesis that the cumulative effects of many common variants underlie complex trait genetics (Kathiresan et al. 2009). While many of these loci occur at genes with known function in lipid metabolism, others are in regions with no a priori evidence. This discovery of new genetic loci associated with plasma lipids in humans thus has the potential to yield new molecular targets for treatment of dyslipidemia. Ongoing efforts to resequence areas of association to find the as-yet elusive “causal” variants and functional studies may yield a mechanistic understanding of these novel associations.
In the short term, human genetics allow assessment of the vascular consequences of lifelong differences in lipids associated with these variants. Conceptually similar studies of monogenic lipid disorders have proven highly useful. Nobel-Prize winning work carried out in the 1970’s first identified mutations in the LDL receptor (LDL-R) as a cause of familial hypercholesterolemia (FH), a rare genetic disorder characterized by dramatically elevated levels of LDL-C and early onset of CHD (Brown and Goldstein 1986). This discovery was a key component in overcoming skepticism that LDL-C played a causal role in the progression of atherosclerosis. Decades later, pharmacologic upregulation of hepatic LDL-R density, with subsequent increased uptake of circulating LDL particles, remains at the core of modern LDL-C reduction strategies.
Will targeting new lipid loci reduce cardiovascular risk? The role of Mendelian randomization
Addressing the question of whether a human genetic variant is associated with CHD can be brought into the high-throughput “-omics” era using a genetic epidemiology tool known as Mendelian randomization (Smith et al. 2008). This process, referred to as a randomized trial of nature, capitalizes on the random collection of genetic variants assigned at birth via meiosis and independent chromosomal assortment (Thanassouli et al. 2009). Groupings of patients by genotype are thus analogous to the treatment groups used in a traditional randomized controlled trial. Successful Mendelian randomization analyses require two key relationships: 1) an association between a SNP and an intermediate phenotype of interest (i.e. LDL-C levels); and 2) a relationship between the intermediate phenotype and manifest disease (i.e. CHD). The critical final step involves assessing whether the genetic variant is associated with disease as might be predicted from its association with the intermediate trait (See Figure 1).
Figure 1. Selected applications of Mendelian randomization.
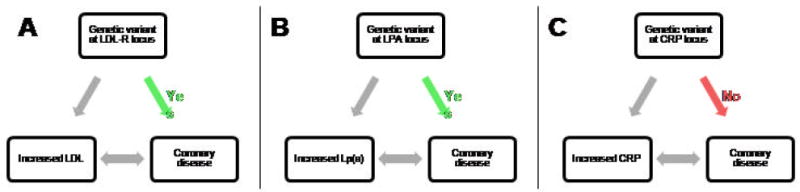
Mendelian randomization analyses require robust associations between 1) a genetic variant and an intermediate phenotype; and 2) the intermediate phenotype and disease. If this relationship is causal, the genetic variant should related to disease as well. This evidence for causality has been confirmed for variants affecting both LDL and Lipoprotein(a) levels (A and B), but not C-reactive protein (CRP) (C).
This approach has several key advantages, particularly with regard to assessment of causality. Relationships in observational epidemiology can be attributed to: 1) a causal relationship between the biomarker and disease; 2) confounding by other factors; or 3) reverse causality, in which the disease onset precedes changes in biomarker levels (Schunkert and Samani 2008). Because inheritance of any one SNP is largely independent of variants at other loci and occurs at conception, Mendelian randomization can help overcome these barriers. It also provides insight into the effects of lifelong exposure to differences in the intermediate phenotype (i.e. LDL-C), which may be important in slowly progressive diseases like atherosclerosis. Finally, by implementing common genetic variants and diseases, the process overcomes concerns about the limited sample sizes in studies of patients with rare genetic disorders.
