

Abstract
The objectives of this work were to express the EC5 domain of E-cadherin and determine its structural characteristics as well as to evaluate the binding properties of HAV and BLG4 peptides to EC5 using spectroscopic methods. Homophilic interactions of E-cadherins are responsible for cell-cell adhesion in the adherens junctions of the biological barriers (i.e., intestinal mucosa and blood-brain barriers). The EC5 domain of E-cadherin has an important role in T-cell adhesion to intestinal mucosa via αEβ7 integrin-E-cadherin interactions. In this study, the expressed EC5 has a high thermal stability (Tm = 64.3 °C); it also has two stable conformations at room temperature, which convert to one conformation at approximately 54.5 °C. NMR and FTIR showed that HAV and BLG4 peptides bind to EC5. HSQC-NMR showed that either Asn or Gln of EC5 was involved in the interactions with HAV and BLG4 peptides. EC5 underwent a conformational change upon interaction with the HAV and BLG4 peptides. Finally, the binding properties of both peptides were modeled by docking experiments, and the results suggest that Asn-46 and Asn-75 of EC5 could be involved during the interaction with the peptides and that the Ser and Trp residues of the HAV and BLG4 peptides, respectively, were important for binding to EC5.
Keywords: E-cadherin, EC5 domain, conformation, spectroscopy, adherens junction, cell-cell adhesion, peptide binding
INTRODUCTION
E-cadherins are glycoproteins that create calcium-dependent cell-cell adhesion at the adherens junctions of biological barriers (i.e., the intestinal mucosa and the blood-brain barrier) (1) and have an important role in cell morphogenesis, tumor invasion and metastasis (2, 3). The extracellular (EC) domain of E-cadherin consists of five repeats, EC1 to EC5 domains that have been suggested to form homophilic interactions. The structures of various repeats of the EC domain of E-, N-, and C-cadherins have been studied by NMR, X-ray, electron microscopy, and surface force apparatus (4–9). The results from various studies generated two possible major mechanisms, one of which involves only EC1-EC1 interactions (10–13) while the other involves interactions of all the EC domains (EC1 through EC5). However, there are no available data that can reconcile these two major mechanisms. The X-ray structure of C-cadherin suggests that the trans-dimer of cadherins is formed by the interaction only between two EC1 domains from the opposing membranes, while the cis-interaction is between the EC1 and EC2 domains of two molecules from the same surface (14). However, surface force measurements and binding studies of truncated domains of C-cadherin suggest that trans-interactions between cadherins from opposing cells are caused by interdigitized multidomain interactions (7, 15–17). If this is the case, it is important to study the structure of the other domains of E-cadherin (i.e., EC3, EC4, and EC5) and their roles in E-cadherin-E-cadherin interactions.
The interaction between E-cadherin and αEβ7-integrin during T-cell adhesion to the intestinal epithelium involves EC1 and EC5 domains of E-cadherins (18, 19). Thus, elucidation of the structure and interaction properties of the EC5 domain could provide information for designing a selective way to modulate E-cadherin-mediated cell-cell adhesion for therapeutic purposes such as improving drug delivery and controlling inflammation. We also have shown that peptides derived from E-cadherin (i.e., HAV peptides) enhance the paracellular permeation of a marker molecule (i.e., 14C-mannitol) through Madin-Darby Canine Kidney (MDCK) cell monolayers (13, 20, 21). The peptide activity may be due the inhibition of E-cadherin interactions in the intercellular junctions. Thus, characterization of the peptide-binding to EC domains is necessary to improve the activity of E-cadherin peptides in modulating E-cadherin-mediated cell-cell adhesion.
In this work, the EC5 domain of E-cadherin was expressed and its structural properties were evaluated using spectroscopic methods (i.e., circular dichroism (CD), UV absorbance, Fourier transform infrared (FTIR), intrinsic fluorescence, and NMR spectroscopy, and differential scanning calorimetry (DSC). EC5 has the potential to inhibit homotypic cell-cell adhesion and heterotypic T-cell adhesion. The unique structure of EC5 with two disulfide bonds is a good model for studying the role of disulfide bonds on its structural stability. The binding properties of the HAV (Ac-SHAVSS-NH2) and BLG4 (Ac-TYRIWRDTAN-NH2) peptides to the EC5 domain of E-cadherin were also studied using FTIR and NMR spectroscopy. These two peptides appear to induce a conformational change in EC5 upon binding. Finally, computational docking experiments were performed to provide working models for the interaction between each peptide and EC5. In the future, these binding models will be used to design small peptide derivatives that effectively modulate the intercellular junctions of the in vitro and in vivo models of the biological barriers (i.e., intestinal mucosa and blood-brain barriers).
EXPERIMENTAL METHODS
Recombinant EC5 plasmid construction
Construction of the pASK-IBA6/EC5 plasmid
The method used to clone the gene for EC5 is similar to that employed for the EC4 domain (22). Full-length human E-cadherin cDNA cloned in pERF-cadherin was provided by Dr. David Rimm of Yale University. Both forward and reverse primers contain the BsaI restriction site. A standard PCR program was used to amplify EC5 cDNA, and the expression vector pASK-IBA6 (Sigma-Genosys, Woodlands, TX) was digested with 15 units of BsaI. The BsaI-digested EC5 cDNA was cloned downstream of the OmpA signal peptide, Strep-Tag II, and the Factor Xa site of the vector using T4 ligase (Promega, Madison, WI). The recombinant plasmid pASK-IBA6/EC5 was transformed into competent E. coli BL21 cells (Stratagene, La Jolla, CA) using a heat pulse at 42 °C for 45 s. The transformed cells were grown overnight at 37 °C on LB plates containing 100 µg/mL ampicillin. The positive clones were screened by PCR, and the EC5 cDNA sequence was confirmed by DNA sequencing.
Construction of the pET24d/EC5 plasmid
To improve the yield of EC5, we developed a new expression plasmid. The forward primer contains an NcoI restriction site and the reverse primer an XhoI site. A standard PCR program was used with the following conditions: 94 °C for 2 min; 30 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 1 min; and, finally, 72 °C for 10 min. The PCR amplification was performed using 5 µL of 5 µM forward and reversed primers, 2 µL of 50 ng/µL full-length cDNA, 200 µM dNTP, 28 µL of distilled water, 5 µL of 10X PCR buffer (Stratagene), and 1 µL Pfu Turbo Polymerase (Promega). After PCR, the purified EC5 cDNA and the pET24d vector (Novogen, Madison, WI) were digested with NcoI and XhoI. The digested products were ligated using T4 ligase (Promega). After ligation, pET24d/EC5 was inserted into the high expression vector pET24d (Novogen). The recombinant plasmid pET24d/EC5 was then transformed into competent E. coli BL21 cells (Stratagene) using a heat pulse at 42 °C for 45 s. The transformed cells were grown overnight at 37 °C on LB plates containing 30 µg/mL kanamycin. The correct EC5 cDNA sequence was confirmed by DNA sequencing.
Expression and purification of recombinant EC5
Production of Strep-Tag II-containing EC5 (pASK-IBA6/EC5)
The expression and purification of EC5 was performed in a manner similar to a previously described method (22). E. coli cells were cultured in 250 mL of 2-X YT medium with 100 mg/L of ampicillin and then induced with 25 µL (200 µg/L) anhydrotetracycline (Sigma-Genosys). After induction, cells were collected every hour and lysed. The whole-cell lysates were analyzed by SDS-PAGE. An intense protein band was produced with the expected size of Strep-Tag II fused-EC5. The optimal expression of EC5 was reached after 3 h of induction. Recombinant EC5 was found in the supernatant after cell lysis. The EC5 protein was subjected to a one-step purification using a Strep-Tactin affinity column. The affinity-purified EC5 fractions were analyzed by 4–20% SDS-PAGE; they produced a single band at the correct molecular weight for EC5 and an optimal yield of 0.5 mg/L.
Production of EC5 without Strep-Tag (pET24d/EC5)
For producing the EC5 domain without a Strep-Tag at the N-terminus, E. coli BL21/pET24d-EC5 cells were inoculated in 500 mL 2-X YT medium with 30 mg/L of kanamycin at 37 °C using a 200 rpm shaking incubator followed by concentration measurements every 1 or 2 h. The optical density (OD) of the culture was measured at 600 nm. When the OD value was between 0.6 and 0.8 (2.5 to 3 h), 0.5 mL of 100 mg/mL isopropyl β-D-thiogalactoside (IPTG) (Sigma-Aldrich, Milwaukee, WI) was added to initiate the over expression. The culture was over expressed for 4 h and harvested by centrifugation at 4000 × g at 4 °C for 10 min. The resulting pellets were kept at −80 °C overnight.
On the following day, the cell pellets were thawed and resuspended in 10 mL of 50 mM Tris-HCl and 100 mM NaCl buffer at pH 7.5, then lysed using a French Press. The cell lysate was centrifuged at 48000 × g and 4 °C in a Beckman JA 20 rotor for 45 min, after which the majority of the EC5 was found in the supernatant. To purify the EC5 domain, a heat/cool cycle was used to remove most of the E. coli proteins. Because there are two intramolecular disulfide bonds, EC5 is relatively thermally stable and its temperature-induced conformational change is reversible. The supernatant was therefore incubated at 80 °C for 10 min and then in ice for 5 min (23). EC5 remained soluble in the supernatant. The supernatant was dialyzed in a 50 mM Tris buffer at pH 7.5 overnight. After dialysis, the protein solution was centrifuged at 48000 × g and 4 °C in a Beckman JA 20 rotor for 30 min. The supernatant was loaded onto a Q-sepharose anion exchange column (Amersham Biosciences, Piscataway, NJ). The following buffers were used for the gradient elution. Buffer A contained 50 mM Tris-HCl at pH 7.5 and buffer B 50 mM Tris-HCl and 1 M NaCl at pH 7.5. After elution from the Q-sepharose column, the fractions containing EC5 were collected, concentrated, and loaded onto a Superdex 200 size-exclusion column (Amersham Biosciences) for final purification. The fractions containing EC5 were collected and loaded onto a 4–20% SDS-PAGE gel. The final purity of the EC5 domain was greater than 99% based on reversed-phase HPLC analysis. A Shimadzu (Shimadzu, Columbia, MD) 10A VP HPLC system containing a Vydac 208TP C8 column (Grace Vydac, Hesperia, CA) with 4.6 mm diameter and 25 cm length was used. The flow rate was 1 mL/min with a 60-min running time. The injection volume was 10 µL. The detection wavelength was 280 nm and the retention time was 52.6 min. The optimal yield of EC5 in 2-X YT medium was 5–10 mg/L.
15N-labeled EC5 was used in binding studies between the peptide and EC5. One set of binding properties was monitored using 1H, 15N 2D-HSQC NMR spectroscopy. To produce 15N-labeled EC5, E. coli cells containing EC5 cDNA were cultured in 15N-labeled minimal medium (M9) containing 13 g Na2HPO4·7H2O, 3 g KH2PO4, 0.5 g NaCl, 1 g 15NH4Cl (Cambridge Isotope, Andover, MA), 2.2 g MgSO4·7H2O, and 0.7 g CaCl2·2H2O per liter. The pH of the final medium was adjusted to 7.4 using 5.0 N HCl. Prior to inoculation, the M9 medium was supplemented with freshly prepared 20% D-glucose, 1 × 10−3 mM thiamine, 1 × 10−3 mM FeSO4·7H2O, and 30 mg/L of kanamycin. Over expression and purification methods for the labeled EC5 were the same as employed for unlabeled EC5. The final purified protein had a concentration of 0.5 mM in 100 mM Tris buffer at pH 7.5.
15N,13C double-labeled EC5 was used to study peptide binding by monitoring changes in the amide I band using FTIR. The same method described above was used to produce 15N,13C double-labeled EC5. In this case, 13C-labeled D-glucose (Spectra Stable Isotopes, Columbia, MD) was utilized instead of unlabeled D-glucose. After purification, 15N,13C double-labeled EC5 was concentrated to 0.5 mM in 100 mM Tris buffer at pH 7.5.
Structural studies of EC5 and binding properties to E-cadherin peptides
Characterization of disulfide bonds
Twenty µL each of 10 µM native and reduced EC5 in 25 mM Tris buffer at pH 7.5 were added to 75 µL of 8.0 M guanidinium hydrochloride (Gdn.HCl). This mixture was shaken for 10 s to unfold the EC5 protein. Five µL of 200 µM ThioGlo-1 was added, and the mixture was incubated for 30 min at room temperature. The amount of free thiol was detected by determining the amount of incorporated fluorescence label in the ThioGlo-1 reaction (24). Twenty µL of 25 mM Tris buffer at pH 7.5 received the same treatment and was used as a blank. The fluorescence intensity was detected using a BioTek FL600 microplate fluorescence reader (BioTek Instruments, Winooski, VT) with an excitation wavelength of 360 nm and emission detected at 530 nm.
Size-exclusion chromatography (SEC)
SEC was performed with a 24 mL Superdex 200 column (Amersham Biosciences) using UV absorbance detection at 280 nm. The mobile phase was 25 mM Tris-HCl (pH 7.5) with a flow rate of 0.4 mL/min. The standard protein kit was used for calibration.
CD spectroscopy
CD spectra of EC5 were obtained from 250 nm to 200 nm using a J-810 CD spectrometer (Jasco, Easton, MD) at room temperature. Four hundred µL of 0.02 mM EC5 in various buffers were loaded into a 0.1 cm path-length cell. The spectra were averaged over three repetitive scans with data acquired every 0.5 nm with a 2-s response time (25, 26). The corresponding buffer was scanned as a blank and subtracted. The secondary structure of EC5 in 0.02 mM EC5 in 25 mM Tris buffer at pH 7.5 was estimated using the Selcon3 program (27). The temperature-induced unfolding of EC5 was monitored at a fixed wavelength of 230 nm. The temperature was varied from 10 to 90 °C with data taken in 0.2 °C increments and a heating rate of 15 °C/h. For temperature studies, EC5 was examined in 25 mM K2HPO4 buffer at pH 7.5. The melting temperature of EC5 was estimated by Sigmoidal fit in Microcal Origin 6.0.
FTIR spectroscopy
EC5 was concentrated to 1.6 mM in 25 mM Tris at pH 7.5 in the presence and absence of 2.5 mM DTT for analysis by FTIR spectroscopy. FTIR spectra were acquired employing a Nicolet Magna-IR 560 spectrometer with a mercury-cadmium-telluride (MCT) detector. The EC5 protein was placed between CaF2 transmittance windows using a 5.6 µm spacer. The reference sample was 25 mM Tris buffer at pH 7.5 and was run under identical conditions. Two hundred and fifty-six consecutive scans were collected for each sample at a resolution of 2 cm−1. The water signal was subtracted from each spectrum using the criterion of a straight baseline between 2000 and 1700 cm−1. Omnic E.S.P. 4.1 software was used to smooth the original spectra. GRAMS/AI (7.00) was used to calculate derivative spectra. A mixed Lorentzian and Gaussian function was used for curve fitting (28). Correlation coefficients (r = ∑ xiyi/√ ∑ xi2∑ yi2) were used to evaluate the difference between the second derivative spectra of native and reduced EC5 (29). xi and yi represent the second derivative values of native and reduced EC5 spectra at each frequency, respectively.
Binding between the peptide (HAV, Ac-SHAVSS-NH2 or BLG4, Ac-TYRIWRDTAN-NH2) and EC5 was evaluated by observing the change in the FITR spectra of 15N,13C-labeled EC5 in the absence and presence of the peptide. Binding experiments were conducted in 100 mM Tris buffer in D2O at pH 7.5. In this case, 50 mM HAV or 25 mM BLG4 was added to a 0.5 mM solution of labeled EC5. The sample was introduced into a calcium fluoride transmittance window with a 0.5 mm pathlength. Spectra were collected using a Nicolet Magna-IR 560 spectrometer with a MCT detector. The detector was cooled with liquid nitrogen during data collection. The FTIR spectrum of unlabeled EC5 was also collected for comparison. Two hundred and fifty-six consecutive scans were collected for each sample with a resolution of 2 cm−1. The buffer signal was subtracted from each spectrum using a straight baseline between 1900 cm−1 and 1700 cm−1. Omnic E.S.P. 4.1 software (Nicolet, Madison, WI) was used to smooth the original spectra. Derivative spectra from 1700 cm−1 to 1500 cm−1 were calculated using GRAMS/AI (7.00). Curve fitting was performed using a mixed Lorentzian and Gaussian function between 1647 cm−1 and 1563 cm−1 in Omnic software (Nicolet) (28). The number and position of the deconvoluted peaks were determined from the second derivative trace of the amide I band.
