

1. Introduction
Most organic molecules are inherently modular in their constitution. With respect to the molecules found in living systems, this modularity is a direct consequence of the fact that nearly all biosynthetic systems are based on the iterative coupling of bifunctional building blocks.i For example, polypeptides are built from amino acids, oligonucleotides are derived from nucleotide monomers, and oligosaccharides are stitched together from individual sugar units. Interestingly, most small molecule natural products are similarly constructed via the iterative coupling of bifunctional building blocks, e.g., polyketides from malonyl-CoA or methylmalonyl-CoA units, non-ribosomal peptides from amino acids, polyterpenes from isopentenyl pyrophosphate or dimethylallyl pyrophosphate, and fatty acids from malonyl-CoA.i Similarly, many man-made pharmaceuticals are also highly modular because they are constructed by using different reactions to assemble collections of small building blocks, typically cyclic and heterocyclic fragments and their associated appendages. Thus, modularity appears to be a remarkably general feature of many of the molecules that are targeted for synthesis in the laboratory.
Despite this common modularity, the strategies utilized for making polypeptides, oligonucleotides, and oligosaccharides are very different than those typically used to prepare small molecules. Specifically, all of the former classes of compounds are almost always constructed via iterative coupling of suitably-protected forms of their consituent monomers.ii Organic polymers are similarly prepared in this fashion.iii Due to the powerfully simple nature of this iterative coupling approach, these processes are now increasingly performed in a fully-automated fashion.ii,iii With peptides and oligonucleotides, the advanced development of such automation has made it possible for even non-chemists to routinely prepare these types of compounds for a wide range of applications.
In stark contrast, it is typical for a synthetic chemist to develop a unique, customized strategy for each small molecule that is targeted for preparation in the laboratory. As a result, the synthesis of small molecules remains a relatively complex, unsystematized, and inflexible process practiced almost exclusively by highly trained specialists. Driven by the hypothesis that the inherent modularity in small molucules remains largely underutilized, we have established a research program that aims to develop a unified strategy for the construction of these compounds via the iterative coupling of bifunctional building blocks.iv,v,vi,vii Specifically, we have targeted the development of building blocks representing substructures that appear frequently in natural products and man-made pharmaceuticals and the chemistry that will enable their precise union via iterative, metal-mediated cross-coupling reactions. In the idealized form of this envisioned “iterative cross-coupling” (ICC) approach, building blocks having all of the required functional groups preinstalled in the correct oxidation state and with the desired stereochemical relationships are iteratively united using only a single, stereospecific cross-coupling reaction (Figure 1). In addition to being simple, efficient, and potentially amenable to automation, the modularity of this approach makes it inherently well-suited for generating diverse collections of compounds simply by substituting modified building blocks into the same synthesis pathway. It is anticipated that the advanced development of this ICC strategy will substantially enable the laboratory synthesis of a wide range of natural products, pharmaceuticals, and organic materials, and may even extend the power of small molecule synthesis to the non-chemist.
Figure 1.

Analogous strategies for the synthesis of peptides and small molecules.
As described in this review, N-methyliminodiacetic acid (MIDA) boronatesviii represent a highly promising platform for this type of synthesis strategy. These building blocks are remarkably convenient to prepare, analyze, purify, and store. The MIDA boronate functional group is also stable to anhydrous cross-coupling conditions but easily hydrolyzed with mild aqueous base, thereby enabling the controlled, ICC of B-protected “haloboronic acids.”iv,v,vi In addition, MIDA boronates are remarkably stable to a wide range of common reaction conditions and chromatography, which makes possible the facile preparation of complex borane building blocks from simple MIDA boronate starting materials via multistep synthesis.vi,vii Importantly, many MIDA boronate building blocks are now commercially-available worldwide from Sigma-Aldrich (www.sigmaaldrich.com/mida). This review aims to enable the effective utilization of this platform and the ICC strategy to promote the simple, efficient, and flexible construction of small molecules.
2. Synthesis of MIDA boronates
N-Methyliminodiacetic acidix (Figure 2) is non-toxic, biodegradable,x and commercially-available. xi It can also be conveniently and efficiently synthesized on large scale for trivial costxii from the commodity chemical iminodiacetic acid.xiii
Figure 2.