Mendelian randomization has already been successfully implemented with traits that extend beyond lipids. A recent analysis of genetically elevated C-reactive protein (CRP) provided the most compelling evidence to date that, while CRP is a predictor of CHD events, it is likely not directly involved in the pathogenesis (Zacho et al. 2008). However, several important limitations of this approach should be noted. Developmental compensation may serve to buffer the metabolic perturbation conferred by a genetic polymorphism, a phenomenon termed “canalization.” Secondly, individual GWAS associations reported to date have only modest effects on lipid levels, thus corresponding to “predicted” CHD odds ratios between 1.0 and 1.3. These effect sizes mandate large CHD cohorts for sufficient power – a barrier increasingly overcome via metaanalyses of existing studies. An additional concern relates to the potential of one genetic variant to have pleiotropic effects not captured by a change in a single biomarker. For example, the analysis assumes that a SNP in the LDL-R gene affects risk of disease via differences in plasma LDL-C levels. However, particularly in the case of SNPs that affect multiple lipid subfractions (i.e. both TG and HDL), this assumption may not hold true. Single cross-sectional assessments of lipids and heart disease may not capture SNP effects that vary with patient age. Finally, strategies to systematically account for gene-gene and gene environment interactions remain largely lacking.
LDL Cholesterol
Plasma LDL-C levels are a strong predictor of coronary risk, even in a recent statin trial that achieved a median on-treatment LDL-C of 55 mg/dl (Ridker et al. 2009). Furthermore, the direct role of LDL particles in atherosclerosis has been well-characterized – LDL is taken up into the intimal space of the vasculature and is ultimately phagocytosed by macrophages to create lipid-laden foam cells (Ross 1999). Nevertheless, the question has been raised whether all mechanisms that lead to LDL-C reduction are necessarily cardioprotective. The Mendelian randomization approach using loci that influence LDL-C levels represents one strategy to address this important question.
GWAS studies published to date have confirmed common variants in at least eleven loci that lead to variation in LDL-C levels (Kathiresan et al. 2009). A myocardial infarction case-control analysis involving over 12,000 patients provides strong evidence that LDL-C- associated SNPs are robustly associated with risk of MI (Willer et al. 2008). Consistent with the effects of FH on premature CHD, a common LDL-R SNP with a much less dramatic effect on LDL-C was nevertheless associated with an OR for MI of 1.29. Furthermore, five LDL-associated SNPs, when assembled into an “LDL-C genotype score” led to a graded increase in risk in prospectively determined cardiovascular events (Kathiresan et al. 2008a).
HMG-CoA reductase (HMGCR) was discovered biochemically over 30 years ago to be the rate-limiting step in cholesterol synthesis, paving the way for the development of statins. Interestingly, a common variant at the HMGCR locus is associated with very modest increases in circulating LDL-C (Kathiresan et al. 2009). Although the common HMGCR SNP has small effects on LDL-C, pharmacologic inhibition of HMGCR with statins leads to much greater reduction in LDL and a pronounced decrease in risk of major coronary events (Larosa et al. 1999). An unbiased interrogation of the entire genome thus pointed to the target of the most effective class of drugs in modern cardioprevention. Furthermore, this example demonstrates that the magnitude of the effect of common SNPs on lipid traits is not a predictor of the potential value of that gene as a new therapeutic target. Ongoing efforts will determine whether this HMGCR SNP is additionally linked to risk of CHD. Given the recent controversy regarding the benefits of LDL-lowering with ezetimibe (Taylor et al. 2009, Kastelein et al. 2008), it will be of substantial interest to determine whether variants at Niemann-Pick C1-Like 1 (NPC1L1), the drug’s molecular target, are associated with LDL-C levels and CHD.
A striking example of how human genetics can serve as validation for a therapeutic lipid target is that of protease proprotein convertase subtilisin/kexin type 9 (PCSK9). Several lines of genetic evidence have converged upon PCSK9 inhibition as a potential therapeutic strategy. Gain-of-function mutations in PCSK9, a rare cause of severe hypercholesterolemia, were first identified in 2003 via classical linkage genetics (Abifadel et al. 2003). Subsequent resequencing efforts in individuals with very low LDL-C led to the 2006 observation that 2-3% of African American patients harbored nonsense mutations in the gene associated with a 28% reduction in LDL-C and a dramatic 88% reduction in risk of CHD. In addition, a low-frequency missense variant in Caucasians was associated with a more modest reduction in LDL-C and CHD risk (Cohen et al. 2006, Kathiresan et al. 2008b). Finally, a GWAS study related a common PCSK9 SNP with a 3 mg/dl decrease in LDL-C and a 13% decreased risk of CHD (Willer et al. 2008). Thus, very rare, rare, and common genetic variants in PCSK9 affect both LDL-C and CHD in a highly consistent fashion.