High-resolution second derivative UV absorbance spectroscopy
UV spectra were recorded using a Hewlett-Packard 8453 UV-visible spectrophotometer (Agilent, Palo Alto, CA). One mL of 0.02 mM purified EC5 in a 25 mM phosphate buffer at pH 7.5 was loaded into a 1-cm pathlength quartz cuvette. Spectra were obtained over the temperature range of 10 to 85 °C at 2.5 °C intervals using a 5-min equilibration period before data acquisition. Agilent UV-visible Chemstation software was used to calculate second derivatives; data were processed as described previously, and Microcal Origin 6.0 was used to plot the data (30, 31).
Differential scanning calorimetry (DSC)
DSC was performed with a Nano-DSC calorimeter (Calorimetry Sciences Corp., American Fork, UT). The pure EC5 protein in a 25 mM phosphate buffer at pH 7.5 (1.2 mL of a 0.1 mM solution) was injected into the sample cell while 1.2 mL of 25 mM phosphate buffer at pH 7.5 was placed in the reference. Thermograms were obtained from 10 to 100 °C with a scan rate of 60 °C/h. Microcal Origin 6.0 software was used for curve fitting (28).
Fluorescence spectroscopy
Intrinsic fluorescence emission spectra were collected with a QuantaMaster spectrofluorometer (PTI, Monmouth Junction, NJ). One and one-half mL of 0.02 mM EC5 in a 25 mM phosphate buffer at pH 7.5 was loaded into a 1-cm pathlength quartz fluorescence cuvette. Spectra were collected from 10 to 80 °C at 2.5 °C intervals. A 5-min equilibration was performed before each collection of spectra. The excitation wavelength was set at 295 nm (>95% Trp emission), and emission was monitored from 305 to 455 nm. The excitation and emission slits were set at 4 nm. The Felix program was used to analyze the data (32, 33).
NMR spectroscopy
To establish conditions for structural analysis by NMR, 15N-labeled EC5 was dissolved in 25 mM Tris buffer at pH 7.5 and 1H-15N 2D HSQC spectra were collected using a Bruker Avance 800 MHz NMR spectrometer (Bruker BioSpin Corp., Billerica, MA) equipped with a TCI triple-axis gradient cryoprobe. D2O (2%) was added to each sample to provide the lock signal. These spectra were acquired at 5 °C temperature intervals from 10 to 65 °C with 5-min equilibration prior to the start of each experiment. Each spectrum was signal averaged over 16 scans in which 1024 points were collected in 1H and 128 points in 15N, and referencing was performed relative to DSS (34). Spectra were processed using nmrPipe (35).
For peptide binding studies, 1H-15N-HSQC spectra of 0.2 mM 15N-labeled EC5 in 5% D2O in the absence or presence of 20 mM HAV or 10 mM BLG4 were acquired in 100 mM Tris buffer at pH 7.5 using a Bruker Avance 800 MHz NMR spectrometer equipped with a TCI triple-axis gradient cryoprobe. Sixteen consecutive scans were collected and averaged for each spectrum. One thousand and twenty four points were obtained in the direct detect dimension (1H) and 128 increments were collected for 15N. Referencing was performed relative to DSS (34). After collection, all data were analyzed by nmrPipe (35).
Computational docking
The structural model of the EC5 domain of E-cadherin was generated by homology modeling using the EC5 domain of C-cadherin (PDB entry 1L3W) as a template (14). In addition, all publically available cadherin crystal structures, including C-cadherin (pdb entry 1L3W), N-cadherin (pdb entries 1NCH, 1NCI, 1NCG) and E-cadherin (pdb entry 1EDH), and a modeled structure of the EC5 domain of E-cadherin were initially used to characterize structural space of cadherins. These structures were minimized and superimposed into a common reference frame is based on similarity of Cα atoms for a common set of residues from the bulge and groove protein regions. The peptide conformations were generated using molecular modeling software MACROMODEL and minimized using AMBER* force field. Conformational search for the low—energy peptide conformations was performed within MACROMODEL modeling tool, utilizing Monte Carlo conformational search with an energy window of 6.0 kcal/mole and up to 1000 Monte Carlo steps. Minimization is performed using AMBER* force field with the Generalized Born/Solvent Accessible Surface Area (GB/SA) salvation term. We have employed a modeling approach, in which structure and energetics of peptide binding can be effectively interrogated using fully automated flexible peptide docking with ensembles of multiple protein structures (36–39). The molecular recognition energetic model includes intramolecular energy terms, given by torsional and nonbonded contributions and the intermolecular energy contributions calculated using the AMBER force field to describe peptide–protein interactions combined with an implicit solvation model (36–39). Molecular docking was performed using simulated tempering sampling technique with the ensemble of multiple protein structures. The peptide conformations and orientations were sampled in a parallelepiped that encompasses the superimposed structures with a 10.0 Å cushion added to every side of the box surrounding the binding interface, which guaranteed an unbiased search of the bound peptide conformation. The rigid body degrees of freedom and the peptide rotatable angles were treated as independent variables during molecular docking (36–39). Peptide docking with the ensemble of multiple protein structures has allowed to propose a putative-consensus binding site(s) on the surface of EC5 and to understand the binding of HAV and BLG4 peptides to the EC5 domain.
RESULTS
EC5 expression and purification
Production and purification of the EC5 domain with and without a Strep-Tag was straightforward. Initially, the EC5 protein was expressed with a Strep-Tag at the N-terminus for ease of purification. One-step purification using a Strep-Tactin affinity column produces a pure EC5 protein with the correct molecular weight and an optimal yield of 0.5 mg/L. The native EC5 protein without the Strep-Tag was purified using a heat/cool step to remove other proteins (23). The soluble protein was then subjected to two chromatographic purification steps. The isolated EC5 protein had a yield of 5–10 mg/L with a final purity greater than 99% based on reversed-phase HPLC analysis. The production of 15N-labeled EC5 was similar to that of unlabeled EC5, and a similar high degree of purity was obtained.
Characterization of disulfide bonds
There are four Cys residues in the primary sequence of EC5 that could form two disulfide bonds as seen in the EC5 domain of C-cadherin (14). To test whether the Cys residues form such disulfide bonds, the content of free thiol groups in the EC5 protein was analyzed using the ThioGlo-1 reagent (40). There was no difference between the blank buffer and native EC5 (not illustrated) after the ThioGlo-1 reaction. This suggests that the over expressed version of EC5 contains the two predicted disulfide bonds. Upon reduction of the EC5 protein, high fluorescence intensity was observed upon treatment with ThioGlo-1, suggesting that both disulfide bonds were reduced to free thiol groups quantitatively (approximate 100%).
Both non-reducing gel electrophoresis and SEC indicated that there was no covalent dimerization or oligomerization via intermolecular disulfide bond formation. In addition, the single peak observed in SEC corresponds to a monomeric unit of EC5 based on a calibration curve using standard proteins. These results also indicate that EC5 does not form intermolecular disulfide bonds or other physical dimers or oligomers in solution.
Structural studies of EC5 and the effect of disulfide bonds on structural stability
To provide a working model of the structure of the EC5 domain of E-cadherin, a model structure was generated using the X-ray structure of the EC5 domain of C-cadherin (Figure 1A). The BLAST program was used to align the sequences of the EC5 domains of E- and C-cadherin; from the alignment study, there are seven high homology regions between the EC5 domains of E- and C-cadherin (data not shown). The degree of identity and similarity between the two EC5 domains is 57%. Several molecular parameters of EC5 were compared to those of the EC1 and EC4 domains of E-cadherin (Table 1). The EC1 structure was determined by X-ray crystallography (6) while the EC4 structure is a model based on the crystal structure of the corresponding domain of C-cadherin (22). EC5 has the largest calculated surface area followed by the EC4 and then EC1 domains. EC5 has the lowest number of H-bonds compared to EC1 and EC4, and the EC1 domain has the greatest number of H-bonds. These results suggest that EC5 has a less compact structure than EC1 or EC4. EC5 has only a slightly higher apolar surface area (59%) than hydrophilic surface area (41%). Although the absolute hydrophobic surface area of EC1 is almost the same as that of EC5, the percent of hydrophobic area in EC1 (85%) is actually much larger than that of EC5 (59%). In contrast, EC4 has the lowest apolar surface area (20%). All of these parameters indicate that, although EC domains have similar overall structures, they differ in several important structural details that might relate to their multiple functions.
Figure 1.
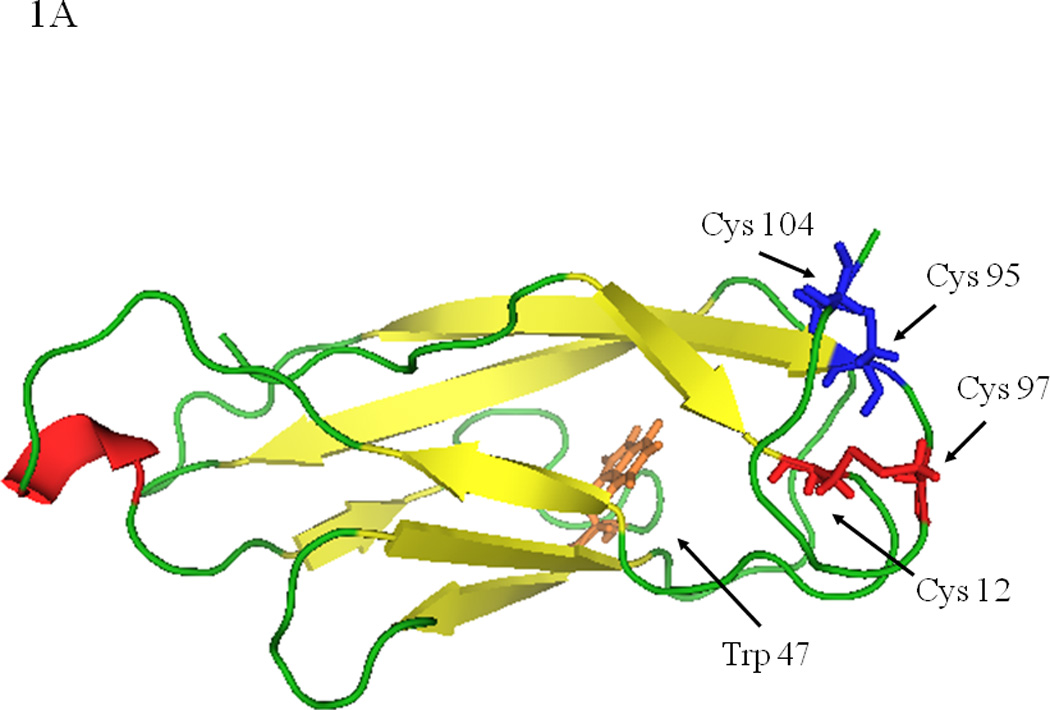
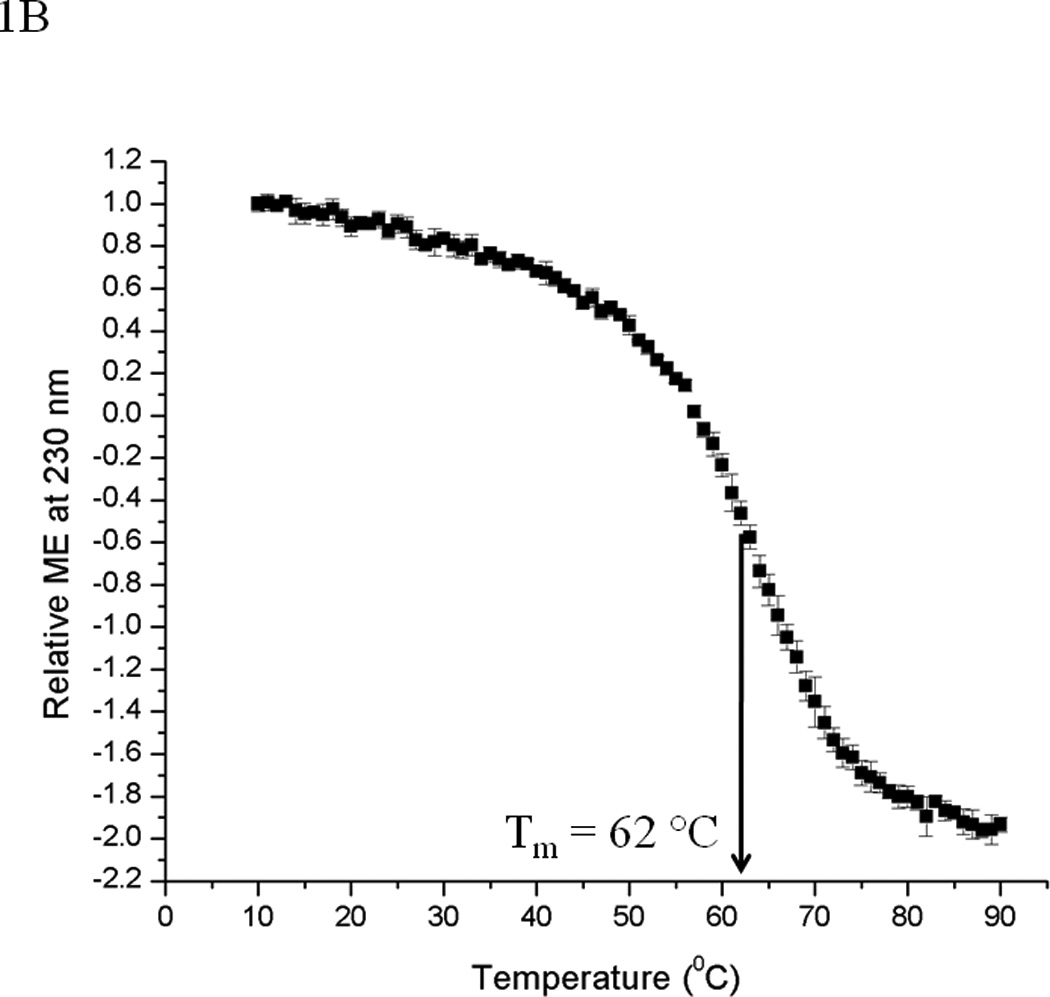
(A) A molecular model of the EC5 domain of E-cadherin derived from the EC5 domain of C-cadherin. The four Cys residues form two intramolecular disulfide bonds. (B) The melting temperatures of the EC5 domain measured by relative molar ellipticity change as a function of temperature in 25 mM phosphate buffer at pH 7.5 at 230 nm. Data are normalized to the ME value at 10 °C.
Table 1.
Comparison of several molecular parameters of the EC1, EC4, and EC5 domains.
| EC1 | EC4 | EC5 | |
|---|---|---|---|
| Atom number | 837 | 851 | 932 |
| aHydrophilic surface area | 914 Å2 (15%) | 5217 Å2 (80%) | 3451.0 Å2 (41%) |
| aHydrophobic surface area | 4998 Å2 (85%) | 1268 Å2 (20%) | 5045.7 Å2 (59%) |
| Total molecular surface area | 5913 Å2 | 6485 Å2 | 8496.7 Å2 |
| H-bond number | 70 | 47 | 35 |
The numbers in parentheses are the percent of hydrophilic or hydrophobic surface areas compared to the total surface area of each EC domain.
The effect of various solution conditions on the secondary structure of EC5 was evaluated using CD spectroscopy. EC5 has high β-sheet and β-turn content because E-cadherin belongs to the immunoglobulin superfamily. An inflection point is observed at 210–215 nm of the native protein from the β-structure (data not shown). The CD spectrum shows a positive peak near 230 nm, which probably originates from one or more aromatic residues or possibly from the disulfide bonds. Upon increasing the temperature, significant spectral changes at 230 nm were observed between 50 and 70 °C with an estimated melting temperature (Tm) of 62 °C (Figure 1B). The relatively high melting temperature is presumably due to the presence of the two intramolecular disulfide bonds that contribute strongly to the conformational rigidity of EC5. Heating from 20 to 90 °C followed by cooling back to 20 °C produced identical spectra (see Supplementary Materials), suggesting that the temperature-induced unfolding is reversible.
Upon reduction of both disulfide bonds in EC5 using DTT, a dramatic change was observed in the CD spectrum, which suggests significant conformational alteration of the protein. The positive peak at 230 nm disappeared (Figure 2A), suggesting that the presence of the two disulfide bonds is important for the structural integrity of the tertiary structure of EC5. Although the secondary structure of EC5 was not completely restored after removal of DTT by dialysis, the positive peak at 230 nm reappeared, indicating the possible re-formation of a significant amount of structure and the disulfide bonds.
Figure 2.