N-Methyliminodiacetic acid (MIDA)
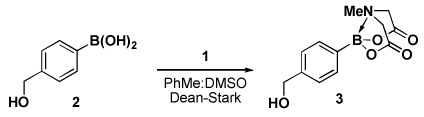
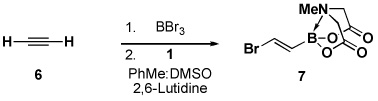
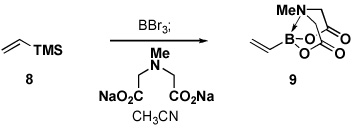
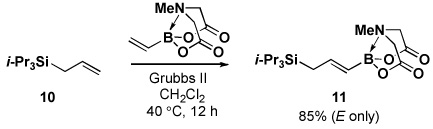
Four different methods for the synthesis of MIDA boronates are shown in Table 1. Many boronic acids can be easily transformed into the corresponding MIDA boronates simply via condensation with MIDA under Dean-Stark conditions (Table 1, entry 1).viii,iv,vi The removal of water by a variety of alternative techniques (e.g., molecular sieves, azeotropic drying with CH3CN, etc.) can also promote full conversion to the MIDA boronate product. Typically this process requires heating to at least 40 °C, and the use of DMSO as a co-solvent is required to partially dissolve the MIDA ligand. Alkenyl MIDA boronates can be synthesized via a bromoboration of an alkyne to form the corresponding dibromoborane followed by trapping with MIDA in the presence of 2,6-lutidine (Table 1, entry 2).v Alternatively, a one-pot procedure has been developed in which organotrimethylsilanes can be converted directly into MIDA boronates via transmetalation with BBr3 followed by trapping with the disodium salt of MIDA (Table 1, entry 3).vii This approach was employed in the efficient synthesis of vinyl MIDA boronate (9) where condensation of MIDA with the related vinyl boronic acid or vinylboronate species failed. In addition, a variety of olefins can be transformed directly to alkenyl MIDA boronates via cross-metathesis with vinyl MIDA boronate (Table 1, entry 4).vii This approach avoids boronic acid intermediates and is notable for its generality, efficiency, and mildness. Moreover, in contrast to previous reports involving the use of vinyl or propenyl pinacol boronic esters,xiv cross-metathesis with vinyl MIDA boronate yields only the E isomer and the products are uniformly compatible with silica gel chromatography (vide infra).
Table 1.
Currently-available methods for the synthesis of MIDA boronates
| General Methods | Specific Examples | |
|---|---|---|
| 1. | 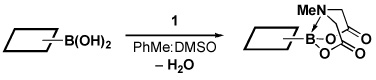 |
 |
| 2. | 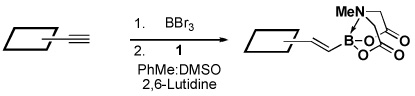 |
 |
| 3. | 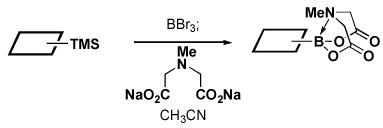 |
 |
| 4. | 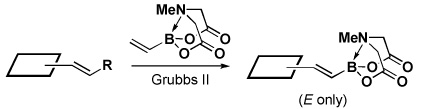 |
 |
3. Physical properties of MIDA boronates
MIDA boronates possess a number of highly enabling physical properties that make them attractive as a platform for ICC and as convenient alternatives to boronic acids and esters for a wide range of other applications. Specifically, MIDA boronates are monomeric, free-flowing, crystalline solids which are stable to storage on the benchtop under air. MIDA boronates are also universally compatible with silica gel chromatography, allowing facile purification and convenient reaction monitoring by TLC.iv,v,vi,vii If the goal is to separate different MIDA boronates of similar polarity, a ternary eluent of hexanes:EtOAc and up to 10% methanol is most effective. With these conditions, we have found that even diastereomeric mixtures of MIDA boronates can be resolved. For the purification of non-polar MIDA boronates hexanes:EtOAc is an effective eluent. Additionally, acetic acid is generally compatible as a co-eluent in most solvent mixtures. Dichloromethane:methanol is a useful eluent for TLC analysis, but some decomposition of the MIDA boronates can occur if this eluent is used for preparative chromatography. Similiarly, MIDA boronates should not be left to stand in solutions containing alcohols for more than an hour.
This compatibility with silica gel chromatography and the facility with which MIDA boronates can be formed from the corresponding boronic acids, makes it possible to utilize MIDA boronate formation/purification as a powerful tool to generate high purity boronic acids from crude mixtures containing many non-boronic acid byproducts. Specifically, we have found that adding a small excess of MIDA to a crude mixture of boronic acid and performing a Dean-Stark complexation leads to the formation of the corresponding MIDA boronate, while the other impurities remain largely unchanged. If the boronic acid is desired, a simple hydrolysis of the purified MIDA boronate with 1M aqueous NaOH followed by extraction of the boronic acid into an organic solvent yields the boronic acid in very pure form.
MIDA boronates are also easily purified by crystallization. A generally effective strategy is to dissolve the crude MIDA boronate in a minimum volume of acetone at 23 °C and then slowly add Et2O until the solution becomes cloudy. Crystals usually begin to form within several minutes. Additional Et2O should be added periodically to the mixture to re-establish the cloud-point. The crystallization is complete when the addition of Et2O no longer clouds the solution. Alternative crystallization solvents include acetonitrile:Et2O and EtOAc:Et2O. X-ray quality crystals are conveniently prepared via vapor diffusion of petroleum ether into an acetone solution of the MIDA boronate.
Another important feature of MIDA boronates is that they are soluble in many organic solvents. Reactions are typically performed using THF, dioxane, dichloromethane, DMF, toluene, DMSO, acetonitrile, acetone, or 1,2-dichloroethane. Prolonged exposure of MIDA boronates to aqueous conditions or alcoholic solvents leads eventually to hydrolysis of the MIDA ligand, and this effect is accelerated with heating or in the presence of base. However, water or alcoholic solvents have been successfully employed as co-solvents in some reactions with MIDA boronates. Further, MIDA boronates are generally compatible with aqueous extractions employing water, brine, aqueous acids (e.g., aq. HCl or NH4Cl), and even some oxidative or reductive aqueous solutions (e.g., aq. H2O2 at pH < 6 or aq. NaS2O3). Remarkably, even saturated aqueous NaHCO3 is tolerated in the absence of alcoholic solvents. Aqueous extractions are typically performed with EtOAc or dichloromethane as an organic phase. For highly polar MIDA boronates, solvent mixtures of EtOAc:acetone or THF:Et2O are convenient. As described below, despite this widespread stability, MIDA boronates are easily hydrolyzed to yield the corresponding boronic acids using very mild aqueous basic reagents at 23 °C.
Interestingly, in contrast to MIDA boronates such as 3, similarly pyramidalized N-methyldiethanolamine adducts such as 12 (Figure 3) are not stable to silica gel. As described below, again in contrast to the MIDA boronates, N-methyldiethanolamine adducts are reactive under cross-coupling and many other common reaction conditions.iv,vi,xv The remarkable (and in many cases unique) stability of MIDA boronates to storage under air, chromatography, aqueous work-ups, as well as cross-coupling and many other reaction conditions (vide infra) is attributed to the extreme conformational rigidity of the fused bicyclic [N-methyliminidiacetate-O,O’,N]borane framework. Specifically, as shown in Figure 3, variable temperature NMR experiments with a DMSO solution of 3 reveal no coalescence of the diastereotopic methylene protons of the MIDA backbone, even at 150 °C.vi,xvi In contrast, the same experiment with 12 reveals coalescence of the diastereotopic methylene protons of the diethanolamine backbone over a temperature range of 23 to 60 °C.vi,xvi These studies are consistent with the conclusion that the potentially reactive boron p orbital and nitrogen lone pair of MIDA boronates are kinetically inaccessible at <150 °C.
Figure 3.