PCSK9 is a secreted protease that binds to LDL receptors on the cell surface and facilitates their degradation, thereby increasing circulating LDL-C levels (Horton et al. 2009). Importantly, statins increase PCSK9 activity, counteracting their upregulation of hepatic LDL-R expression. This provides a potential explanation for the modest incremental LDL-reduction seen with increasing statin doses and suggests that inhibiting PCSK9 may help overcome this barrier. Consistent with this hypothesis, both siRNA and antibody-mediated targeting of PCSK9 have been shown to increase LDL-R and reduce LDL-C levels in animal models (Frank-Kamenetsky et al. 2008, Chan et al. 2009). Ongoing efforts will determine whether conceptually similar approaches are viable in humans.
Among SNPs at loci with novel LDL-C associations, those at chromosomal region 1p13 were among the first for which the association was extended to a CHD phenotype (Willer et al. 2008) and indeed achieved genome-wide significance in a case-control study of early-onset myocardial infarction (Myocardial Infarction Genetics Consortium et al. 2009). Discovering the mechanistic basis of the association of this locus with LDL-C and CHD is of major interest to the field. However, SNPs at the 1p13 locus are frequently inherited together as part of a haplotype block (termed linkage disequilibrium) that spans at least three distinct candidate genes, namely CELSR2, PCRC1, and SORT1. Hepatic expression of all three genes varies according to genotype at the associated SNPs, suggesting a potential for coordinated transcriptional regulation (Kathiresan et al. 2008c). Resequencing across the region, as was done for PCSK9, may prove enlightening in finding the “smoking gun.” Furthermore, systematic manipulation of all three genes using model organisms and in vitro analyses holds the potential for providing additional functional clues. The ultimate inclusion of one or more of these genes in lipoprotein physiology networks would provide much-needed proof-of-concept of this aspect of GWAS follow-up and a potentially interesting new validated molecular target for reducing LDL-C and risk of CHD.
These findings have important implications for drug development targeted toward LDL-C. While various loci likely contribute to LDL variation via several different mechanisms, most or all of them appear to be associated with CHD risk. The precise mechanism of LDL-lowering thus appears less important than the magnitude of LDL reduction, assuming the drug does not have adverse off-target effects. This supports the concept that LDL-reduction should continue to be used as a surrogate for clinical utility early in the development process.
Triglycerides
It is now recognized that plasma triglyceride concentrations are an independent predictor of coronary risk, even after treatment with intensive statin therapy (Sarwar et al. 2007, Miller et al. 2008). Nonfasting measures of triglyceride have been an even stronger predictor of CHD in some studies, potentially indicative of increased atherogenic remnant lipoproteins (Nordestgaard et al. 2007). Hypertriglyceridemia frequently reflects the accumulation of chylomicrons, VLDL, and IDL particles. These particles, along with the remnants formed by hydrolysis, may directly accelerate CHD by entering existing intimal lesions and promoting a proinflammatory state (Yu and Cooper, 2001). However, as with HDL, attempts to prove causality have been limited by substantial confounding with measures of obesity and insulin resistance.
A panel of eleven SNPs serves as a powerful predictor of plasma triglyceride concentrations (Kathiresan et al. 2009). However, individual SNP-CHD associations vary according to specific loci and interpretation thus requires a nuanced understanding of lipid metabolism. For example, lipoprotein lipase (LPL) is widely expressed on the vascular endothelium of skeletal muscle and adipose tissues and hydrolyses triglyceride-rich VLDL particles derived from the liver and intestine. Systemic overexpression of LPL in has been associated with decreased atherosclerosis in mouse models (Shimada, et al. 1996, Yagyu et al. 1999). Rare human cases of genetic LPL deficiency cause marked elevations in circulating chylomicrons and triglycerides, and have been associated with premature atherosclerosis in at least one small study (Benlian et al. 1996). Common variants at the LPL locus now have confirmed associations with decreased TG, increased HDL, and decreased risk of CHD, providing strong evidence that LPL serves a net benefit in atheroprotection (Samani et al. 2007).