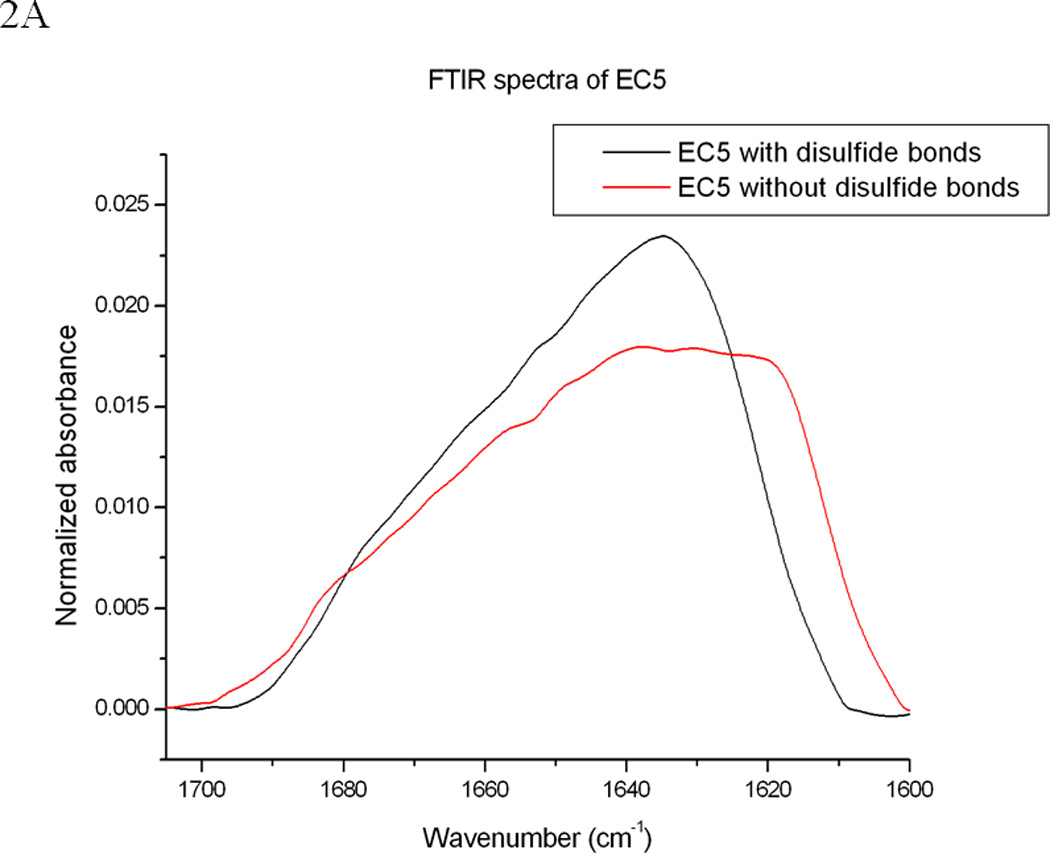
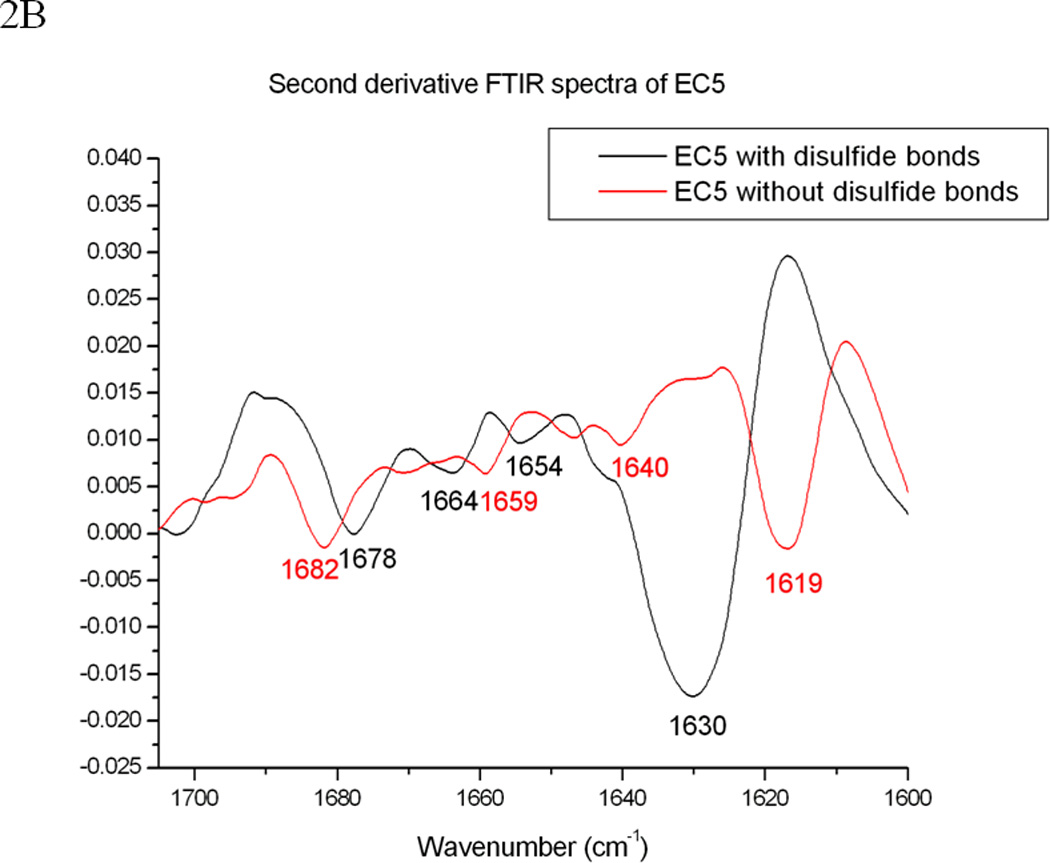
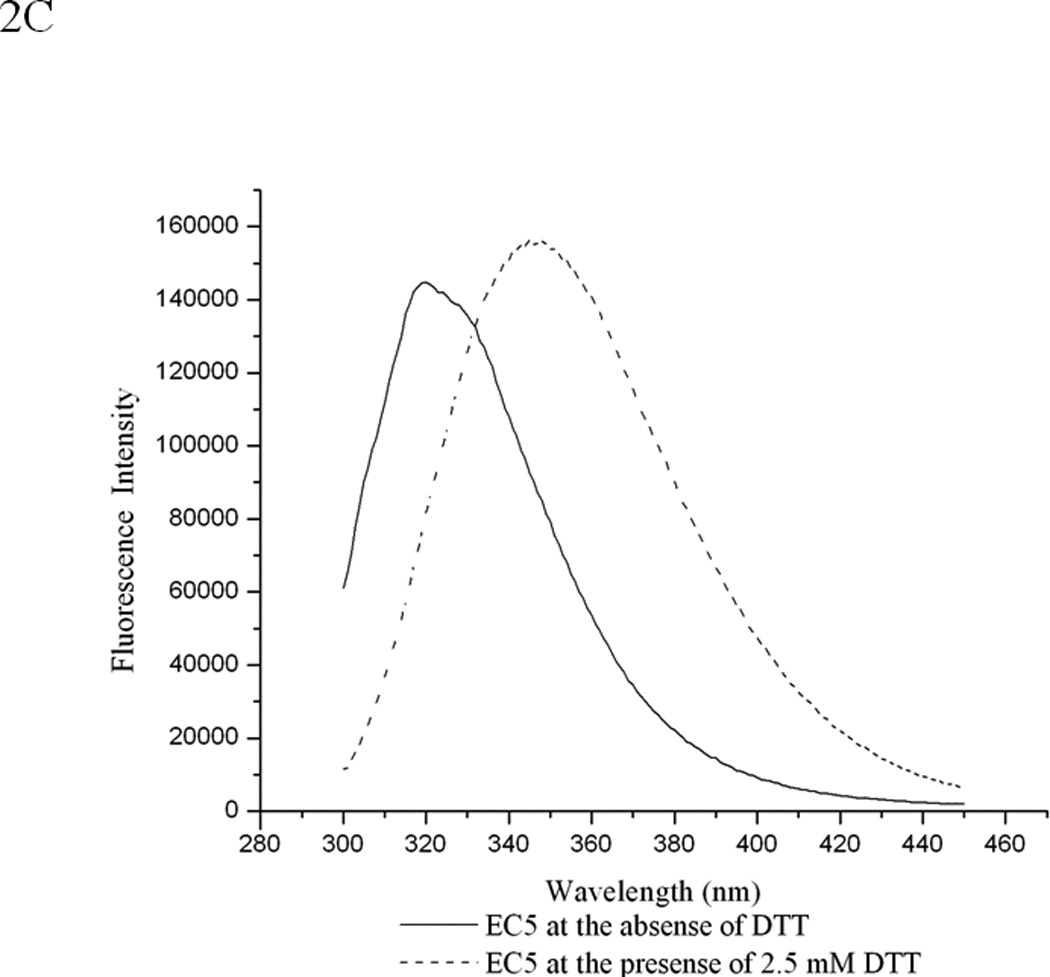
Comparison of the FTIR and fluorescence spectra of EC5 in the absence and presence of 2.5 mM DTT. (A) The amide-1 FTIR spectra of EC5 in the native (black line) or reduced (red line) forms. (B) The second derivative FITR spectra of EC5 in the native (black line) and reduced (red line) forms. (C) Comparison between the emission peaks of the native EC5 (solid line) and reduced EC5 (dashed line).
The secondary structure and the effect of disulfide bonds on the structure of EC5 were also evaluated by FTIR and fluorescence spectroscopy. The FTIR spectra show strong amide I absorption bands at 1630 and 1678 cm−1, signifying that EC5 has a high content of β-sheet (Figures 2A and B); this is similar to the finding from CD spectroscopy and consistent with the molecular model (Figure 1). After reducing the disulfide bonds, the IR absorption spectrum shifted to lower frequency, consistent with an increase in disordered structure (Figures 2A and 2B). The FTIR spectra of the native and reduced EC5 proteins were compared to evaluate conformational changes upon reduction (Figure 2B). Upon reduction, the major negative peaks at 1630 and 1678 cm−1 of native EC5 appear to be shifted to new peaks near 1619 and 1682 cm−1 for the reduced form. The appearance of a signal near 1619 cm−1 is usually attributed to the presence of intermolecular β-structure. The low correlation between the two spectra (r = 0.289) indicates a major difference between the secondary structures of the native and reduced forms (29).
The tertiary structure and thermal unfolding properties of EC5 were evaluated by observing the intrinsic fluorescence of the single Trp residue. The emission spectrum of the Trp residue shows a major peak at 320 nm and a shoulder at 332 nm (Figure 2C), suggesting the presence two different microenvironments of the Trp residue and, consequently, two different stable conformations. Reduction of the two disulfide bonds present in EC5 dramatically shifts the emission peak of the Trp residue from 320/332 nm to a single peak at 346 nm (Figure 2C), which indicates that the reduced form has a more disordered structure with the Trp residue more exposed to aqueous solvent. Thermal studies of EC5 show a first transition with a shift of the major peak at 320 nm to a peak at 332 nm (Figure 3A). Upon further temperature increase, a second transition was observed with a shift of the peak at 332 nm to a new signal at 346 nm, corresponding to a much greater exposure of the indole side chain. The effect of temperature on the wavelength of the maxima defines three possible transition temperatures (i.e., at 35 °C, 55 °C, and 65 °C, Figure 3B).
Figure 3.
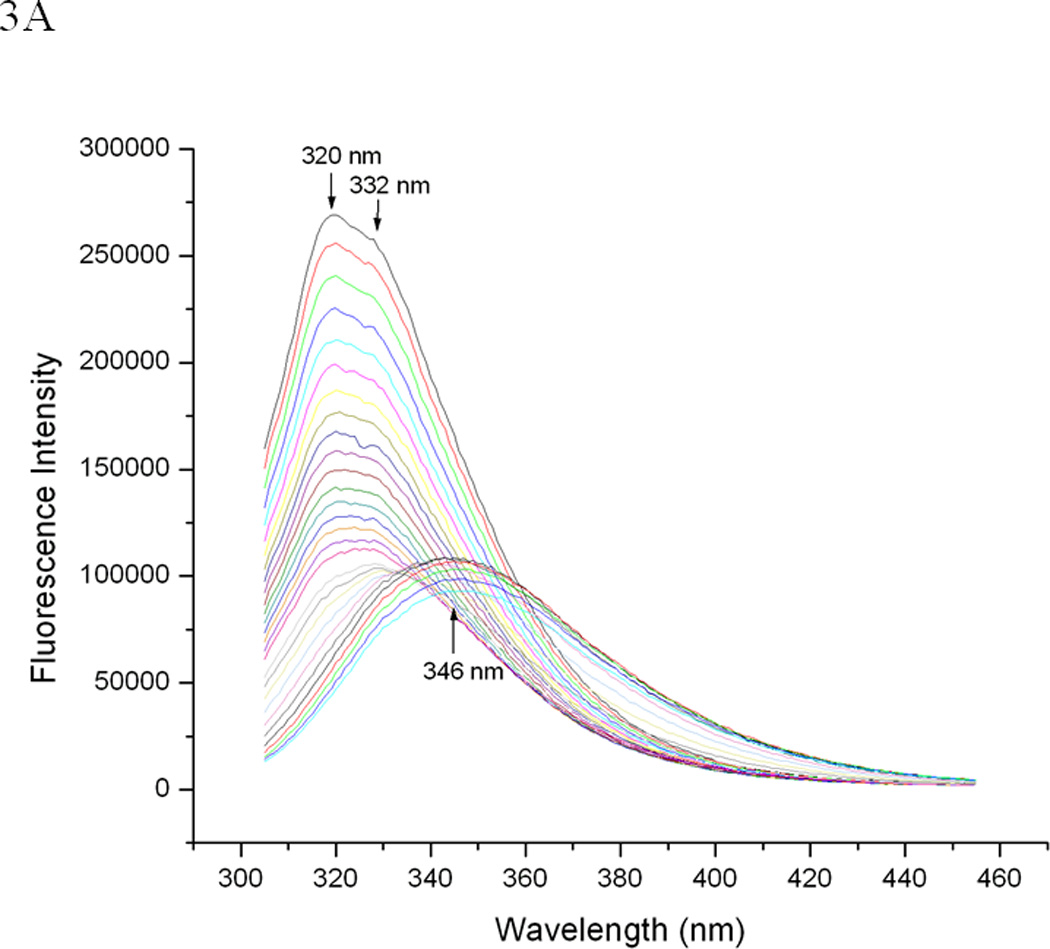
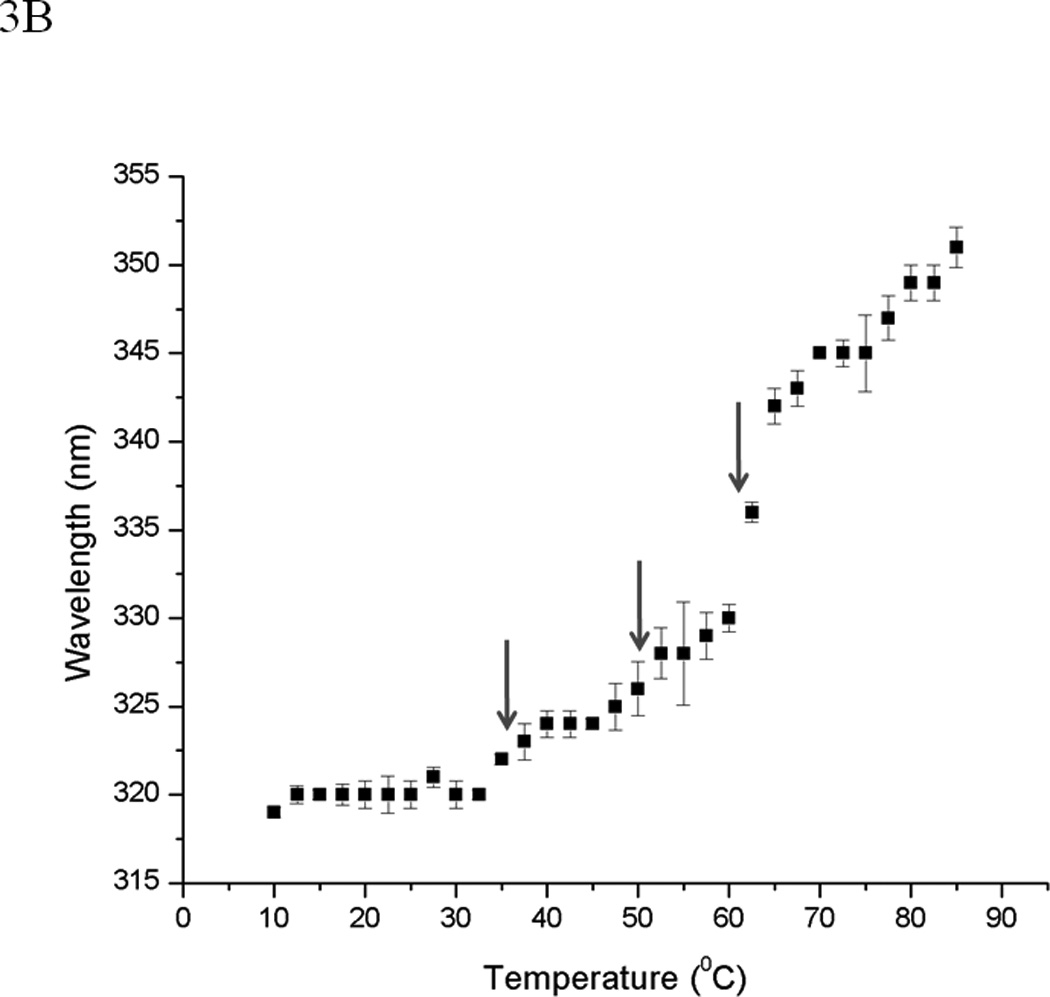
EC5 tertiary structure monitored by intrinsic fluorescence spectra. (A) The temperature-dependent shift of the fluorescence emission peak from the Trp residue at 0.02 mM EC5 in 25 mM phosphate buffer at pH 7.5. (B) The change in emission wavelengths of the same EC5 as a function of temperature.