Variable temperature NMR studies with MIDA boronate and N-methyldiethanolamine adducts that demonstrate the unique and remarkable conformational rigidity of the MIDA boronate framework.
4. Iterative Cross-Coupling (ICC) with halo MIDA boronates
The now routinely automated process of iterative peptide couplingiia represents an inspiring benchmark for a potentially general strategy for making molecules in the laboratory. It is interesting to note that peptides are quite complex in structure, having many different functional groups with varied oxidation states and a large number of stereogenic centers. However, the synthesis of peptides is very simple, involving the use of a single reaction to iteratively assemble a collection of bifunctional building blocks having all of the required functional groups and stereochemistry preinstalled. With the goal of developing an analogous process for the laboratory construction of small molecules, we decided to focus on the Suzuki-Miyaura reaction and the ICC of bifunctional “haloboronic acids” (Figure 1).
To avoid random oligomerization of a haloboronic acid under cross-coupling conditions, it was necessary to find a way to reversibly attenuate the reactivity of one end of this type of bifunctional reagent, analogous to the use of a protective group to control the reactivity of the amine terminus of an amino acid.xvii Toward this goal, we chose to focus on controlling the reactivity of a boronic acid.
It is hypothesized that a vacant and Lewis acidic boron p-orbital is required for transmetallation of a boronic acid under Suzuki-Miyaura cross-coupling conditions (Figure 4A).xviii Consistent with this, complexation with electron-donating, Lewis basic ligands is known to attenuate the reactivity of boronic acids towards cross-coupling (Figure 4B).xviiia For example, pinacol boronic esters can be less reactive than the corresponding boronic acids under anhydrous coupling conditions.xix This reactivity attenuation can be attributed to the decreased Lewis acidity of the boron p-orbital via conjugation with the ligand heteroatom lone pairs.xviiia This same approach has been utilized with a variety of other divalent heteroatomic ligands.xx There is an inherent limitation, however, that precludes the general utilization of this approach for complex small molecule synthesis. Specifically, conjugation between the heteroatom lone pairs and the boron p orbital creates relatively strong boron-heteroatom bonds, creating a high kinetic barrier for bond cleavage. Moreover, the equilibrium between the boronic acid and the corresponding boronic ester typically lies strongly towards the latter, creating an additional thermodynamic barrier to hydrolysis. As a result, cleaving these types of ligands to regenerate the boronic acid typically requires harsh conditionsxviii,xix,xx and/or additional reagents to destroy the divalent ligand after it has been cleaved.xxi These types of conditions can be problematic in the context of complex small molecule synthesis.
Figure 4.

A. Transmetalation in the Suzuki-Miyaura reaction requires a vacant and Lewis acidic p-orbital. B. Strongly electron-donating divalent ligands can attenuate boronic acid reactivity but typically require relatively harsh conditions for cleavage. C. The reactivity of a boronic acid can be reversibly attenuated with a trivalent ligand.
Recognizing the inherent limitations of this approach, we focused on an alternative strategy (Figure 4C).iv Given that the boron p-orbital is predicted to be critical for transmetalation of a boronic acid, we hypothesized that removing this p-orbital via rehybridizing an sp2-hybridized boronic acid to its sp3-hybridized boronate via complexation with a trivalent heteroatomic ligand would eliminate reactivity towards cross-coupling. Further increasing our interest in this approach, it is known that boron-heteroatom bonds in tetrahedral adducts are weaker than those in their tricoordinate counterparts.xxii For example, the pyramidalization of trimethyl borate via complexation with ammonia actually weakens the boron-oxygen bonds by about 10–12 kcal/mol.xxiii Thus, we felt that it might be possible to find relatively mild conditions that could hydrolyze this type of pyramidalized boronate and regenerate the reactive boronic acid. After surveying a series of trivalent heteroatomic ligands, we discovered that MIDA boronates embody all of these expectations and represent a powerful platform for ICC chemistry.
Specifically, in a competition experiment between tolylboronic acid (14b) and para-n-butylphenyl MIDA boronate under Buchwald-typexxiv anhydrous Suzuki-Miyaura cross-coupling conditions with p-bromoanisaldehyde, we observed a 24:1 ratio of products 15 and 16, consistent with a strong preference for cross-coupling of the sp2-hybridized boronic acid.iv Interestingly, a wide range of non-aryl substituents were tolerated on the nitrogen atom (e.g. Table 2, entry 2). However, the corresponding diethanolamine adduct (14d), lacking the carbonyl units of MIDA, was equally reactive as the boronic acid (Table 2, entry 4). As described above, this difference in reactivity is attributed to differences in conformational flexibilty of these two complexes.
Table 2.
Trivalent N-alkyliminodiacetic acid ligands attenuate the reactivity of boronic acids.
| Entry |  |
15 : 16* | |
|---|---|---|---|
| 1 | 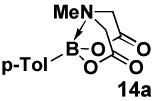 |
24 : 1.0 | 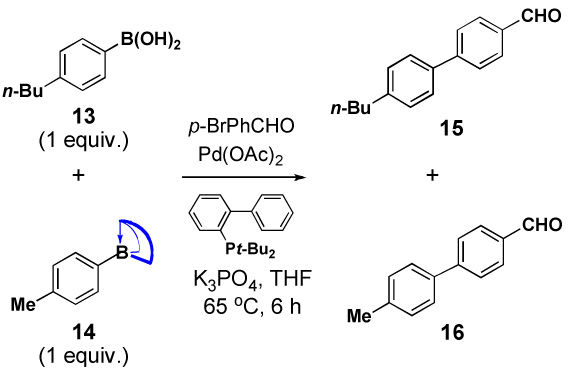 |
| 2 |  |
1.0 : 1.0 | |
| 3 | 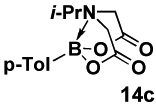 |
25 : 1.0 | |
| 4 | 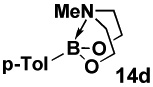 |
1.0 : 1.0 | |
HPLC, average of three runs.
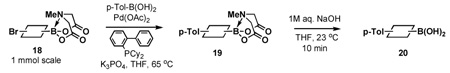
Encouraged by these results, we set out to prepare a series of bifunctional B-protected haloboronic acids and explore their capacity to undergo selective cross-coupling at the halide terminus. The efficient synthesis of aryl heteroaryl, alkenyl and alkyl derivatives was achieved via simple condensation of the corresponding boronic acids with MIDA under Dean-Stark conditions (Scheme 1). As shown in Table 3, this approach proved to be remarkably general, with the same ligand similarly protecting aryl, heteroaryl, alkenyl and alkyl boronic acids. Moreover, consistent with our initial hypothesis, the MIDA boronate products of these reactions can all be hydrolyzed under mild aqueous basic conditions (1N aq. NaOH/THE, 23 °C, 10 min) to generate the corresponding reactive boronic acid.
Scheme 1.
The general preparation of halo MIDA boronates.