Another complex example is that of the APOA1/APOC3/APOA4/APOA5 gene locus, which is highly significantly associated with both TG and HDL-C levels (Kathiresan et al. 2009). Dissecting the specific relationships of this locus with plasma lipids and CHD risk will require considerable effort. As one intriguing example, resequencing efforts in an Amish population identified an ApoC-III premature stop codon associated with lower circulating TG levels, higher HDL-C levels, and decreased atherosclerotic burden as assessed by coronary calcification (Pollin et al. 2008). One physiologic property of ApoC-III is inhibition of LPL. Thus, in combination, these findings suggest that strategies to promote the rapid clearance of TG-rich particles by LPL may prove useful in the treatment of hypertriglyceridemia and CHD.
A number of novel loci have been discovered via GWAS to be associated with plasma TG levels. It remains to be fully determined which of these loci are definitively associated with CHD risk. Interestingly, a variant at the Tribbles homolog 1 (TRIB1) locus is significantly associated with increased TGs, decreased HDL, increased LDL, as well as with increased CHD (Willer et al. 2008). Understanding the mechanism underlying this association is of considerable interest to the field, and could help establish TRIB1 as a novel pharmacologic target.
HDL Cholesterol
An inverse relationship between high density lipoprotein cholesterol levels and risk of CHD is among the most robust in modern epidemiology. An increment of even 1-mg/dl in HDL-C has been associated with a 3-4% reduction in mortality from cardiovascular disease (Gordon 1989). HDL is thought to have a wide range of antiatherogenic properties, including the promotion of reverse cholesterol transport and counteracting proinflammatory and prothrombotic factors. Several therapies, including nicotinic acid, fibric acid derivatives, and statins are known to increase HDL levels, but definitive association with risk reduction has proven challenging. Epidemiologic studies of HDL are limited by extensive confounding – HDL levels are strongly linked to obesity, triglyceride levels, insulin resistance, exercise, and alcohol consumption. Future Mendelian randomization analyses may play a key role in overcoming these barriers to demonstrating causality.
Recent advances in understanding of HDL metabolism lend credence to the notion that increases in HDL levels may be a poor surrogate for clinical effectiveness. Several known monogenic disorders have dramatic effects on HDL concentrations but limited vascular consequences. Patients harboring mutations in ApoA-I (i.e. ApoA-IMilano), ABCA1 (Tangier disease), and LCAT (familial lecithin cholesterol acylytranferase deficiency) all have markedly decreased HDL levels, but their risk of premature CHD is not definitively increased (Qasim and Rader 2006; Frikke-Schmidt et al. 2008; Calabresi et al. 2009). Conversely, patients with cholesteryl ester transfer protein (CETP) deficiency have elevated HDL levels, but reports on predisposition to atherosclerosis have been inconclusive (Zhong et al. 1996, Curb et al. 2004, Thompson et al. 2008).
The cumulative effects of several common variants associated with HDL-C, as computed in a “HDL-C genotype score,” were shown to increase risk of cardiovascular events (Kathiresan et al. 2008a). However, data regarding associations between individual SNPs associated with decreased HDL and increased CHD are largely lacking. SNPs at some loci, such as CETP, likely influence HDL via differential rates of transfer of cholesteryl ester between HDL and LDL/VLDL particles. Others, such as those near endothelial lipase, presumably confer variable degrees of HDL remodeling via phospholipid hydrolysis. Still others, such as a SNP in GALNT2, play an unknown role in lipid biology. Thus, the likely heterogeneity with regard to CHD risk reinforces the concept that the mechanistic basis of increased HDL is critically important. Unlike the case of LDL, pharmacologically-induced change in HDL-C quantity is likely to remain a poor surrogate for clinical effectiveness.