The presence of at least two different conformations for EC5 in the fluorescence experiments was confirmed by differential scanning calorimetry (DSC) (Figure 4). Deconvolution of the experimental thermograms provides two melting transitions located at approximately 54.5 and 64.3 °C. These transition temperatures are similar to those found using fluorescence spectroscopy as shown in Figure 3B, except that the first transition at 35 °C is absent. This suggests that the first transition may be due to a local dynamic effect rather than a global unfolding transition.
Figure 4.
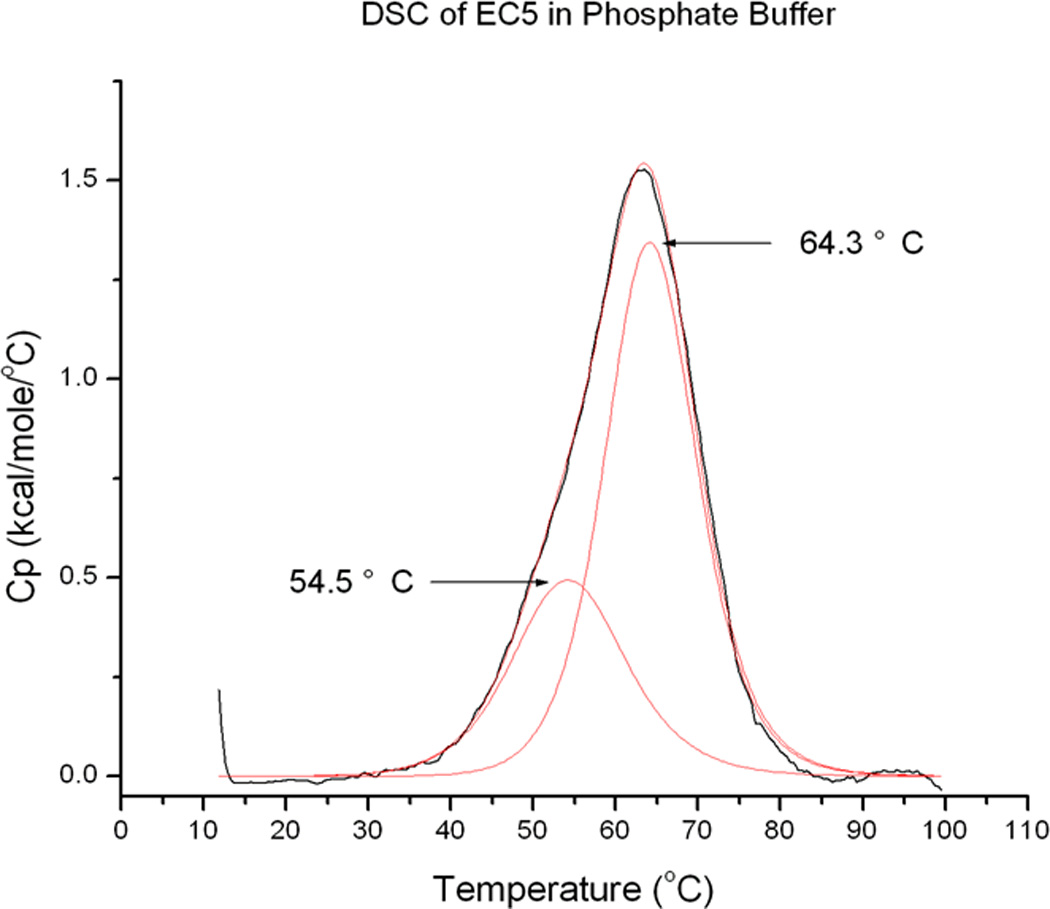
Thermally induced unfolding of 0.1 mM EC5 in 25 mM phosphate buffer at pH 7.5 monitored by DSC.
The second derivative UV spectrum of EC5 shows six peaks that are contributed by six aromatic chromophores (i.e., one Trp, two Tyr, and three Phe residues) (Figure 5A). The second derivative UV spectra can be used to probe the effect of environmental changes on EC5 conformation around the aromatic residues (41–43). Peaks 1, 2, and 3 could possibly be due to the three Phe residues. Peak 4 could possibly be derived from the two Tyr residues while peak 5 could represent a joint contribution from the Tyr and Trp residues. Peak 6 could be generated by the single Trp residue alone. The effect of temperatures on the wavelength of peaks 5 and 6 (Figure 5B) shows typical melting curves for thermal unfolding with a melting temperature of 65 °C as also seen by CD and fluorescence spectroscopy. These peaks exhibit blue shifts during the thermal unfolding process, indicating that the Trp and Tyr residues are more exposed to the aqueous solvent at higher temperatures. The lower wavelength peaks (i.e., peaks 1–4) display a much smaller spectral change upon temperature elevation. This is no doubt due to the nature of the Phe residues, which are buried in the more hydrophobic interior regions of the protein and therefore do not undergo dramatic changes in the polarity of their environment (30). Thus, it appears that the core structure remains in EC5 even at high temperature. This is also consistent with the less than complete shifting of the Trp residue to a completely exposed state. This is expected due to the presence of the disulfide bonds that prevent complete unfolding. In the reduced state of the protein, both peaks 5 and 6 lose their distinctive melting curves (Figure 5C).
Figure 5.
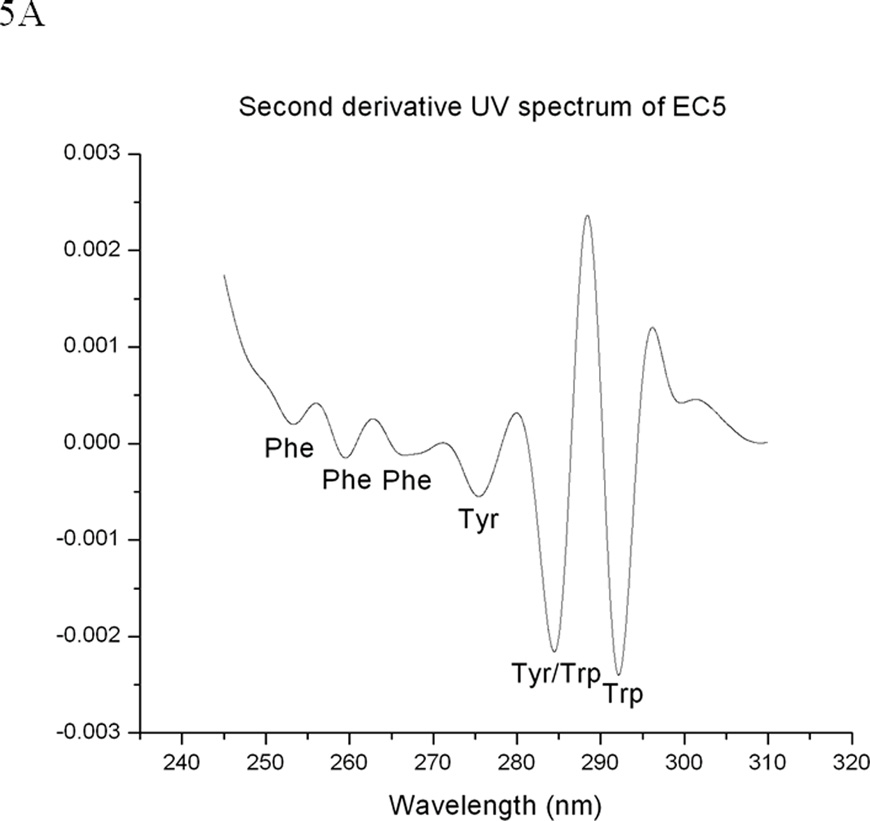
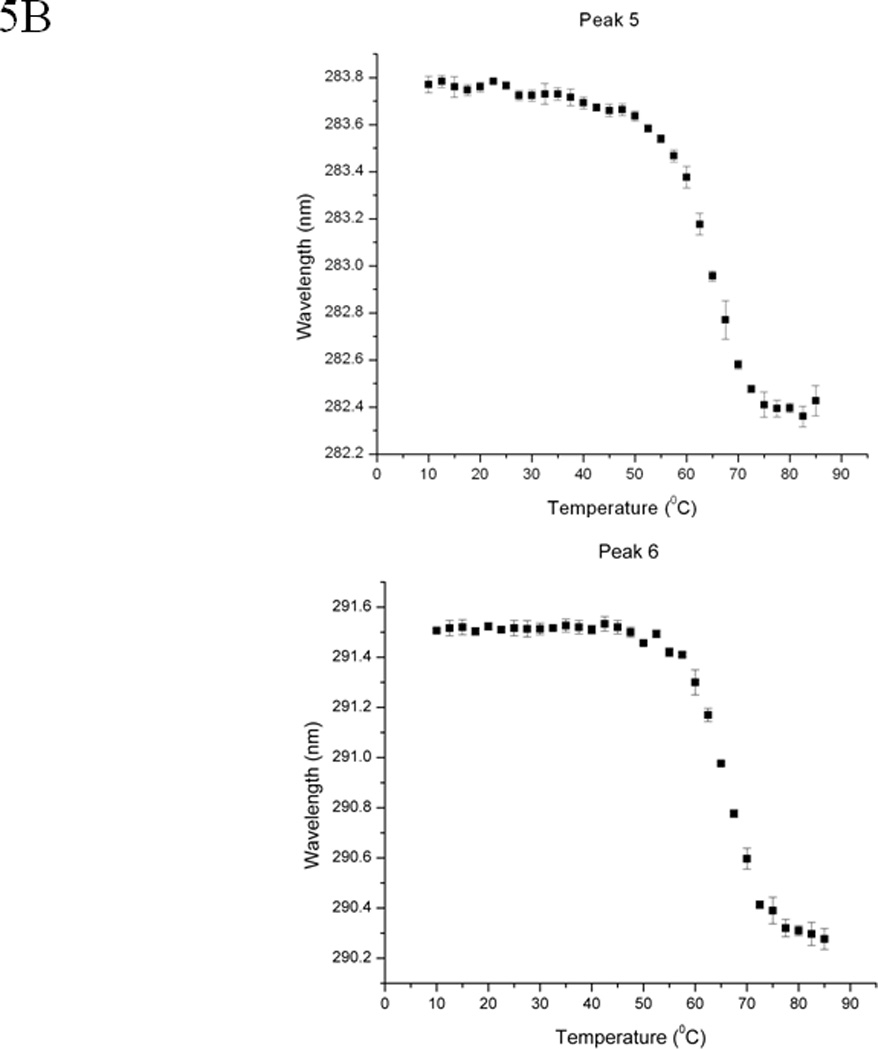
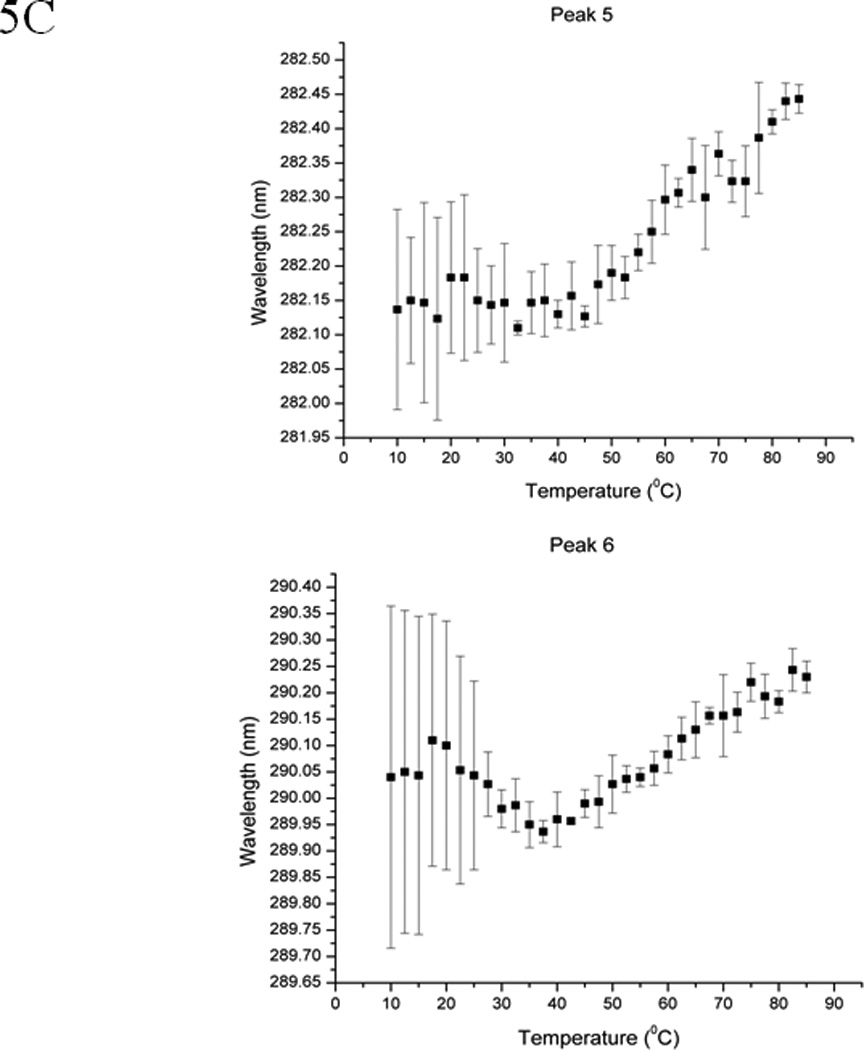
EC5 tertiary structure monitored by second derivative UV spectra. (A) 0.02 mM EC5 in 25 mM phosphate buffer at pH 7.5. (B) The effect of temperature on the wavelengths of two aromatic residues (peaks 5 and 6) of native EC5. (C) The effect of temperature on peaks 5 and 6 of the aromatic residues from the reduced form of EC5.
1H-15N-HSQC NMR spectra also imply the presence of multiple conformations of the EC5 protein. At 30 °C, the NMR spectrum of EC5 shows good dispersion of cross-peaks between 8.5 and 10 ppm, indicating a well-folded protein (Figure 6A); however, more than 200 peaks are observed. The expected number of peaks in the HSQC spectrum is 135 because EC5 has 116 residues with 11 Pro and 15 Asn/Gln. The higher number of peaks compared to the expected number suggests the presence of multiple conformers. In addition, the indole ring NH of the only Trp residue gives two distinct peaks (9.96/128.52 ppm for 1H/15N and 9.92/128.4 ppm for 1H/15N) at low temperature (see arrows on Figure 6A). The NMR spectrum is simplified upon heating to 65 °C as the protein partially unfolds and the Trp indole NH displays only a single resonance (see arrow in Figure 6B). The chemical shifts of the side chains of Asn and Gln are also collapsed into a single pair of peaks around 6.6/7.3 ppm for the 1H and 111 ppm for the 15N upon structural disruption at 65 °C (Figure 6B). When the protein is cooled to 30 °C or 40 °C for about 30 min, the NMR spectrum returns to its original state, suggesting that the protein can be refolded to the original structure (see Supplementary Materials). In this case, previous NMR studies (data not shown) indicated that the EC5 conformation does not change significantly within the pH range of 5.0 to 7.5. Thus, the spectral difference between 30 °C and 65 °C is not induced by the potential change of pH.
Figure 6.