Table 3.
The general use of halo MIDA boronates in ICC
 | |||||
|---|---|---|---|---|---|
| Entry | 18 | Protected product | % Yield | Deprotected product | % Yield |
| 1 | 18a | 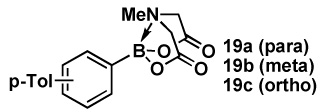 |
87 | 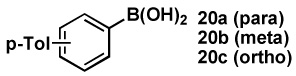 |
86 |
| 2 | 18b | 85 | 92 | ||
| 3 | 18c | 80 | 97a | ||
| 4b | 18d | 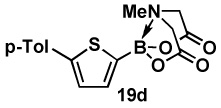 |
81 | 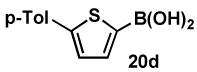 |
88 |
| 5 | 18e | 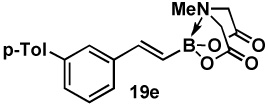 |
82 | 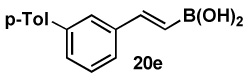 |
83 |
| 6 | 18f | 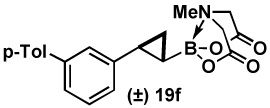 |
94 | 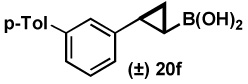 |
91 |
B-Deprotection was also achieved via treatment with saturated aq. NaHCO3/MeOH, 23 °C, 6 hours (85%).
2-(Dicyclohexylphosphino)-2',4',6'-triisopropyl-1,1'-biphenyl (X-Phos) was used as the phosphine ligand.
Polyenes are especially challenging synthetic targets because of the sensitivity of this framework to light, oxygen, and acid. It is also critical to control the stereochemistry of each double bond. The ICC approach is particularly well-suited to making these types of compounds due to the mild and stereospecific nature of metal-mediated cross-coupling. Given the prevalence of akenyl and polyenyl subunits in both natural products and pharmaceutical targets, we developed a collection of bifunctional building blocks specifically designed to enable polyene synthesis via ICC.v
As described in Table 1, trans-(2-bromovinyl) MIDA boronate 7 can be prepared via bromoboration of acetylenexxv followed by complexation with MIDA in the presence of 2,6-lutidine. An alternative and more convenient procedure involves transmetalation of 1-bromo-2-(trimethylsilyl)ethylene with BBr3xxvi followed by trapping with MIDA2−Na+2.vii This building block has proven to be a remarkably versatile cross-coupling partner (Scheme 2).v Specifically, Suzuki-Miyaura, Stille, and Heck couplings were all achieved at the bromide terminus without perturbing the MIDA boronate. A series of bis-metallated lynchpin-type reagents were also created via Sonagashira coupling with TMS acetylene, Miyaura borylation with pinacolatodiborane 25, and a triply metal-selective (Zn vs. Sn and B) Negishi coupling with bismetalleted olefin 27.
Scheme 2.

In the first example of boron-selective coupling of a differentially-ligated bis-borylated building block, 26 was selectively coupled with trans-1-chloro-2-iodoethylene at the sp2-hybridized pinacol boronic ester terminus to generate chlorodienyl MIDA boronate 29 (Scheme 3). This type of boron-selective coupling of differentially-ligated diboron reagents has the potential to be a generally useful strategy.xxvii A more well-precedented Sn vs B couplingxxviii between bis-metallated diene 28 and trans-1-chloro-2-iodoethylene generated chlorotrienyl MIDA boronate 30.
Scheme 3.

The olefin cross-metathesis route to MIDA boronates is remarkably tolerant to a wide range of functional groups, including halogens. Thus, this method is particularly well suited for preparing various haloalkenyl MIDA boronates, as shown for a series of bromostyrene derivatives in Scheme 4.vii
Scheme 4.

The capacity to prepare and selectively couple bifunctional halo MIDA boronates enables one to envision the synthesis of natural products or pharmaceuticals using only a single reaction iteratively to assemble a collection of pre-assembled building blocks. This was first demonstrated with the total synthesis of ratanhine,iv a complex neolignan isolated from the Ratanhiae radix by Arnone and coworkers in 1990.xxix This natural product was retrosynthetically fragmented using recursive Suzuki-Miyaura transforms to generate four simpler building blocks 35 – 38 (Scheme 5). There were several challenges associated with this plan that were expected to test the limits of the MIDA-based ICC methodology. First, couplings of alkenyl boronic acids tend to be less efficient than those with aryl derivatives, making the selective coupling between 35 and aryl MIDA boronate 36 unsecured. In addition, 2-substituted heterocyclic boronic acids such as 36 are notoriously unstable and difficult to purify, store, and cross-couple.xxx Finally, the coupling of highly deactivated bromoaryl MIDA boronate 37 was expected to demand more forcing reaction conditions that would test the limits of stability of the MIDA boronate functionality.
Scheme 5.

Despite these challenges, the first total synthesis of ratanhine was achieved via ICC as shown in Scheme 6. Specifically, a selective coupling between propenylboronic acid 35 and bromobenzofuranyl MIDA boronate 36 proceeded smoothly to generate 2-substituted benzofuranyl MIDA boronate 39. Remarkably, while the corresponding benzofuranyl boronic acid decomposed over the course of several days, MIDA boronate 39 was stored on the benchtop under air without noticeable decomposition for more than 6 months. This MIDA boronate was hydrolyzed under mild conditions and the resulting boronic acid was immediately utilized in a cross-coupling reaction with bromoaryl MIDA boronate 37. As expected, this coupling required increased temperature (80 °C in a sealed tube) and extended reaction time (28 h). Remarkably, however, the MIDA boronate functional group was stable to these forcing conditions, yielding the highly conjugated MIDA boronate product 40. A final sequence of boronic acid deprotection and coupling with alkenyl bromide 38 and MOM-ether deprotection completed the first total synthesis of this natural product. More importantly, to the best of our knowledge, this represents the first total synthesis of any natural product in which a single reaction was utilized iteratively to assemble all of the required building blocks.
Scheme 6.

This same ICC strategy has proven to be highly effective in the synthesis of polyene natural products.v Specifically, as shown in Scheme 7, all-trans-retinalxxxi was prepared simply via ICC of boronic acid 42, bromoalkenyl MIDA boronate 7, and alkenyl bromide 44. In a similar vein, β-parinaric acidxxxii was prepared via the ICC of butenylboronic acid 46, chlorodienyl MIDA boronate 29, and alkenyliodide 48 (Scheme 8). Finally, the notoriously challenging heptaene framework of the polyene natural product amphotericin B was prepared using only the Suzuki-Miyaura reaction to assemble a collection of bifunctional haloalkenyl MIDA boronates (Scheme 9).
Scheme 7.

Scheme 8.

Scheme 9.