Moving forward, future Mendelian randomization studies that robustly extend SNP-HDL associations to cardiovascular disease phenotypes may help prioritize therapeutic strategies. Ongoing optimization of assays that quantify HDL “functionality” may have utility beyond measurement of static HDL levels (deGoma et al. 2008). Finally, the creative use of animal models to interrogate the effects of genetic or pharmacologic modulation on HDL metabolism can provide important preclinical information. For example, a validated murine assay that quantifies flux through the reverse cholesterol transport pathway has proven a better predictor of atherosclerosis measures than HDL quantity (Rader et al. 2009). It is clear that HDL targets will need to be carefully chosen to maximize the likelihood that the development of a novel therapeutic will not only modulate HDL-C levels but also reduce risk of CHD events.
Lipoprotein(a)
Lipoprotein(a) concentrations, both highly variable and highly heritable in humans, have been linked with CHD across a wide spectrum of epidemiological studies (Emerging Risk Factors Collaboration et al. 2009). Lp(a) is a large, LDL-like particles that contains an apolipoprotein B-100 covalently bound to an apolipoprotein(a) molecule. The size of this apolipoprotein(a) component contains multiple “Kringle-like” domain repeats, the number of which depend on the highly polymorphic LPA gene. Two independent genetics studies in the last year have combine to provide strong evidence that Lp(a) is causally linked to CHD. A genome wide haplotype association study strongly implicated a gene cluster containing LPA in CHD risk (Trégouët et al. 2009). A Mendelian randomization exercise linked genetic elevations in Lp(a), as determined by number of Kringle-like repeats, to the predicted increased risk of myocardial infarction (Kamstrup et al. 2009).
The molecular basis of Lp(a)’s atherogenicity is poorly understood and worthy of additional focus (Deb and Caplice, 2004). Past studies have provided evidence for Lp(a) effects on inhibition of endothelial function, entry into the intima and subsequent foam cell formation, and plaque rupture. Structural homology to plasminogen suggests that Lp(a) may play an important role in thrombosis as well. However, it remains unclear which, if any, of these are operative in humans in vivo. Of the medicines currently widely used in clinical practice, niacin has the largest effect on Lp(a) levels, with documented reductions of 20% (Guyton et al. 2000). Statins notably do not reduce Lp(a) concentrations, raising the possibility that a compound specifically targeting Lp(a) could be combined with a statin to achieve further gains (Cobbaert et al. 1997).
Conclusion
Lipids clearly play a major role in the progression of atherosclerosis and have appropriately received substantial interest from experts in basic science, genetics, and drug development. Recently published and upcoming GWAS results have the potential to serve as a major advance in the field, particularly if the mechanistic basis for gene-lipid associations can be determined. Furthermore, the findings offer the opportunity to rapidly implement the principles of Mendelian randomization to prioritize ongoing research and development efforts.