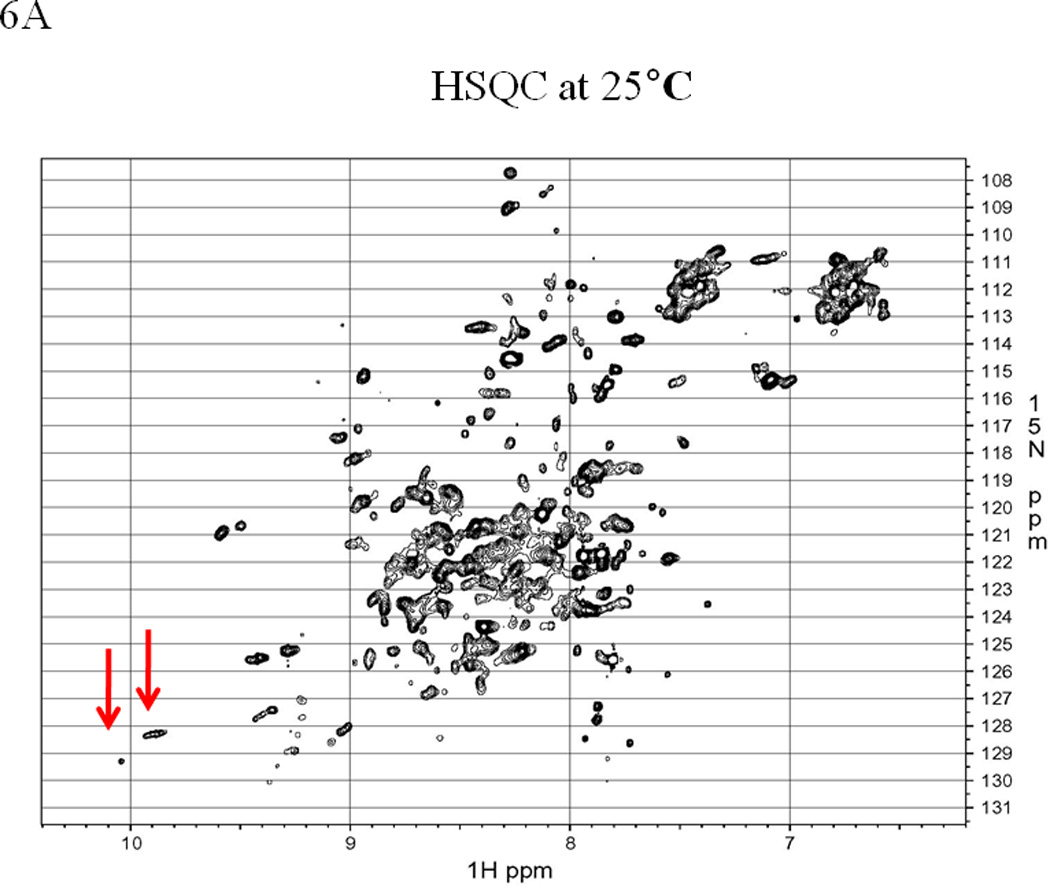
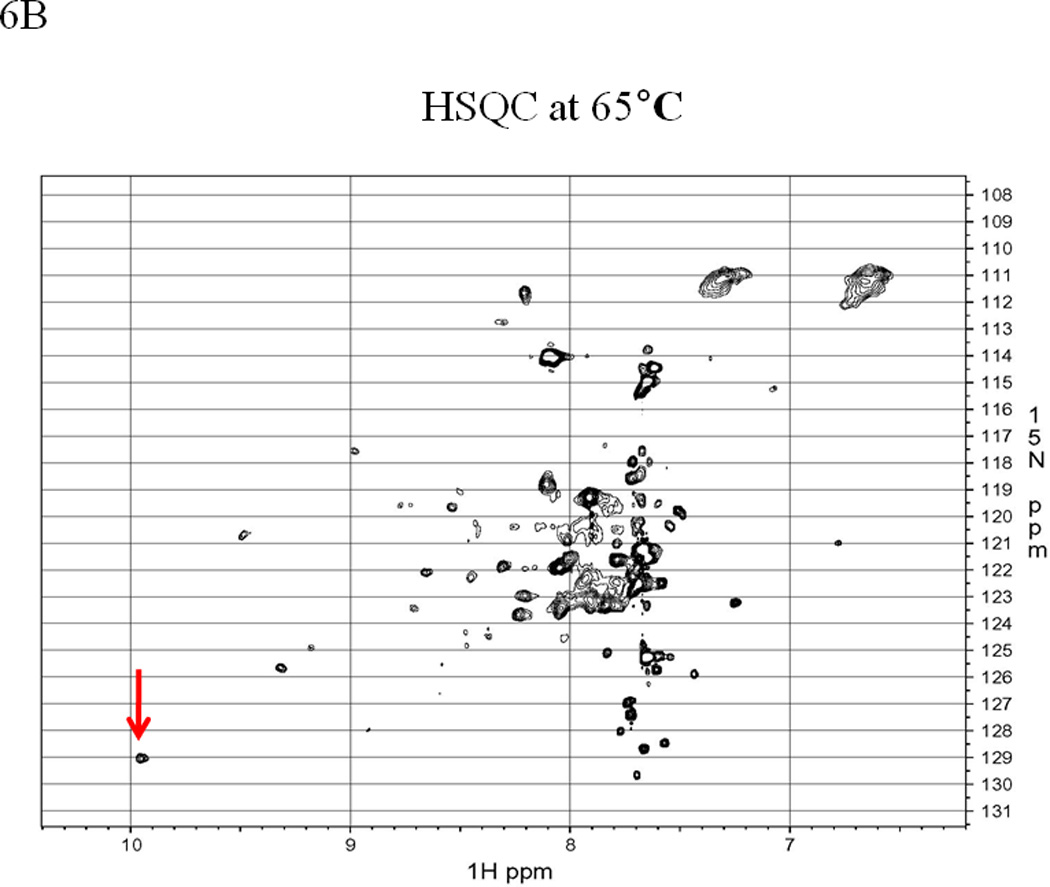
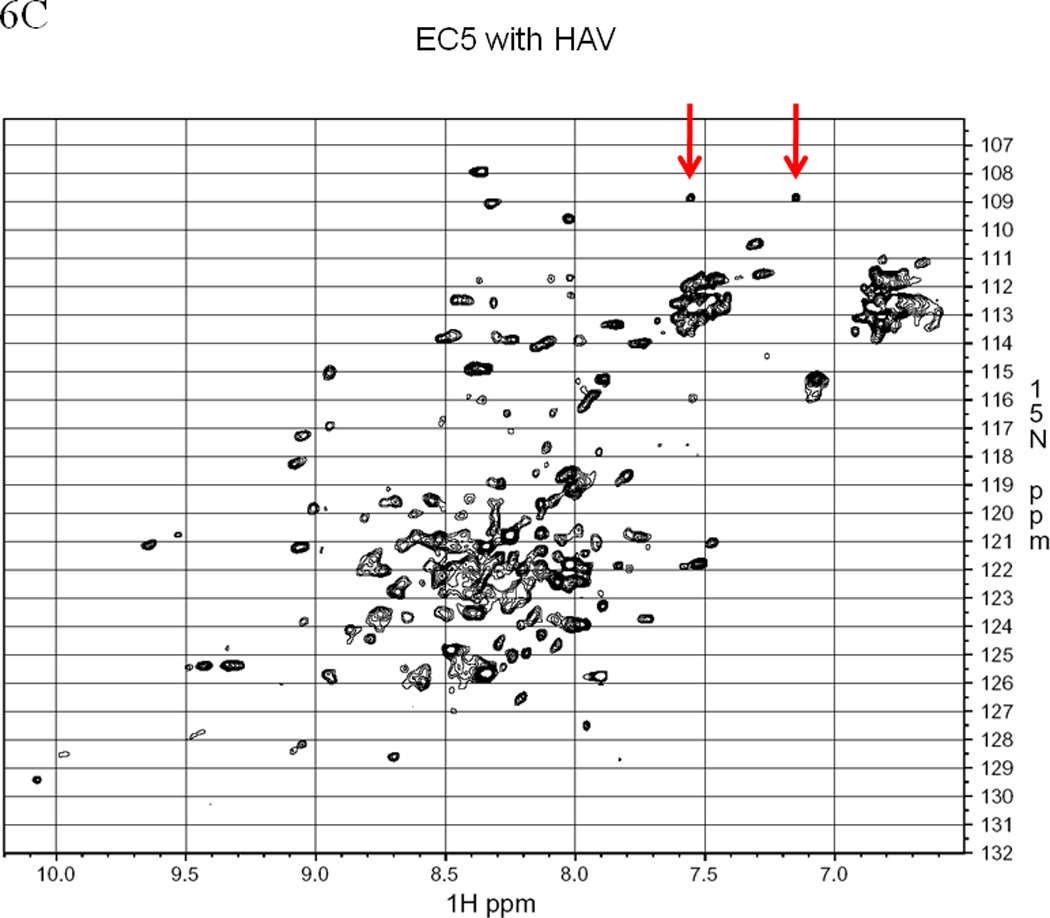
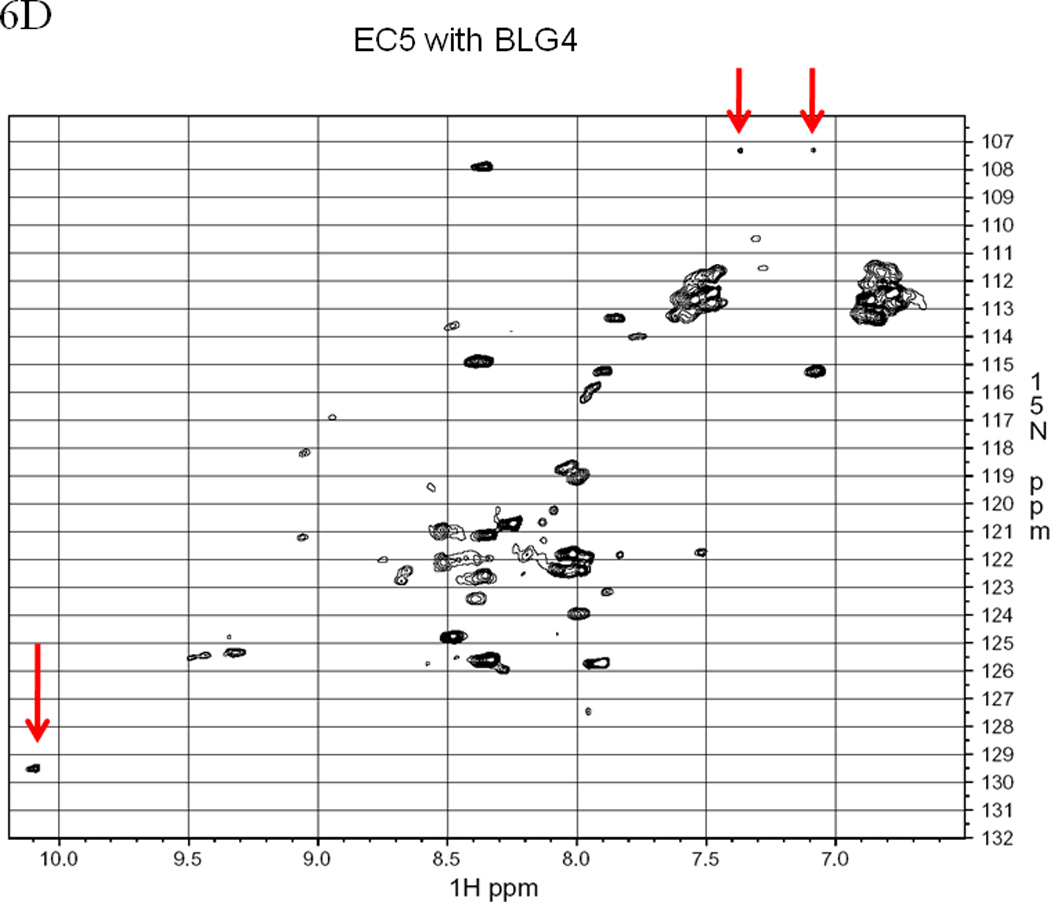
EC5 tertiary structure monitored by NMR with arrows indicating the resonance(s) for the indole NH of the Trp residue. The 1H-15N HSQC NMR spectrum of 0.5 mM EC5 domain in 25 mM Tris buffer with 10% D2O in pH 7.5 at (A) 30 °C and (B) 65 °C. 1H-15N-HSQC NMR spectrum of 0.2 mM 15N labeled EC5 in 100 mM Tris buffer containing 5% D2O at pH 7.5 and room temperature. (C) EC5 in the presence of 20 mM HAV peptide. The red arrows indicate the chemical shift changes seen after peptide binding. (D) The NMR spectrum of EC5 upon addition of 10 mM BLG4 peptide and the chemical shift changes upon peptide addition are indicated by red arrows.
The NMR data concur with the fluorescence spectroscopy results and are consistent with the hypothesis that the protein exists in two distinct conformations. NMR is well-suited for detecting dynamical properties and changes in dynamics. Movements that occur on a fast timescale relative to data acquisition generate an averaged peak, whereas more than one peak may emerge from the same residue when exchange is slow. It appears that at least two sets of resonances are present in the spectra of EC5 at lower temperatures, as peaks having distinctly different intensities are apparent (Figure 6). Many peaks, particularly those in unique chemical shift positions, appear to be doubled, reflecting a major and a minor population for each pair. Despite a difference in signal strength, the linewidths for the two populations are similar, suggesting either that the conversion between states is slow or that two slightly different forms of the protein are present in the sample, as would be the case if disulfide bond mixing occurred. When the protein is examined at 65 °C, the linewidth sharpens, reflecting the increased mobility of individual residues in the unfolded state. However, retention of a few peaks above 8.5 ppm, indicates that the structure of EC5 has not become a completely random coil. The peak at 10 ppm is generated by the Trp indole, but the other peaks, particularly the peak at 9.3 ppm, likely correspond to backbone amides near the disulfide bonds, where the local structure is better preserved at elevated temperatures. Although intensive analyses can provide residue-specific information in well-behaved systems, only a cursory analysis of the peak-width was used to assess the structural integrity of EC5 due to the complexity of the spectrum.
In an attempt to convert EC5 to a single conformation for NMR structural studies, various solution conditions were investigated, including alterations in pH, buffers, and salt concentration (data not shown). Addition of KCl or CaCl2 or a change to low pH improved the NMR spectra and, in some cases, enhanced the signal-to-noise ratio; unfortunately, none of these changes resulted in convergence to a single set of peaks. Thus, complete assignment of the resonances and site-specific dynamics analysis remain difficult at this time.
Binding studies of the HAV and BLG4 peptides to EC5 using NMR and FTIR
The interactions of the HAV and BLG4 peptide with 15N-labeled EC5 were investigated using 1H-15N-HSQC NMR experiments. There are two clusters of peaks at 6.8/112.5 and 7.5/112.5 ppm, which originate from the NH2 groups of the side-chains of the Asn and Gln residues. Each side-chain generates two peaks in the HSQC spectrum with distinct 1H but identical 15N chemical shifts. This occurs because the peaks emanate from each of the two 1H atoms attached to the same 15N atom. These peak clusters suggest that the side-chains of the Asn and Gln residues are solution exposed and in virtually identical chemical environments. This makes them difficult to distinguish from each other. Adding the HAV peptide to 15N-labeled EC5 induces a change in its HSQC spectrum, causing the appearance of two new peaks at 7.55/108.85, and 7.15/108.85 ppm (see the red arrows in Figure 6C). These peaks are separated from the clusters at 6.8/112.5 and 7.5/112.5 ppm. Because changes in the chemical shifts reflect changes in the environment when the environment around an atom is altered by reducing its exposure to solvent/solution or participating in hydrogen bonds, these two shifted peaks suggest that one of the Asn or Gln residues might be interacting with the HAV peptide and altering the chemical shift of a side-chain NH2 group. In addition, the intensity of one of the NH indole peaks of EC5 (9.96/128.5 ppm) decreases in the presence of the HAV peptide. These changes may be due to a change in the conformation of EC5 upon binding to the HAV peptide.
Addition of the BLG4 peptide to 15N-labeled EC5 causes even more dramatic changes in the spectrum of EC5 (data not shown). The overall spectrum of EC5 is simplified significantly in the presence of the BLG4 peptide, i.e., addition of this peptide substantially reduces the total number of peaks from 252 to 161. The two peaks of the NH indole ring from the single Trp residue (9.96/128.5 and 10.08/129.5 ppm) merge into one peak at 10.1/129.4 ppm in the presence of the BLG4 peptide. These results suggest that binding of BLG4 to EC5 stabilizes one of the two equilibrium conformations of EC5. In addition, two peaks from the NH2 side-chain of one of the Asn or Gln residues are again shifted from the original clusters at 6.8/112.5 and 7.5/112.5 ppm on EC5 alone to 7.37/107.3 and 7.09/107.3 ppm in the complex. Thus, like the HAV peptide, BLG4 may also bind to EC5 in a region containing an Asn and/or Gln residue.