Demonstrated by these examples, the ICC approach has substantial potential to enable the simple, efficient, and flexible construction of small molecules.
6. Multistep synthesis of complex boronic acids from simple MIDA boronates
To avoid a general incompatibility with synthetic reagents, it is typically necessary to introduce the boronic acid functional group just prior to its utilization in a cross-coupling or other type of reaction. However, most of the methods that are available for achieving this have poor functional group tolerance. Collectively, these limitations can render the synthesis of complex boronic acids very challenging. This can preclude the use of boronic acids in complex molecule synthesis and represents a potential bottleneck for the development of a truly general ICC-based approach.
Some sterically-bulky boronic esters are known to be more tolerant to synthetic reagents;xxxiii however, removing these ligands to generate a targeted boronic acid usually requires harsh conditions that are generally incompatible with sensitive building blocks. Trifluoroborate salts represent highly useful surrogates for boronic acidsxxxiv and are compatible with many synthetic reagents.xxxv These features have provided novel access to many new organoborane building blocks. However, the incompatibility of the trifluoroborate salts with column chromatography can limit the utilization of these reagents in multistep synthesis, which is often necessary for accessing structurally and/or stereochemically complex building blocks.
Overcoming all of these limitations, we have recently found that the MIDA boronate functional group is stable to a wide range of common synthetic reagents.vi Combined with the general compatibility of MIDA boronates with chromatography and the capacity to release the corresponding boronic acids under very mild conditions, this stability enables the first reliable approach for the multistep synthesis of complex boronic acids from simple organoborane starting materials.
Due to the absence of a reactive p-orbital on boron, we hypothesized and confirmed experimentally that the MIDA boronate functional group would be stable to mild synthetic reagents. For example, hydroxymethylphenyl MIDA boronate 3 can be smoothly oxidized under Swern conditions to generate the corresponding benzaldehyde (Scheme 10). However, we were very surprised to find that MIDA boronates are even stable to the very strongly acidic and oxidizing Jones conditions (H2SO4/CrO3). This latter stability is highly unique; i.e., under these same conditions, the corresponding boronic acid (56a), pinacolboronic ester (56b), 1,8-diaminonaphthalene adduct (56c), trifluoroborate salt (56d), and N-methyldiethanolamine boronatexxxvi (56e) all decomposed. Similar to that which we observed under cross-coupling conditions, the remarkable difference in reactivity between the MIDA and diethanolamine boronates is likely related to the differences in conformational flexibility of the two complexes (Figure 3).
Scheme 10.

This unique compatibility with strong acid and oxidants suggested that MIDA boronates could be stable to a wide range of reaction conditions. In fact, even triflic acid (pKa -14) was tolerated, enabling the p-methoxybenzylation of 3 and the reversal of this transformation with DDQ (Scheme 11). Similarly silation and desilation were well-tolerated, as was transformation into the corresponding benzyliodide with PPh3/I2.
Scheme 11.

This latter reaction suggested compatibility with soft nucleophiles. In this vein, benzaldehyde 54 was successfully utilized in a series of carbon-carbon bond forming reactions including the Evans aldol, Horner-Wadsworth-Emmons, and Takai olefination reactions. Reductive amination and aldehyde reduction were also well-tolerated (Scheme 12).
Scheme 12.

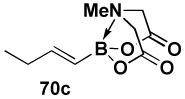
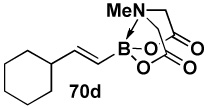
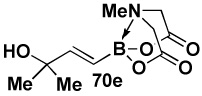
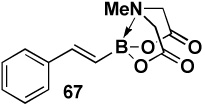
In another study, broad compatibility was also observed using vinyl MIDA boronate 9 as a versatile starting material (Scheme 13).vii Specifically, cyclopronation of 9 produced cyclopropyl MIDA boronate in excellent yield.xxxvii Remarkably, epoxidation of this olefinxxxviii with mCPBA was also well-tolerated and even this product was stable to column chromatography and benchtop storage under air. This same versatile building block 9 was successfully engaged in the Heck reactionxxxix to yield styrenyl derivative 66 as a single stereo-and regioisomer. Similarly, the White catalystxl promoted an efficient oxidative Heckxli-type reactionxlii to yield 67. As described above (Scheme 4), this same vinyl MIDA boronate building block is also an excellent substrate for olefin cross-metathesis (analogous to tert-butyl ethylene), yielding E-octenyl MIDA boronate 68 as air- and chromatographically stable crystalline solid and a single stereoisomer (Scheme 13). Fortunately, this approach has proven to be quite general and represents a very useful new approach to prepare a range of (E)-alkenyl MIDA boronates (Table 4).
Scheme 13.

Table 4.
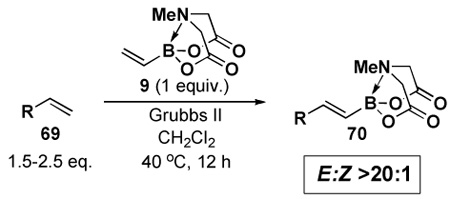 | |||
|---|---|---|---|
| Entry | cross partner | cross product | isolated yield (%) |
| 1 | 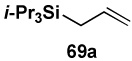 |
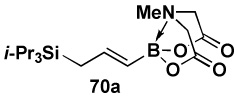 |
85 |
| 2 | 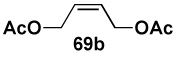 |
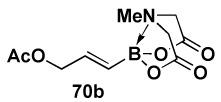 |
84 |
| 3 | 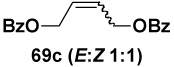 |
 |
98 |
| 4 | 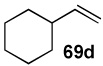 |
 |
96 |
| 5 | 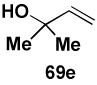 |
 |
94 |
| 6 | 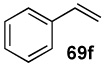 |
 |
93 |
This broad compatibilty of the MIDA boronate functional group can enable the transformation of simple MIDA boronate starting materials into otherwise difficult to access complex boronic acids for use in a variety of synthesis applications. Specifically regarding the ICC strategy, this approach can also provide access to a wide range of structurally complex B-protected haloboronic acids.
To explore the enabling potential of this approach, we targeted the total synthesis of the natural product crocacin Cxliii via ICC. As shown in Scheme 14, this molecule was retrosynthesized via recursive cross-coupling reactions into known building blocks 72 and 74 as well as the unknown, complex iodoalkenyl MIDA boronate 73. Preparation of the latter represents a significant challenge that we hypothesized could be overcome via multistep synthesis starting with simple MIDA boronate 75 (Scheme 15).
Scheme 14.

Scheme 15.