Acknowledgments
AVK gratefully acknowledges funding from the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Smith GD, Timpson N, Ebrahim S. Strengthening causal inference in cardiovascular epidemiology through Mendelian randomization. Ann Med. 2008;40:524–541. doi: 10.1080/07853890802010709. [DOI] [PubMed] [Google Scholar]
- Abifadel M, Varret M, Rabès JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- Barter P, Gotto AM, LaRosa JC, et al. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med. 2007a;357:1301–1310. doi: 10.1056/NEJMoa064278. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007b;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- Benlian P, De Gennes JL, Foubert L, et al. Premature atherosclerosis in patients with familial chylomicronemia caused by mutations in the lipoprotein lipase gene. N Engl J Med. 1996;335:848–54. doi: 10.1056/NEJM199609193351203. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- Calabresi L, Baldassarre D, Castelnuovo S, et al. Functional lecithin: cholesterol acyltransferase is not required for efficient atheroprotection in humans. Circulation. 2009;120:628–635. doi: 10.1161/CIRCULATIONAHA.108.818143. [DOI] [PubMed] [Google Scholar]
- Chan JC, Piper DE, Cao Q, et al. A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci U S A. 2009;106:9820–9825. doi: 10.1073/pnas.0903849106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbaert C, Jukema JW, Zwinderman AH, Withagen AJ, Lindemans J, Bruschke AV. Modulation of lipoprotein(a) atherogenicity by high density lipoprotein cholesterol levels in middle-aged men with symptomatic coronary artery disease and normal to moderately elevated serum cholesterol. Regression Growth Evaluation Statin Study (REGRESS) Study Group. J Am Coll Cardiol. 1997;30:1491–1499. doi: 10.1016/s0735-1097(97)00353-7. [DOI] [PubMed] [Google Scholar]
- Cohen JC, Boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- Curb JD, Abbott RD, Rodriguez BL, et al. A prospective study of HDL-C and cholesteryl ester transfer protein gene mutations and the risk of coronary heart disease in the elderly. J Lipid Res. 2004;45:948–953. doi: 10.1194/jlr.M300520-JLR200. [DOI] [PubMed] [Google Scholar]
- Deb A, Caplice NM. Lipoprotein(a): new insights into mechanisms of atherogenesis and thrombosis. Clin Cardiol. 2004;27:258–264. doi: 10.1002/clc.4960270503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deGoma EM, deGoma RL, Rader DJ. Beyond high-density lipoprotein cholesterol levels evaluating high-density lipoprotein function as influenced by novel therapeutic approaches. J Am Coll Cardiol. 2008;51:2199–2211. doi: 10.1016/j.jacc.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerging Risk Factors Collaboration. Erqou S, Kaptoge S, et al. Lipoprotein(a) concentration and the risk of coronary heart disease,stroke, and nonvascular mortality. JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford ES, Ajani UA, Croft JB, et al. Explaining the decrease in U.S. deaths from coronary disease, 1980-2000. N Engl J Med. 2007;356:2388–98. doi: 10.1056/NEJMsa053935. [DOI] [PubMed] [Google Scholar]
- Frank-Kamenetsky M, Grefhorst A, Anderson NN, et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci U S A. 2008;105:11915–11920. doi: 10.1073/pnas.0805434105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frikke-Schmidt R, Nordestgaard BG, Stene MC, et al. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA. 2008;299:2524–2532. doi: 10.1001/jama.299.21.2524. [DOI] [PubMed] [Google Scholar]
- Garber AML. An uncertain future for cardiovascular drug development? N Engl J Med. 2009;360:1169–71. doi: 10.1056/NEJMp0808414. [DOI] [PubMed] [Google Scholar]
- Gordon DJ. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79:8–15. doi: 10.1161/01.cir.79.1.8. [DOI] [PubMed] [Google Scholar]
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- Guyton JR, Blazing MA, Hagar J, et al. Extended-release niacin vs gemfibrozil for the treatment of low levels of high-density lipoprotein cholesterol. Niaspan-Gemfibrozil Study Group. Arch Intern Med. 2000;160:1177–1184. doi: 10.1001/archinte.160.8.1177. [DOI] [PubMed] [Google Scholar]
- Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50:S172–177. doi: 10.1194/jlr.R800091-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- Kastelein JJ, Akdim F, Stroes ES, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–1443. doi: 10.1056/NEJMoa0800742. [DOI] [PubMed] [Google Scholar]
- Katan MB. Apolipoprotein E isoforms, serum cholesterol, and cancer. Lancet. 1986;327:507–8. doi: 10.1016/s0140-6736(86)92972-7. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, Melander O, Anevski D, et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008a;358:1240–1249. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- Kathiresan S Myocardial Infarction Genetics Consortium. A PCSK9 missense variant associated with a reduced risk of early-onset myocardial infarction. N Engl J Med. 2008b;358:2299–300. doi: 10.1056/NEJMc0707445. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, Melander O, Guiducci C, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008c;40:189–97. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiresan S, Willer CJ, Peloso GM, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–1861. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- LaRosa JC, He J, Vupputuri S. Effect of statins on risk of coronary disease: a meta-analysis of randomized controlled trials. JAMA. 1999;282:2340–2346. doi: 10.1001/jama.282.24.2340. [DOI] [PubMed] [Google Scholar]
- Miller M, Cannon CP, Murphy SA, et al. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol. 2008;51:724–730. doi: 10.1016/j.jacc.2007.10.038. [DOI] [PubMed] [Google Scholar]
- Myocardial Infarction Genetics Consortium. Kathiresan S, Voight BF, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordestgaard BG, Benn M, Schnohr P, et al. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308. doi: 10.1001/jama.298.3.299. [DOI] [PubMed] [Google Scholar]
- Pharmaceutical Research and Manufacturers of America. 2009 report: Medicines in development for heart disease and stroke. [June 27, 2009]; at http://www.phrma.org/files/Heart%202009.pdf.
- Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–1705. doi: 10.1126/science.1161524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qasim A, Rader DJ. Human genetics of variation in high-density lipoprotein cholesterol. Curr Atheroscler Rep. 2006;8:198–205. doi: 10.1007/s11883-006-0074-0. [DOI] [PubMed] [Google Scholar]
- Rader DJ, Alexander ET, Weibel GL, et al. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009;50:S189–194. doi: 10.1194/jlr.R800088-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Danielson E, Fonseca FA, et al. Reduction in C-reactive protein and LDL cholesterol and cardiovascular event rates after initiation of rosuvastatin: a prospective study of the JUPITER trial. Lancet. 2009;373:1175–1182. doi: 10.1016/S0140-6736(09)60447-5. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarwar N, Danesh J, Eiriksdottir G, et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115:450–458. doi: 10.1161/CIRCULATIONAHA.106.637793. [DOI] [PubMed] [Google Scholar]
- Schunkert H, Samani NJ. Elevated C-reactive protein in atherosclerosis--chicken or egg? N Engl J Med. 2008;359:1953–1955. doi: 10.1056/NEJMe0807235. [DOI] [PubMed] [Google Scholar]
- Shimada M, Ishibashi S, Inaba T, et al. Suppression of diet-induced atherosclerosis in low density lipoprotein receptor knockout mice overexpressing lipoprotein lipase. Proc Natl Acad Sci U S A. 1996;93:7242–7246. doi: 10.1073/pnas.93.14.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GD, Timpson N, Ebrahim S. Strengthening causal inference in cardiovascular epidemiology through Mendelian randomization. Ann Med. 2008;40:524–541. doi: 10.1080/07853890802010709. [DOI] [PubMed] [Google Scholar]
- Taylor AJ, Villines TC, Stanek EJ, et al. Extended-release niacin or ezetimibe and carotid intima-media thickness. N Engl J Med. 2009;361:2113–22. doi: 10.1056/NEJMoa0907569. [DOI] [PubMed] [Google Scholar]
- Thanassoulis G, O’Donnell CJ. Mendelian randomization: nature’s randomized trial in the post-genome era. JAMA. 2009;301:2386–2388. doi: 10.1001/jama.2009.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Di Angelantonio E, Sarwar N, et al. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008;299:2777–88. doi: 10.1001/jama.299.23.2777. [DOI] [PubMed] [Google Scholar]
- Trégouët DA, König IR, Erdmann J, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- Willer CJ, Sanna S, Jackson AU, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagyu H, Ishibashi S, Chen Z, et al. Overexpressed lipoprotein lipase protects against atherosclerosis in apolipoprotein E knockout mice. J Lipid Res. 1999;40:1677–1685. [PubMed] [Google Scholar]
- Yu KC, Cooper AD. Postprandial lipoproteins and atherosclerosis. Front Biosci. 2001;6:D332–354. doi: 10.2741/yu. [DOI] [PubMed] [Google Scholar]
- Zacho J, Tybjaerg-Hansen A, Jensen JS, et al. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. 2008;359:1897–1908. doi: 10.1056/NEJMoa0707402. [DOI] [PubMed] [Google Scholar]
- Zhong S, Sharp DS, Grove JS, et al. Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J Clin Invest. 1996;97:2917–2923. doi: 10.1172/JCI118751. [DOI] [PMC free article] [PubMed] [Google Scholar]