13C,15N-double-labeled EC5 was also used to determine the binding properties of HAV and BLG4 peptides to EC5. The presence of heavier isotopes (i.e., 13C or 15N) in double-labeled EC5 shifts the amide I band to a lower frequency (44) from the band of unlabeled EC5 with the assumption that both of the labeled and unlabeled EC5 have identical conformations. The FTIR amide I bands unlabeled EC5, double-labeled EC5, and a mixture of double-labeled EC5 and the peptides were analyzed to determine the effects of peptide binding on EC5 (Figure 7). The amide I band of unlabeled EC5 was shifted from 1637 cm−1 to 1594 cm−1 in the double-labeled EC5 (Figure 7A). Addition of the unlabeled HAV or BLG4 peptide to double-labeled EC5 altered the amide I band. Deconvolution of the amide I band of the unlabeled EC5 resolves several bands, including the 1608 cm−1 and 1585 cm−1 signals (Figures 7A–D). Both of these bands were used to estimate the amount of α-helix and β-sheet in EC5, respectively (Table 2) (45). Unfortunately, unambiguous assignments for the other peaks are unavailable. The estimated amount of α-helical structure in EC5 was increased from 13% to 22% and 24% upon binding to HAV and BLG4, respectively (Figures 7C and D; Table 2). A small decrease in β-sheet structure in EC5 was also observed when binding to either peptide. A new band was seen at 1676 cm−1 in the spectrum of the mixture of EC5 and BLG4; this band is presumably derived from the β-sheet structure of the BLG4 peptide itself (46). Thus, upon binding to EC5, the BLG4 peptide also appeared to change its conformation from disordered to β-sheet structure.
Figure 7.
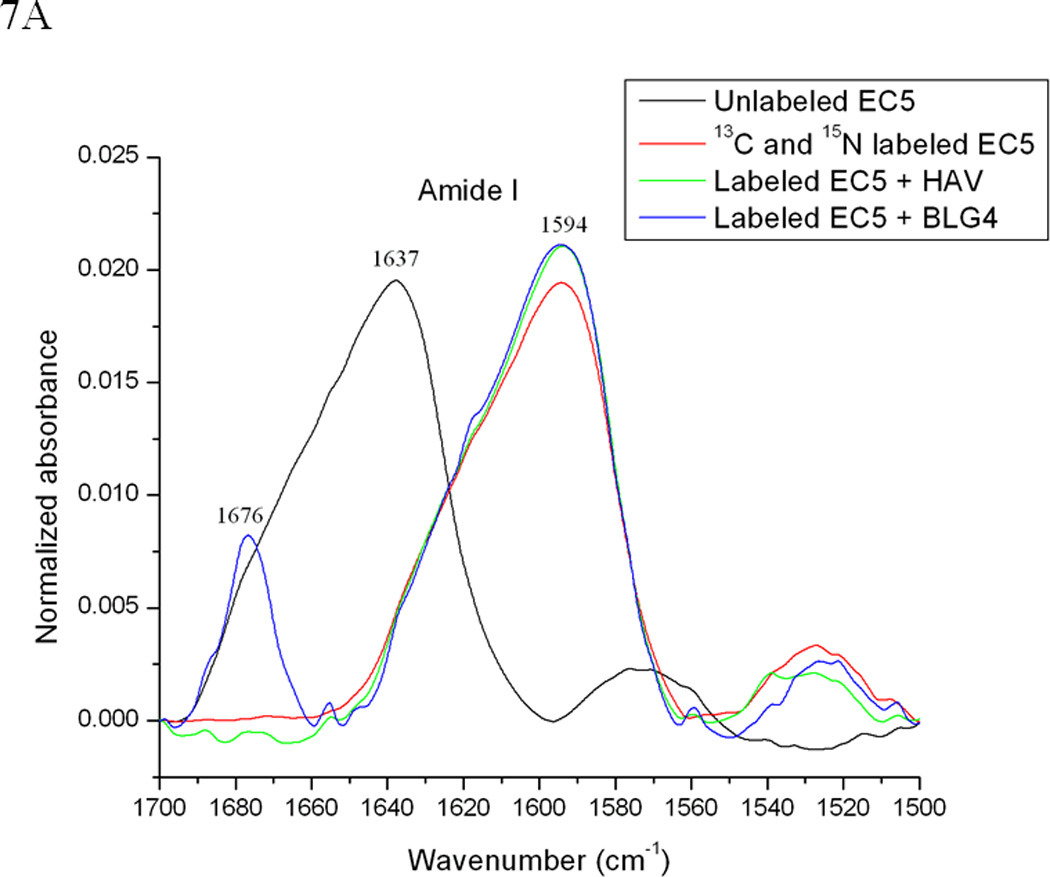
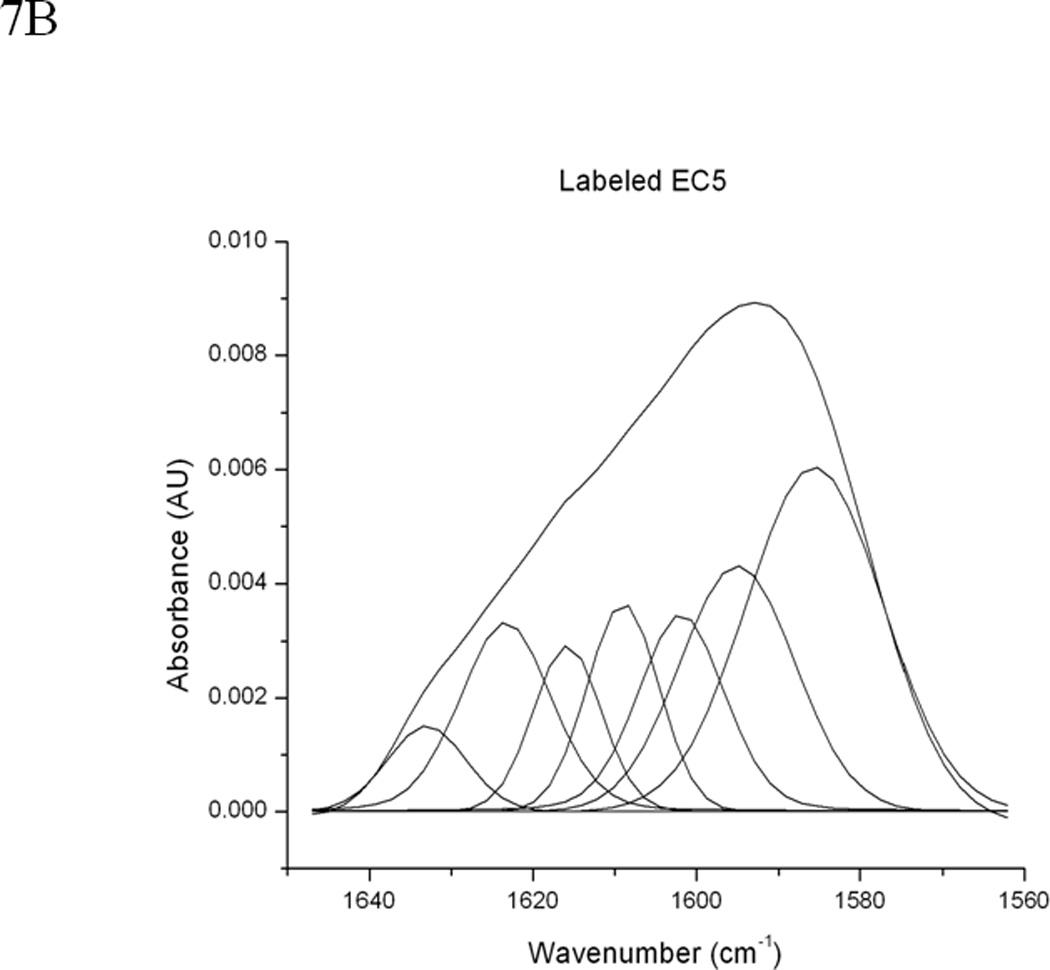
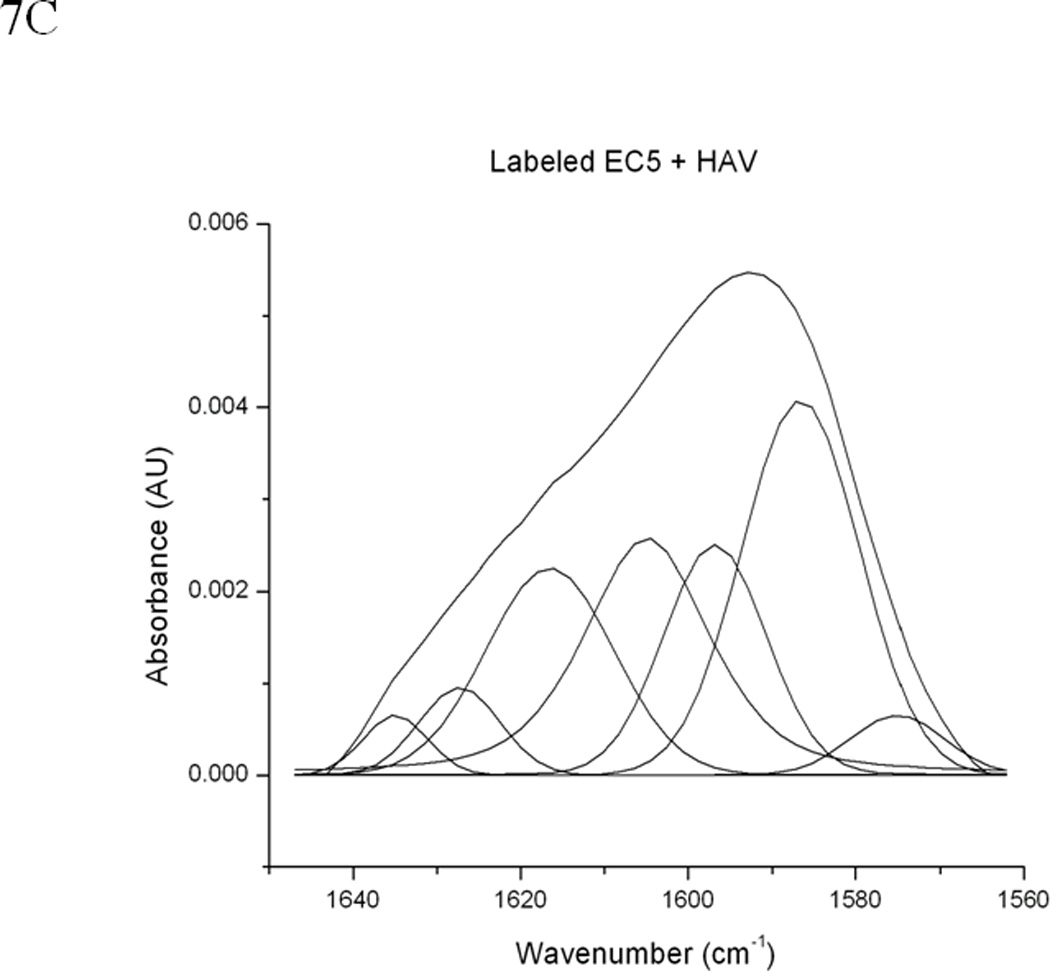
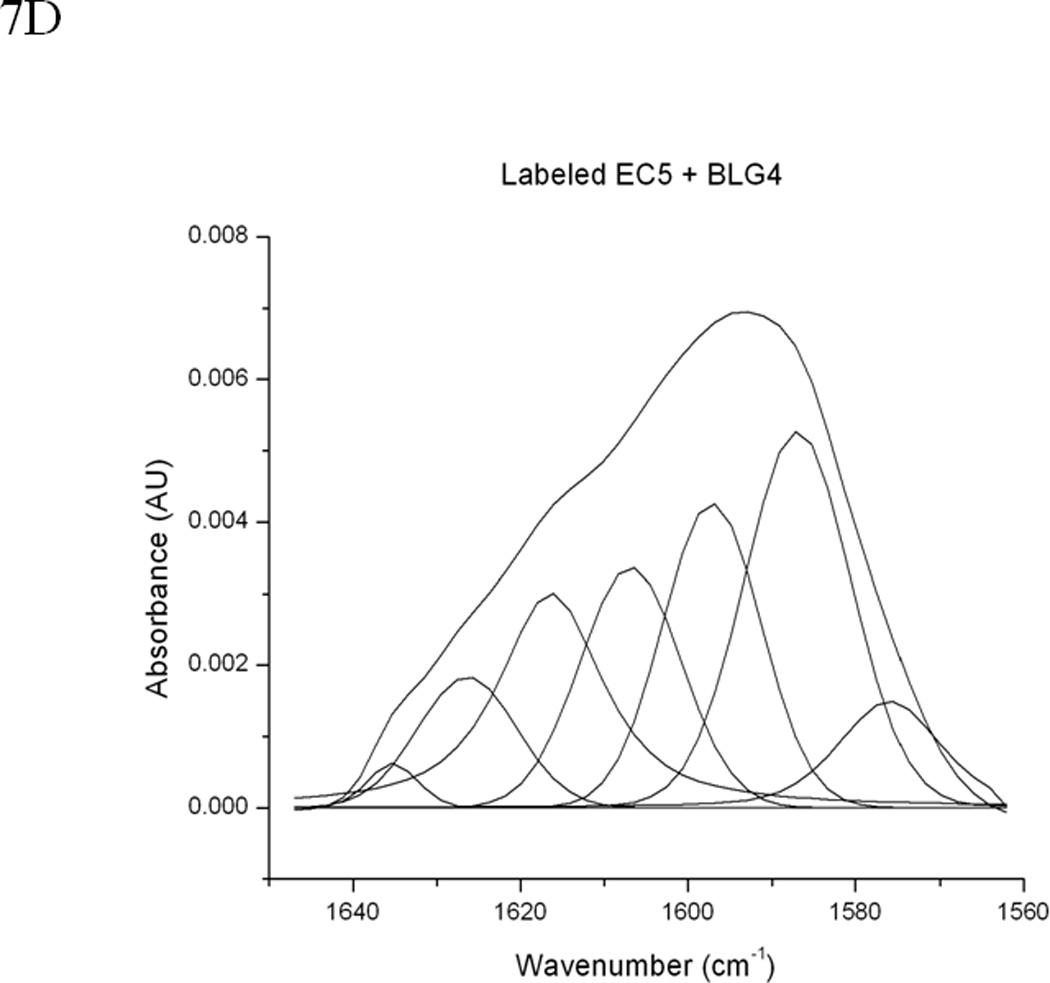
(A) 0.5 mM unlabeled or 13C and 15N double-labeled EC5 in the absence or presence of 50 mM HAV or 25 mM BLG4 peptides in 100 mM Tris-D2O buffer at pH 7.5 and room temperature. (B) Deconvolution of the amide I band of 13C and 15N double-labeled EC5 in the absence of peptide. (C) Deconvolution of the amide I band of 13C and 15N double-labeled EC5 in the presence of HAV peptide. (D) Deconvolution of the amide I band of 13C and 15N double-labeled EC5 in the presence of BLG4 peptide.
Table 2.
Secondary structure changes of 0.5 mM 15N and 13C double-labeled EC5 after addition of 50 mM HAV or 25 mM BLG4 in 100 mM Tris-D2O buffer at pH 7.5 and room temperature.
| α-helix | β-Sheet | |
|---|---|---|
| EC5 | 13% ± 5% | 34% ± 7% |
| EC5 + HAV | 22% ± 2% | 32% ± 3% |
| EC5 + BLG4 | 24% ± 7% | 26% ± 5% |
DISCUSSION
The structure and dynamic properties of EC5 may play an important role during the homophilic and heterophilic interactions of E-cadherin; therefore, the structural and binding properties of EC5 were investigated. Based on SEC, EC5 exists in a monomeric form with two intramolecular disulfide bonds, which play an important role in the structural stability of EC5 (47, 48). Reducing these disulfide bonds lowers EC5 structural stability, and the change in EC5 conformation is reflected in the disappearance of the CD positive peak at 230 nm, which is due to a combination of different structural features (i.e., β-turns, aromatic residues, and disulfide bonds). The second derivative FTIR spectrum of the reduced form has a new absorption band at 1619 cm−1 (Figure 2B), which suggests the presence of some intermolecular β-sheet structure, perhaps produced by aggregation. In addition, reduction of the disulfide bond shifts the fluorescence λmaxEM from a dual peak at 320 and 332 nm to a single signal at 346 nm, indicating a dramatic change in the tertiary structure of EC5 (Figure 2C). The fluorescence λmaxEM at 346 nm of reduced EC5 is much closer to the λmaxEM of free indole (typically 350–355 nm) in water, and is similar to that of the thermally disrupted form of EC5. In summary, the relatively high thermal stability of EC5 probably is due to the existence of the two intramolecular disulfide bonds.
The fluorescence and NMR spectroscopy data suggest the presence of two distinct EC5 conformations. Although EC5 contains only one Trp residue, the intrinsic fluorescence of this residue showed two peaks at 320 and 332 nm at room temperature. These two peaks merged into a single peak at 332 nm during temperature elevation and, finally, the peak shifted to 346 nm upon further unfolding at high temperature (Figure 3A). The existence of two conformers is also indicated by the presence of two indole ring NH cross-peaks in the 1H-15N-HSQC NMR spectrum at 30 °C in the 15N-labeled protein (Figure 6A). At 65 °C, these two cross-peaks collapsed to a single cross-peak at 9.95 and 129 ppm for 1H and 15N resonances, respectively (Figure 6B). The two DSC transitions observed at 54.5 °C and 64.3 °C also support the possible presence of two different stable conformation of EC5 (Figure 4).