In practice, a Paterson aldol reaction between 75 and 76 followed by diastereoselective reduction of the resulting β-hydroxyketone yielded diol 77. Importantly, the small amounts of diastereomeric byproducts that are typically generated in these types of transformations were readily removed by taking advantage of the compatibility of the MIDA boronate functional group to silica gel chromatography. A subsequent sequence of permethylation with Meerwein’s salt, oxidative cleavage of the PMB ether, oxidation of the resulting primary alcohol, and Takai olefination yielded the targeted complex halo MIDA boronate 73. Importantly, 75, 73, and all intermediates were compatible with chromatography and storage on the benchtop under air. With B-protected haloboronic acid 73 in hand, the synthesis of crocacin C was readily achieved via ICC (Scheme 16).
Scheme 16.

7. Prospectus
As described herein, the inherent modularity found in many of the small molecules targeted for synthesis in the laboratory stands to be more effectively harnessed via the ICC-based approach. Analogous to the synthesis of peptides, oligonucleotides, and oligosaccharides, this strategy has the potential to enable the preparation of a wide range of small molecules via the simple, iterative union of pre-assembled, bifunctional building blocks. Due to their ease of synthesis, putification, characterization, and storage, their reversibly attenuated reactivity under cross-coupling conditions, and their compatibility with a wide range of common synthetic reagents, MIDA boronates represent a powerful platform for the development of this type of synthesis strategy.
Looking forward, the ever-expanding scope of the Suzuki-Miyaura coupling suggests that the potential generality of this approach could be substantial. Particularly critical to realizing this potential will be to find a way to form Csp3–Csp2 and even Csp3–Csp3 bonds with the same efficiency and stereospecificity that is now routinely achieved with Csp2–Csp2 linkages. The discovery of additional methods to prepare MIDA boronates that do not proceed through the intermediacy of a difficult to access and/or unstable boronic acid will also be vital. Moreover, to realize the ultimate goal of developing a machine with the capacity for fully-automated ICC, it will be important to identify Suzuki-Miyaura cross-coupling conditions that are maximally generalxxiv (to avoid the requirement for ad hoc optimization of conditions for each combination of coupling partners) and amenable to translation to the solid-phase or some other form of iterative synthesis enabling technology. While these challenges are admittedely considerable, we are convinced that they each can be solved. Achieving these goals could have a substantial impact on the synthesis of small molecules in the laboratory and may ultimately even expand the power of this discovery engine to the non-chemist.
References
- i.(a) Fischbach MA, Walsh CT. Chem. Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]; (b) Garret, Grisham . Biochemistry. Saunders College Publishing; 1995. [Google Scholar]
- ii.(a) Merrifield RB. Angew Chem. Int. Ed. 1985;24:799. [Google Scholar]; (b) Caruthers MH. Science. 1985;230:281. doi: 10.1126/science.3863253. [DOI] [PubMed] [Google Scholar]; (c) Plante OJ, Palmacci MR, Seeberger PH. Science. 2001;291:1523. doi: 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- iii.(a) Zhang J, Moore JS, Xu Z, Aguirre RA. J. Am. Chem. Soc. 1992;114:2273–2274. [Google Scholar]; (b) Young JK, Nelson JC, Moore JS. J. Am. Chem. Soc. 1994;116:10841–10842. [Google Scholar]; (c) Feuerbacher N, Vogtle F. Topics in Current Chemistry. 1998;197:1–16. [Google Scholar]
- iv.Gillis EP, Burke MD. J. Am. Chem. Soc. 2007;129:6716–6717. doi: 10.1021/ja0716204. [DOI] [PubMed] [Google Scholar]
- v.Lee SJ, Gray KC, Paek JS, Burke MD. J. Am. Chem. Soc. 2008;130:466–468. doi: 10.1021/ja078129x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vi.Gillis EP, Burke MD. J. Am. Chem. Soc. 2008;130:14084–14085. doi: 10.1021/ja8063759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vii.Uno BE, Gillis EP, Burke MD. Tetrahedron. 2009 In Press (DOI 10.1016/j.tet.2008.11.010) [Google Scholar]
- viii.(a) Mancilla T, Contreras R, Wrackmeyer B. J. Organomet. Chem. 1986;307:1–6. [Google Scholar]; (b) Mancilla T, coworkers Arkivoc. 2005:366–376. [Google Scholar]
- ix.(a) Berchet GJ. Org. Syn. 1938;18:56. [Google Scholar]; (b) Childs AF, Goldsworthy LJ, Harding GF, King FE, Nineham AW, Norris WL, Plant SGP, Selton B, Tompsett ALL. J. Chem. Soc. 1948:2174–2177. [Google Scholar]; (c) Chase BH, Downes AM. J. Chem. Soc. 1953:3874–3877. [Google Scholar]
- xx.Warren CB, Malec EJ. Science. 1972;176:277–279. doi: 10.1126/science.176.4032.277. [DOI] [PubMed] [Google Scholar]
- xi.Aldrich: M51008
- xii.Ballmer SG, Gillis EP, Burke MD. submitted [Google Scholar]
- xiii.Tens of thousands of metric tons of iminodiacetic acid is produced annualy worldwide: Yangong Z. China Chemical Reporter. 2005:16..
- ivx. Blackwell HE, O’Leary DJ, Chatterjee AK, Washenfelder RA, Bussmann AA, Grubbs RH. J. Am. Chem. Soc. 2000;122:58–71. Morrill C, Grubbs RH. J. Org. Chem. 2003;68:6031–6034. doi: 10.1021/jo0345345. Njardarson JT, Biseas K, Danishefsky SJ. Chem. Commun. 2002;23:2759–2761. doi: 10.1039/b209941a. Nicolaou KC, Li A, Edmonds DJ. Angew. Chem. Int. Ed. 2006;45:7086–7090. doi: 10.1002/anie.200603892. Jankowska M, Pietraszuk C, Marciniec B, Zaidlewicz M. SynLett. 2006;11:1695–1698. Dakas PY, Barluenga S, Totzke F, Zirrgiebel U, Winssinger N. Angew. Chem. Int. Ed. 2007;46:6899–6902. doi: 10.1002/anie.200702406. Wu T, Gao J. Org. Lett. 2008;10:1533–1536. doi: 10.1021/ol800137f. Ring Closing Metathesis reactions: Ashe AJ, III, Fang X, Kampf JW. Organometallics. 2000;19:4935–4937. Ashe AJ, III, Hong Y, Fang X, Kampf JW. Organometallics. 2002;21:4578–4580..
- xv. For some important exceptions, see: Ebdrup S, Jacobsen P, Farrington AD, Vedso P. Bioorg. Med. Chem. 2005;13:2305–2312. doi: 10.1016/j.bmc.2004.12.042. Vedso P, Olesen PH, Hoeg-Jensen T. Synlett. 2004;5:892–894. Kirihata M, Morimoto T, Mizuta T, Ichimoto I. Bioscience, Biotechnology, and Biochemistry. 1995;59:2317–2318. Kirihara M, Morimoto T, Ichimoto I. Bioscience, Biotechnology, and Biochemistry. 1993;57:1940–1941. doi: 10.1271/bbb.57.69. Yamamoto Y, Seko T, Rong FG, Nemoto H. Tetrahedron Lett. 1989;30:7191–7194..
- xvi.These results are consistent with previous studies: (a) Ref 8, Psarras TG, Zimmerman HK, Rasiel Y, Weidman H. Liebigs Ann. Chem. 1962;655:48–54. Contreras R, García C, Mancilla T, Wrackmeyer B. J. Organomet. Chem. 1983;246:213–217..
- xvii.(a) Bergmann M, Zervas L. Ber. Dtsch. Chem. Ges. 1932;65:1192. [Google Scholar]; (b) Carpino LA, Han GY. J. Am. Chem. Soc. 1970;92:5748. [Google Scholar]; (c) Tarbell DS, Yamamoto Y, Pope BM. P{roc. Nat. Acad. Sci. USA. 1972;69:730–732. doi: 10.1073/pnas.69.3.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xviii.(a) Matos K, Soderquist J. J. Org. Chem. 1998;63:461–470. doi: 10.1021/jo971681s. [DOI] [PubMed] [Google Scholar]; (b) Miyaura N. J. Organomet. Chem. 2002;653:54–57. [Google Scholar]; (c) Braga AAC, Morgon NH, Ujaque G, Maseras F. J. Am. Chem. Soc. 2005;127:9298–9307. doi: 10.1021/ja050583i. [DOI] [PubMed] [Google Scholar]
- xix.(a) Holmes D, Chotana GA, Maleczka RE, Jr, Smith MR., III Org. Lett. 2006;8:1407–1410. doi: 10.1021/ol060205y. [DOI] [PubMed] [Google Scholar]; (b) Gillis EP, Burke MD. unpublished results [Google Scholar]
- xx.(a) Deng X, Mayeux A, Cai C. J. Org. Chem. 2002;67:5279–5283. doi: 10.1021/jo0257750. [DOI] [PubMed] [Google Scholar]; (b) Hohn E, Pietruszka J. Adv. Synth. Catal. 2004;346:863–866. [Google Scholar]; (c) Noguchi H, Hojo K, Suginome M. J. Am. Chem. Soc. 2007;129:758–759. doi: 10.1021/ja067975p. [DOI] [PubMed] [Google Scholar]
- xxi.(a) Nakamura H, Fujiwara M, Yamamoto Y. Bulletin of the Chemical Society of Japan. 2000;73:231–235. [Google Scholar]; (b) Falck JR, Bondlela M, Venkataraman SK, Srinivas D. J. Org. Chem. 2001;66:7148–7150. doi: 10.1021/jo015838z. [DOI] [PubMed] [Google Scholar]; (c) Murphy JM, Tzschucke CC, Hartwig JF. Org. Lett. 2007;9:757–760. doi: 10.1021/ol062903o. [DOI] [PubMed] [Google Scholar]
- xxii.Hall DG. Boronic Acids. Germany: Wiley-VCH; 2005. pp. 3–14. [Google Scholar]
- xxiii.Matteson DS. Stereodirected Synthesis with Organoboranes. Berlin: Springer; 1995. pp. 1–20. [Google Scholar]
- xxiv.(a) Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]; (b) Billingsley K, Buchwald SL. J. Am. Chem. Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]; (c) Ruben M, Buchwald SL. Acc. Chem. Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxv.Hyuga S, Chiba Y, Yamashina N, Hara S, Suzuki A. Chem. Lett. 1987:1757–1760. [Google Scholar]
- xxvi.Singleton DA, Leung SW. J. Organomet. Chem. 1997;544:157–161. [Google Scholar]
- xxvii. For some recent additional examples, see: Molander GA, Sandrock DL. J. Am. Chem. Soc. 2008;130:15792–15793. doi: 10.1021/ja807076d. Noguchi H, Shioda T, Chou C-M, Suginome M. Org. Lett. 2008;10:377–380. doi: 10.1021/ol702420x..
- xxviii.(a) Coleman RS, Walczak MC. Org. Lett. 2005;7:2289–2291. doi: 10.1021/ol050768u. [DOI] [PubMed] [Google Scholar]; (b) Coleman RS, Lu X, Modolo I. J. Am. Chem. Soc. 2007;129:3826–3827. doi: 10.1021/ja070265e. [DOI] [PubMed] [Google Scholar]
- xxix.Arnone A, coworkers Gazzetta Chimica Italiana. 1990;120:397. [Google Scholar]
- xxx.Tyrell E, Brookes P. Synthesis. 2003;4:469–483. [Google Scholar]
- xxxi.Previous cross-coupling-based syntheses of retinoids: Negishi E, Owczarczyk Z. Tetrahedron Lett. 1991;32:6683–6686. (b) ref 13b. Uenishi J, Kawahama R, Yonemitsu O, Wada A, Ito M. Angew. Chem. Int. Ed. 1998;37:320–323. doi: 10.1002/(SICI)1521-3773(19980216)37:3<320::AID-ANIE320>3.0.CO;2-4. Uenishi J, Matsui K, Wada A. Tetrahedron Lett. 2003;44:3093–3096..
- xxxii.Some prior syntheses of β-parinaric acid: Kuklev DV, Smith WL. Chem. Phys. Lipids. 2004;131:215–222. doi: 10.1016/j.chemphyslip.2004.06.001. Goerger MM, Hudson BS. J. Org. Chem. 1988;53:3148–3153. Hayashi T, Oishi T. Chem. Lett. 1985:413–416..