Typically, the interpretation of the observed spectroscopic and calorimetric data would support two possible mechanisms (A and B, Figure 8) for the conformational change in EC5 upon temperature increase. Mechanism A suggests the presence of two conformations (C1 and C2) at room temperature. As the temperature increases, C1 converts to a more stable C2 conformation at 54.5 °C. Conformation C2 then unfolds at a melting temperature of 64.3 °C. Mechanism B postulates the existence of three different conformational states (C1, C2, and C3); at room temperature, EC5 exists in two conformations, C1 and C2. As the temperature is increased, both the C1 and C2 states convert to C3 at 54.5 °C, and C3 is the intermediate state prior to unfolding. Conformation C3 unfolds more extensively at 64.3 °C.
Figure 8.
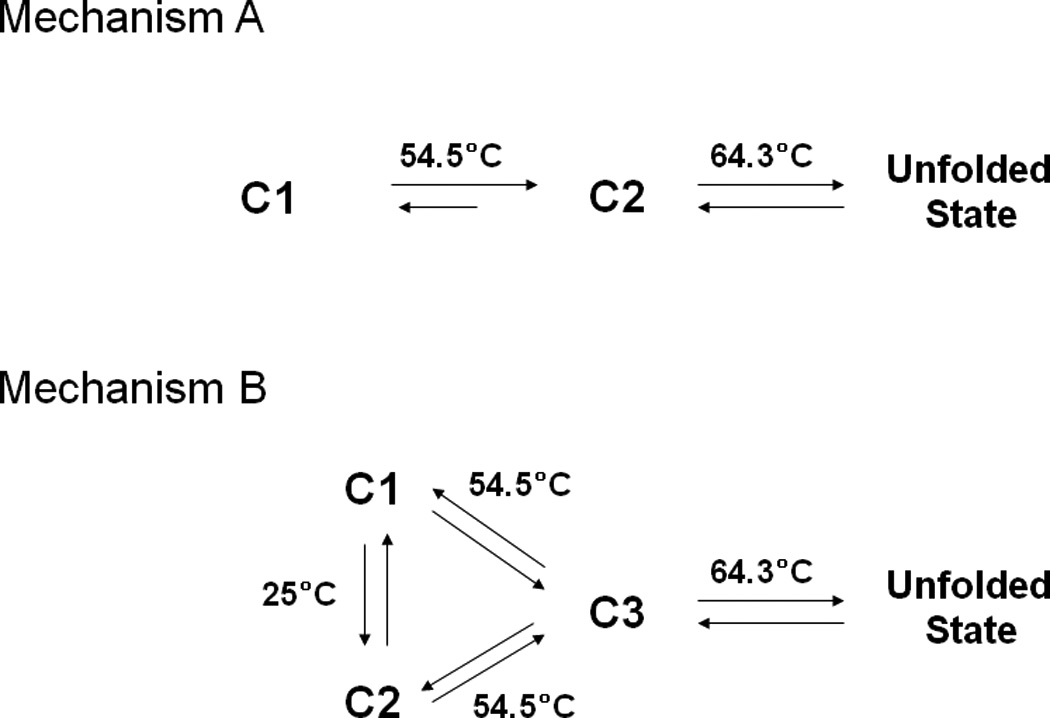
Proposed multiple conformations of EC5 and their pathways of structural alteration upon increases in temperature.
In support of mechanism A, the Trp intrinsic fluorescence peaks at 320 and 332 nm can be assigned to the C1 and C2 conformations, respectively. Increasing the temperature shifts the peak at 320 nm to the peak at 332 nm, reflecting the conversion of conformation C1 to C2. Thus, most of the molecules in C1 have converted to C2 at 54.5 °C. As the temperature reaches 64.3 °C, the C2 conformation is more unfolded. The two conformations (C1 and C2) of EC5 at room temperature are also reflected by the presence of the two NH indole cross-peaks with chemical shifts of 9.96/128.52 ppm (C1) and 9.92/128.4 ppm (C2) in the 1H/15N HSQC spectrum. At 60 °C, these two cross-peaks merge into one cross-peak at 9.95/129 ppm for 1H/15N nuclei. Although the proton resonance (9.95 ppm) of this final cross-peak is similar to the proton resonance of the C2 conformation at room temperature, the 15N resonance (129 ppm) more closely resembles the 15N resonance of C1 (129.2 ppm). Thus, the NMR results cannot completely rule out the possibility of Mechanism B. To explain the data in terms of Mechanism B, the intrinsic fluorescence peaks at 320 and 332 nm would represent C1 and C2, respectively, similar to Mechanism A. Furthermore, a low-temperature transition at 35 °C was observed in the fluorescence spectra (Figure 3B), which suggests the possible existence of C1 and C2 at low temperature. Upon increases in temperature above 54.5 °C, both C1 and C2 are converted to C3. In this case, the intrinsic fluorescence of the Trp residue of C3 is similar to that of C2. As mentioned above, the final chemical shift of the NH indole of the Trp residue may resemble that of an intermediate between those NH groups of the C1 and C2 states, suggesting the presence of C3. Therefore, it is difficult to conclusively differentiate between the two mechanisms.
It is not yet clear whether these two conformations are present in the intact E-cadherin molecules and if they are involved in the function of E-cadherin in its role of mediating cell-cell adhesion. It is known, however, that the entire extracellular domain of E-cadherin forms a globular structure in the absence of calcium and a rod-like unit in its presence. It is possible that the two different conformations of EC5 may be involved in the conversion between the globular and rod-like structures upon addition of calcium. The interconversion between these two conformers could also be involved in the cis- and trans- interactions of E-cadherin during the cell-cell adhesion process. In another cell adhesion receptor, lymphocyte function-associated antigen 1 (LFA-1), the I-domain of its α-subunit can convert between “open” and “closed” forms upon binding to its ligands (i.e., allosteric inhibitors) (49).
One of the goals of this project was to search for small peptides (i.e., hexapeptide or smaller) with good biological activity to modulate the intercellular junctions. The HAV hexapeptide was discovered by initially examining a large peptide of 24 amino acid residues (24-mer) that includes the His-Ala-Val (HAV) sequence derived from the groove region of the EC1 domain (50). The 24-mer peptide was reduced to several decapeptides with overlapping sequences; one of the decapeptides, HAV10 (Ac-LFSHAVSSNG-NH2), containing the His-Ala-Val sequence was found to have the best ability to inhibit E-cadherin-mediated cell-cell adhesion (50). Finally, HAV10 was reduced to several hexapeptides and one of these, HAV (Ac-SHAVSS-NH2), was more effective than the others (20).
The BLG4 peptide sequence was found by homology study between the EC1 and EC4 of E-cadherin; it was from the bulge region of EC4 similar to the ADT peptide from EC1 (51). BLG4 was also found to inhibit E-cadherin-mediated cell-cell adhesion and the resealing of intercellular junctions of Caco-2 cell monolayers.
The binding properties of the HAV and BLG4 peptides to the EC5 domain were evaluated using NMR and FTIR. Both peptides induce a dramatic shift of the double-labeled EC5 amide I band (about 43 cm−1 to a lower frequency), suggesting structural changes in EC5 upon peptide binding (44). The β-sheet structure (i.e., the 1676 cm−1 band) of BLG4 was detected when a complex is formed between BLG4 and EC5. In contrast, no new amide I signal was observed in the complex between HAV and EC5, suggesting no major structural change in HAV during binding. The apparent difference in secondary structural change seen between HAV and BLG4 during binding may also be related to the different binding modes of each peptide. Although both peptides bind to EC5, it is still possible that these peptides bind to other EC domains as well (i.e., EC1 to EC4) during the inhibition of E-cadherin-mediated cell-cell adhesion; thus, the potential interaction between the peptides and other EC domains will also be examined.
NMR spectroscopy indicates that EC5 changes its tertiary structure when it binds to each peptide. The two HSQC spectra of EC5 in the presence of HAV or BLG4 are very distinct from each other. The total peak number compared to that of EC5 alone (252) is increased upon HAV addition (284) but decreased by BLG4 addition (161). In addition, a distinct new isolated peak pair from either Asn or Gln is observed in each spectrum upon peptide addition (i.e., they do not display the same chemical shifts). This alone does not establish that the peptides bind to different sites on EC5, but, in light of the numerous other changes, it may indicate that different residues are involved. In the case of BLG4 binding, the two conformations of EC5 merge into a single structure (data not shown), as shown by the behavior of the resonances of the NH-indole ring of the Trp residue of EC5. Combined with the FTIR results, this suggests that both BLG4 and EC5 have to change their conformations significantly during complexation. During binding of HAV to EC5, the increased intensity of the NMR peaks at 7.55/115.9 and 7.7/123.7 ppm and the appearance of a new peak at 8.2/109.6 ppm suggest a possible small increase in α-helical content (Figure 6C), which is consistent with the statistically marginal FTIR results.
Molecular docking simulations were carried out to evaluate the interaction between the HAV peptide and EC5. The HAV peptide binds to EC5 with a favorable binding energy (Figures 9A and B) with a network of interactions, including: (i) a hydrogen bond between the backbone carbonyl oxygen of Ser-5 on the peptide and the amide side-chain of the Asn-46 residue on EC5; (ii) a hydrogen bond between the carbonyl group of Asn-46 and the side-chain hydroxyl group of Ser-6 on the peptide; (iii) two hydrogen bonds from both the side-chain oxygen and the NH backbone of the Ser-1 residue with the side-chain carboxyamide of Asn-75; (iv) a hydrogen bond between the backbone carbonyl oxygen of Ser-1 and the amide side-chain of Asn-75; and (v) a hydrogen bond between the backbone NH group of Ala-3 on the peptide and the backbone carbonyl oxygen of His-41 on EC5. Our previous Ala scanning experiment also indicated that both Ser-5 and Ser-6 have an important role in the activity of the HAV peptide (20).
Figure 9.
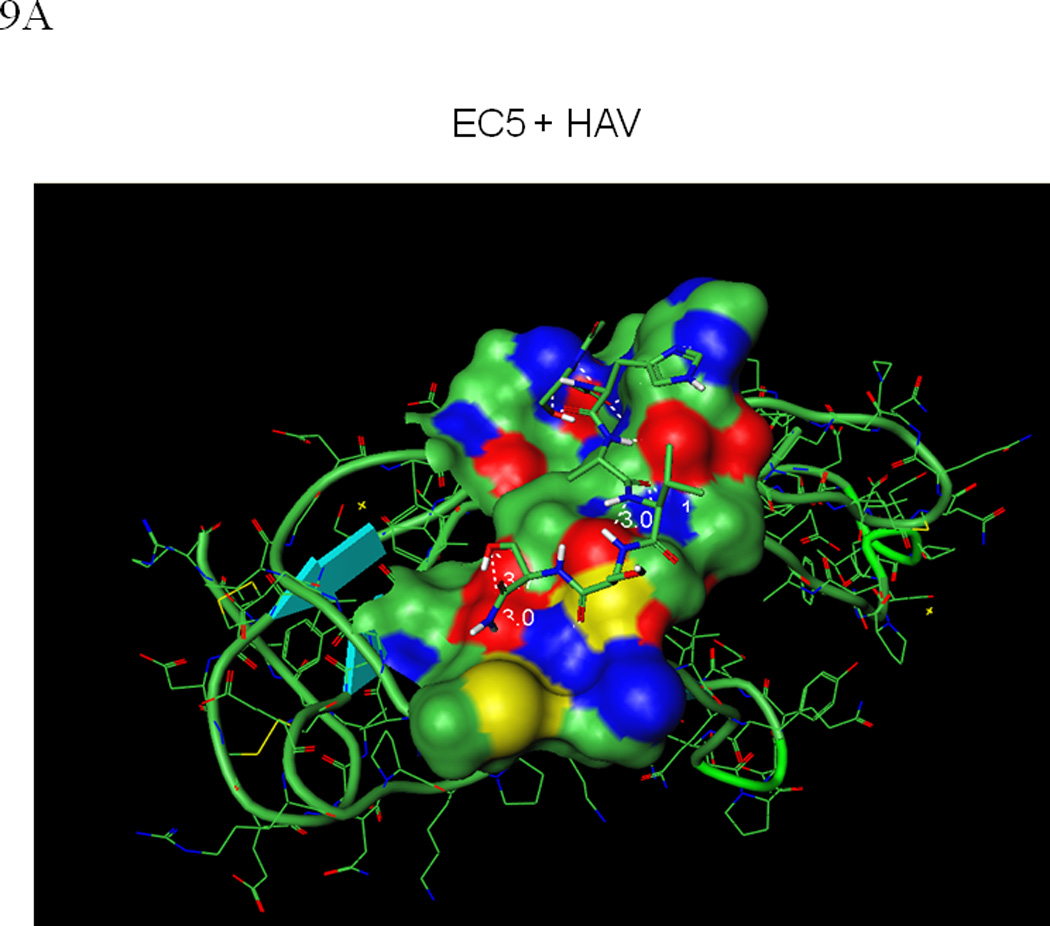
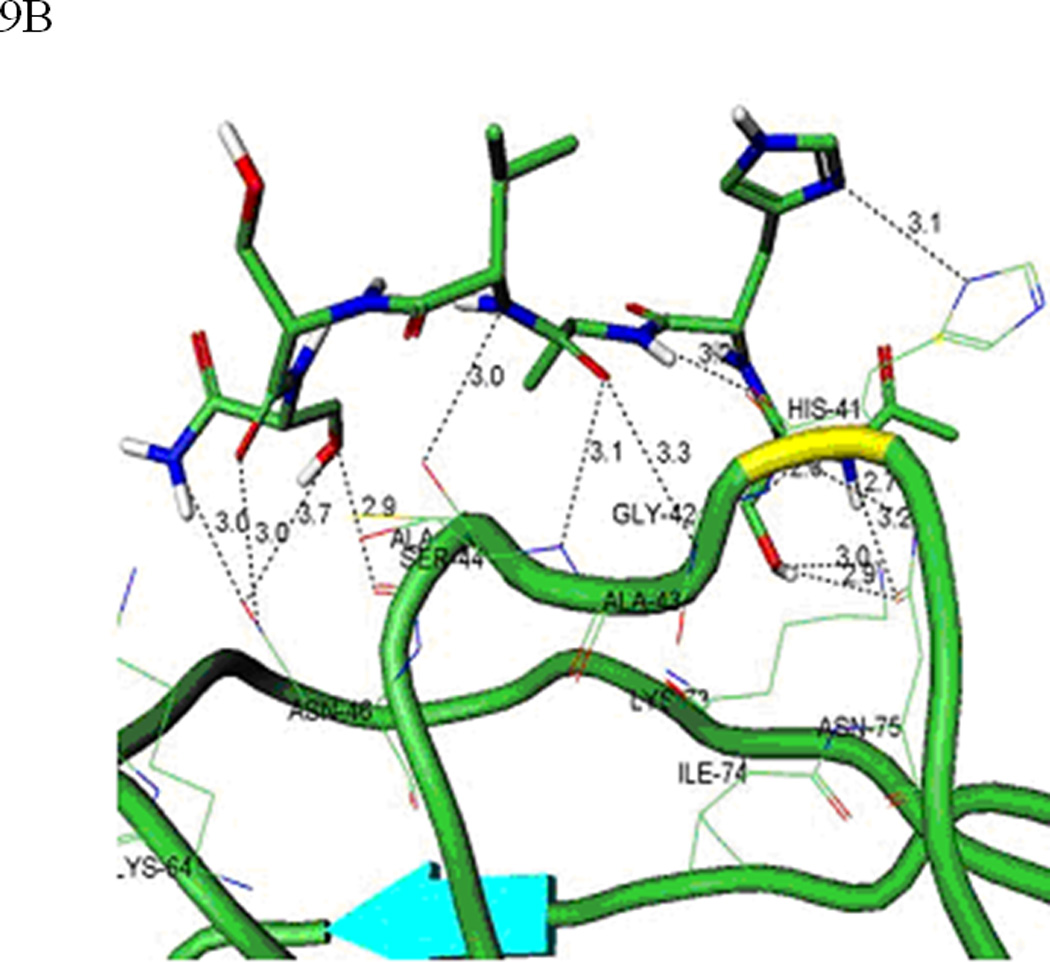

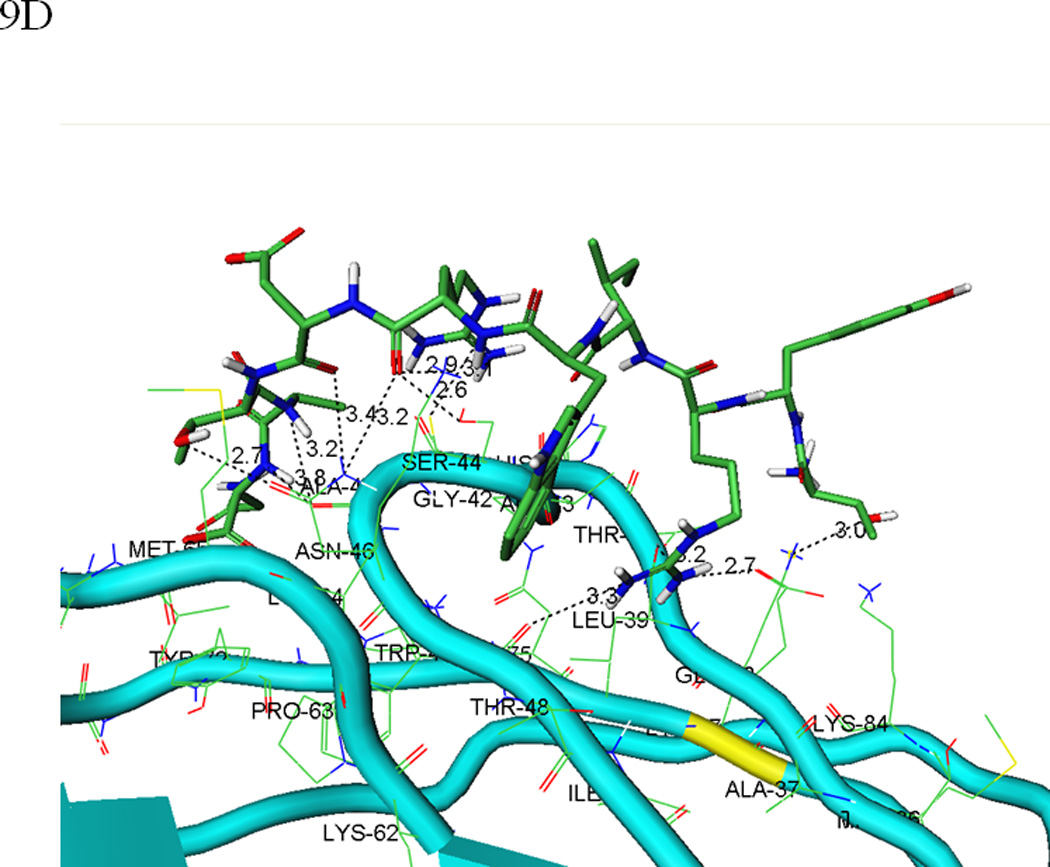
(A) The predicted conformation of the HAV peptide bound to the homology modeled structure of the EC5 domain of E-cadherin. (B) A close-up of the hydrogen bond network formed by the HAV peptide bound to EC5. (C) The predicted conformation of the BLG4 peptide bound to EC5. (D) A close-up of the hydrogen bond network formed by the BLG4 peptide bound to EC5.