- xxxiii.(a) Matteson DS, Kandil AA, Soundararajan R. J. Am. Chem. Soc. 1990;112:3964–3969. [Google Scholar]; (b) Matteson DS, Man H-W, Ho OC. J. Am. Chem. Soc. 1996;118:4560–4566. [Google Scholar]; (c) Luithle JEA, Pietruszka J. J. Org. Chem. 2000;65:9194–9200. doi: 10.1021/jo0056601. [DOI] [PubMed] [Google Scholar]; (d) Hilt G, Bolze P. Synthesis. 2005;13:2091–2115. [Google Scholar]; (e) Jin B, Liu Q, Sulikowski GA. Tetrahedron. 2005;61:401–408. [Google Scholar]; (f) Gopalarathnam A, Nelson SG. Org. Lett. 2006;8:7–10. doi: 10.1021/ol051861l. [DOI] [PubMed] [Google Scholar]
- xxxiv.(a) Molander GA, Ellis N. Acc. Chem. Res. 2007;40:275–286. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]; (b) Darses S, Genet J-P. Chem. Rev. 2008;108:288–325. doi: 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- xxxv.(a) Molander GA, Petrillo DE. J. Am. Chem. Soc. 2006;128:9634–9635. doi: 10.1021/ja062974i. [DOI] [PubMed] [Google Scholar]; (b) Molander GA, Ribagorda M. J. Am. Chem. Soc. 2003;125:11148–11149. doi: 10.1021/ja0351140. [DOI] [PubMed] [Google Scholar]; (c) Molander GA, Petrillo DE. Org. Lett. 2008;10:1795–1798. doi: 10.1021/ol800357c. [DOI] [PubMed] [Google Scholar]; (d) Molander GA, Gormisky PE, Sandrock DL. J. Org. Chem. 2008;73:2052–2057. doi: 10.1021/jo800183q. [DOI] [PubMed] [Google Scholar]; (e) Pucheault M, Darses S, Genet J-P. J. Am. Chem. Soc. 2004;126:15356–15357. doi: 10.1021/ja044749b. [DOI] [PubMed] [Google Scholar]
- xxxvi.(a) Vedso P, Olesen PH, Hoeg-Jensen T. Synlett. 2004:892–894. [Google Scholar]; (b) Gravel M, Thompson KA, Zak M, Bérubé C, Hall DG. J. Org. Chem. 2002;67:3–15. doi: 10.1021/jo0106501. [DOI] [PubMed] [Google Scholar]
- xxxvii.Previous cyclopropanations with vinyl boranes: Fontani P, Carboni B, Vaultier M, Carrié R. Tetrahedron Lett. 1989;30:4815–4818. Fontani P, Carboni B, Vaultier M, Maas G. Synthesis. 1991;8:605–609.
- xxxviii.Previous epoxidation of 1,2-disubstituted alkenyltrifluoroborate salts: Molander GA, Ribagorda M. J. Am. Chem. Soc. 2003;125:11148–11149. doi: 10.1021/ja0351140.
- xxxix.Heck reactions with vinyl boranes: Hunt AR, Stewart SK, Whiting A. Tetrahedron Lett. 1993;482:3599–3602. Stewat SK, Whiting A. J. Organomet. Chem. 1994;482:293–300. Lightfoot AP, Maw G, Thirsk C, Twiddle S, Whiting A. Tetrahedron Lett. 2003;44:7645–7648. Stewart SK, Whiting A. Tetrahedron Lett. 1995;36:3925–3928. Hénaff N, Whiting A. Org. Lett. 1999;1:1137–1139. Hénaff N, Whiting A. Tetrahedron. 2000;56:5193–5024. Lightfoot AP, Twiddle SJR, Whiting A. Org. Biomol. Chem. 2005;3:3167–3172. doi: 10.1039/b507900d. Batsanov AS, Knowles JP, Whiting A. J. Org. Chem. 2007;72:2525–2532. doi: 10.1021/jo0626010. Itami K, Tonogaki K, Nokami T, Ohashi Y, Yoshida J. Angew. Chem. Int. Ed. 2006;45:2404–2409. doi: 10.1002/anie.200600063. Itami K, Tonogaki K, Ohashi Y, Yoshida J. Org. Lett. 2004;6:4093–4096. doi: 10.1021/ol048217b..
- xl.(a) Chen MS, White MC. J. Am. Chem. Soc. 2004;126:1346–1347. doi: 10.1021/ja039107n. [DOI] [PubMed] [Google Scholar]; (b) Chen MS, Prabagaran N, Labenz NA, White MC. J. Am. Chem. Soc. 2005;127:6970–6971. doi: 10.1021/ja0500198. [DOI] [PubMed] [Google Scholar]; (c) Fraunhoffer KJ, Prabagaran N, Sirois LE, White MC. J. Am. Chem. Soc. 2006;128:9032–9033. doi: 10.1021/ja063096r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fraunhoffer KJ, White MC. J. Am. Chem. Soc. 2007;129:7274–7276. doi: 10.1021/ja071905g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Reed SA, White MC. J. Am. Chem. Soc. 2008;130:3316–3318. doi: 10.1021/ja710206u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Young A, White MC. J. Am. Chem. Soc. 2008 ASAP. [Google Scholar]
- xli.(a) Cho CS, Uemura S. J. Organomet. Chem. 1994;465:85–92. [Google Scholar]; (b) Du X, Suguro M, Hirabayashi K, Mori A, Nishikata T, Hagiwara N, Kawata K, Okeda T, Wang H, Fugami K, Kosuhi M. Org. Lett. 2001;3:3133. doi: 10.1021/ol016529y. [DOI] [PubMed] [Google Scholar]; (c) Yoo KS, Yoon CH, Jung KW. J. Am. Chem. Soc. 2006;128:16384. doi: 10.1021/ja063710z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lindh J, Enquist PA, Pilotti A, Nilsson P, Larhed M. J. Org. Chem. 2007;72:7957–7962. doi: 10.1021/jo701434s. [DOI] [PubMed] [Google Scholar]
- xlii.(a) Delcamp JH, White MC. J. Am. Chem. Soc. 2006;128:15076–15077. doi: 10.1021/ja066563d. [DOI] [PubMed] [Google Scholar]; (b) Delcamp JH, Brucks AP, White MC. J. Am. Chem. Soc. 2008;130:11270–11271. doi: 10.1021/ja804120r. [DOI] [PubMed] [Google Scholar]
- xliii. Kunze B, Jansen R, Hofle G, Reichenbach H. J. Antibiot. 1994;47:881–886. doi: 10.7164/antibiotics.47.881. Previous syntheses: Dias LC, de Oliviera LG. Org. Lett. 2001;3:3951–3954. doi: 10.1021/ol016845c. Chakraborty TK, Jayaprakash S. Tetrahedron Lett. 2001;42:497–499. Feutrill JT, Lilly MJ, Rizzacasa MA. Org. Lett. 2000;2:3365–3367. doi: 10.1021/ol0064664. Sirasani G, Paul T, Andrade RB. J. Org. Chem. 2008;73:6386–6388. doi: 10.1021/jo800906p..