In a similar manner, molecular docking simulations of BLG4 to EC5 identified a structurally consistent binding pose of the bound peptide that interacts with both the bulge and groove regions of EC5 (Figures 9C and D). The results indicated a larger degree of structural change upon BLG4 than HAV binding; the backbone and side-chain of the bulge and groove regions fluctuate with a root mean square deviation (RMSD) of up to 1.5 Å from the initial structure. The BLG4 peptide binds to EC5 in an extended structure with the β-sheet conformation forming another extensive hydrogen-bond network (Figure 9D). In particular, these interactions include numerous favorable contacts between BLG4 and Asn-46 on EC5: (i) hydrogen bonds formed by the carbonyl oxygens of the BLG4 Arg-6 and Asp-7 backbone with the amide group of the Asn-46 side-chain; and (ii) hydrogen bonding between the hydroxyl group of the BLG4 Thr-8 side-chain and the carbonyl oxygen of the side-chain of Asn-46 in EC5. These interactions are further strengthened by additional hydrogen bonds between: (i) the Arg-6 guanidine side-chain of BLG4 with the Glu-38 carboxylic side-chain and the Leu-39 backbone carboxyl oxygen of EC5; and (ii) the Thr-1 side-chain of BLG4 and the amino group of the Lys-77 side-chain on the groove region of EC5. This model also supports the FTIR data in which BLG4 forms a stable β-sheet conformation when it binds to EC5 as well as the NMR data that show BLG4 binds to EC5 in a region containing an Asn residue, (i.e., Asn-46).
In conclusion, we have shown that EC5 appears to have at least two different stable conformations with high thermal stability. The presence of disulfide bonds is essential to maintaining its conformational and thermal stability. The HAV and BLG4 peptides bind to EC5, which induces a conformational change in EC5. Molecular modeling studies suggest that HAV and BLG4 bind to a common bulge region but have distinct groove region interactions. Using the results from this study, we will attempt to design small peptides that have higher binding affinity to EC5 than the parent peptides investigated here (i.e., HAV and BLG4) for enhancing drug permeation through the intestinal mucosa and the blood-brain barrier.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from NIH (EB-00226). TJS would like to acknowledge Self Faculty Scholar funds from The University of Kansas and Pfizer Faculty Scholar funds for financial support. We are grateful to Andria Skinner and Dr. David VanderVelde for their assistance with the NMR experiments. We also thank Nancy Harmony for proofreading this manuscript.
REFERENCES
- 1.Zheng K, Trivedi M, Siahaan TJ. Structure and function of the intercellular junctions: barrier of paracellular drug delivery. Curr Pharm Des. 2006;12:2813–2824. doi: 10.2174/138161206777947722. [DOI] [PubMed] [Google Scholar]
- 2.Takeichi M. Cadherins in cancer: implications for invasion and metastasis. CurrOpin Cell Biol. 1993;5:806–811. doi: 10.1016/0955-0674(93)90029-p. [DOI] [PubMed] [Google Scholar]
- 3.Takeichi M. Cadherin cell adhesion receptors as a morphologic regulator. Science. 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 4.Pertz O, Bozic D, Koch AW, Fauser C, Brancaccio A, Engle J. A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J. 1999;18:1738–1747. doi: 10.1093/emboj/18.7.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annual review of cell and developmental biology. 2007;23:237–261. doi: 10.1146/annurev.cellbio.22.010305.104241. [DOI] [PubMed] [Google Scholar]
- 6.Nagar B, Overduin M, Ikura M, Rini JM. Structural basic of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–364. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- 7.Leckband D, Sivasankar S. Mechanism of homophilic cadherin adhesion. Curr Opin Cell Biol. 2000;12:587–592. doi: 10.1016/s0955-0674(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 8.He W, Cowin P, Stokes DL. Untangling desmosomal knots with electron tomography. Science. 2003;302:109–113. doi: 10.1126/science.1086957. [DOI] [PubMed] [Google Scholar]
- 9.Overduin M, Harvey T, Bagby S, Tong K, Yau P, Takeichi M, Ikura M. Solution structure of the epithelial cadherin domain responsible for selective cell-adhesion. Science. 1995;267:386–389. doi: 10.1126/science.7824937. [DOI] [PubMed] [Google Scholar]
- 10.Tomschy A, Fauser C, Landwehr R. Homophilic adhesion of E-cadherin occurs by a co-operative two-step interaction of N-terminal domains. EMBO J. 1996;15:3507–3514. [PMC free article] [PubMed] [Google Scholar]
- 11.Takeda H, Shimoyama Y, Nagafuchi A, Hirohashi S. E-cadherin functions as a cis-dimer at the cell-cell adhesive interface in vivo. Nat Struct Biol. 1999;6:310–312. doi: 10.1038/7542. [DOI] [PubMed] [Google Scholar]
- 12.Kitagawa M, Naroti M, Murase S, Hirano S, Taketani S, Suzuki ST. Mutation analysis of cadherin-4 reveals amino acid residues of EC1 important for the structure and function. Biochem Biophys Res Commun. 2000;271:358–363. doi: 10.1006/bbrc.2000.2636. [DOI] [PubMed] [Google Scholar]
- 13.Makagiansar IT, Ikesue A, Nguyen PD, Kuczera K, Dentler W, Urbauer JL, Galeva N, Alterman M, Siahaan TJ. Disulfide bond formation promotes the cis- and trans-dimerization of the E-cadherin derived first repeat. J Biol Chem. 2002;277:16002–16010. doi: 10.1074/jbc.M200916200. [DOI] [PubMed] [Google Scholar]
- 14.Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- 15.Sivasankar S, Brieher W, Lavrik N, Gumbiner B, Leckband D. Direct molecular force measurements of multiple adhesive interactions between cadherin ectodomains. Proc Natl Acad Sci USA. 1999;96:11820–11824. doi: 10.1073/pnas.96.21.11820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu B, Chappuis-Flament S, Wong E, Jensen IE, Gumbiner BM, Leckband D. Functional analysis of the structural basis of homophilic cadherin adhesion. Biophys J. 2003;84:4033–4042. doi: 10.1016/S0006-3495(03)75129-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chappuis-Flament S, Wong E, Hicks LD, Kay CM, Gumbiner BM. Multiple cadherin extracellular repeats mediate homophilic binding and adhesion. J Cell Biol. 2001;154:231–243. doi: 10.1083/jcb.200103143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiraishi K, Tsuzaka K, Yoshimoto K, Kumazawa C, Nozaki K, Abe T, Tsubota K, Takeuchi T. Critical role of the fifth domain of E-cadherin for heterophilic adhesion with alpha E beta 7, but not for homophilic adhesion. J Immunol. 2005;175:1014–1021. doi: 10.4049/jimmunol.175.2.1014. [DOI] [PubMed] [Google Scholar]
- 19.Karecla PI, Green SJ, Bowden SJ, Coadwell J, Kilshaw PJ. Identification of a binding site for integrin alphaEbeta7 in the N-terminal domain of E-cadherin. Journal of Biological Chemistry. 1996;271:30909–30915. doi: 10.1074/jbc.271.48.30909. [DOI] [PubMed] [Google Scholar]
- 20.Makagiansar IT, Avery M, Hu Y, Audus KL, Siahaan TJ. Improving the selectivity of HAV-peptides in modulating E-cadherin-E-cadherin interactions in the intercellular junction of MDCK cell monolayers. Pharm Res. 2001;18:446–453. doi: 10.1023/a:1011094025008. [DOI] [PubMed] [Google Scholar]
- 21.Calcagno AM, Fostel JM, Orchekowski RP, Alston JT, Mattes WB, Siahaan TJ, Ware JA. Modulation of cell adhesion molecules in various epithelial cell lines after treatment with PP2. Mol Pharm. 2005;2:170–184. doi: 10.1021/mp0499003. [DOI] [PubMed] [Google Scholar]
- 22.Zheng K, Makagiansar IT, Wang M, Urbauer JL, Kuczera K, Siahaan TJ. Expression, purification, and structural study of the EC4 domain of E-cadherin. Protein Expr Purif. 2004;33:72–79. doi: 10.1016/j.pep.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 23.Prasad A, Housley NA, Pedigo S. Thermodynamic stability of domain 2 of epithelial cadherin. Biochemistry. 2004;43:8055–8066. doi: 10.1021/bi049693c. [DOI] [PubMed] [Google Scholar]
- 24.Wright SK, Viola RE. Evaluation of methods for the quantitation of cysteines in proteins. Anal Biochem. 1998;265:8–14. doi: 10.1006/abio.1998.2858. [DOI] [PubMed] [Google Scholar]
- 25.Tam JP, Wu C, Yang JL. Membranolytic selectivity of cystine-stabilized cyclic protegrins. Eur J Biochem. 2000;267:3289–3300. doi: 10.1046/j.1432-1327.2000.01359.x. [DOI] [PubMed] [Google Scholar]
- 26.Greenfield N, Vijayanathan V, Thomas TJ, Gallo MA, Thomas T. Increase in the stability and helical content of estrogen receptor alpha in the presence of the estrogen response element: analysis by circular dichroism spectroscopy. Biochemistry. 2001;40:6646–6652. doi: 10.1021/bi002846l. [DOI] [PubMed] [Google Scholar]
- 27.Lees JG, Miles AJ, Wien F, Wallace BA. A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics. 2006;22:1955–1962. doi: 10.1093/bioinformatics/btl327. [DOI] [PubMed] [Google Scholar]
- 28.Kueltzo LA, Middaugh CR. Structural characterization of bovine granulocyte colony stimulating factor effect of temperature and pH. J Pharm Sci. 2003;92:1793–1804. doi: 10.1002/jps.10440. [DOI] [PubMed] [Google Scholar]
- 29.Prestrelski SJ, Tedeschi N, Arakawa T, Carpenter JF. Dehydration-induced conformational transitions in proteins and their inhibition by stabilizers. Biophys J. 1993;65:661–671. doi: 10.1016/S0006-3495(93)81120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kueltzo LA, Ersoy B, Ralston JP, Middaugh CR. Derivative absorbance spectroscopy and protein phase diagrams as tools for comprehensive protein characterization: a bGCSF case study. J Pharm Sci. 2003;92:1805–1820. doi: 10.1002/jps.10439. [DOI] [PubMed] [Google Scholar]
- 31.Kueltzo LA, Osiecki J, Barker J, Picking WL, Ersoy B, Picking WD, Middaugh CR. Structure-function analysis of invasion plasmid antigen C (IpaC) from Shigella flexneri. J Biol Chem. 2003;278:2792–2798. doi: 10.1074/jbc.M208383200. [DOI] [PubMed] [Google Scholar]
- 32.Rexroad J, Wiethoff CM, Green AP, Kierstead TD, Scott MO, Middaugh CR. Structural stability of adenovirus type 5. J Pharm Sci. 2003;92:665–678. doi: 10.1002/jps.10340. [DOI] [PubMed] [Google Scholar]
- 33.Derrick TS, Kashi RS, Durrani M, Jhingan A, Middaugh CR. Effect of metal cations on the conformation and inactivation of recombinant human factor VIII. J Pharm Sci. 2004;93:2549–2557. doi: 10.1002/jps.20167. [DOI] [PubMed] [Google Scholar]
- 34.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 35.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 36.Verkhivker GM. Computational analysis of ligand binding dynamics at the intermolecular hot spots with the aid of simulated tempering and binding free energy calculations. J Mol Graph Model. 2004;22:335–348. doi: 10.1016/j.jmgm.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Verkhivker GM. Imprint of evolutionary conservation and protein structure variation on the binding function of protein tyrosine kinases. Bioinformatics. 2006;22:1846–1854. doi: 10.1093/bioinformatics/btl199. [DOI] [PubMed] [Google Scholar]
- 38.Verkhivker GM. In silico profiling of tyrosine kinases binding specificity and drug resistance using Monte Carlo simulations with the ensembles of protein kinase crystal structures. Biopolymers. 2007;85:333–348. doi: 10.1002/bip.20656. [DOI] [PubMed] [Google Scholar]
- 39.Verkhivker GM. Computational proteomics of biomolecular interactions in the sequence and structure space of the tyrosine kinome: Deciphering the molecular basis of the kinase inhibitors selectivity. Proteins. 2007;66:912–929. doi: 10.1002/prot.21287. [DOI] [PubMed] [Google Scholar]
- 40.Tyurin VA, Tyurina YY. Oxidative stress following traumatic brain Injury in rats: quantitation of biomarkers and detection of free radical Intermediates. J Neurochem. 2000;75:2178–2189. doi: 10.1046/j.1471-4159.2000.0752178.x. [DOI] [PubMed] [Google Scholar]
- 41.Ragone R, Colonna G, Balestrieri C, Servillo L, Irace G. Determination of tyrosine exposure in proteins by second-derivative spectroscopy. Biochemistry. 1984;23:1871–1875. doi: 10.1021/bi00303a044. [DOI] [PubMed] [Google Scholar]
- 42.Ragone R, Colonna G, Bismuto E, Irace G. Unfolding pathway of myoglobin: effect of denaturants on solvent accessibility to tyrosyl residues detected by second-derivative spectroscopy. Biochemistry. 1987;26:2130–2134. doi: 10.1021/bi00382a010. [DOI] [PubMed] [Google Scholar]
- 43.Torrent J, Alvarez-Martinez MT, Liautard J, Balny C, Lange R. The role of the 132–160 region in prion protein conformational transitions. Protein Sci. 2005;14:956–967. doi: 10.1110/ps.04989405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang M, Fabian H, Mantsch HH, Vogel HJ. Isotope-edited Fourier transform infrared spectroscopy studies of calmodulin's interaction with its target peptides. Biochemistry. 1994;33:10883–10888. doi: 10.1021/bi00202a006. [DOI] [PubMed] [Google Scholar]
- 45.Tatulian SA, Chen B, Li J, Negash S, Middaugh CR, Bigelow DJ, Squier TC. The inhibitory action of phospholamban involves stabilization of alpha-helices within the Ca-ATPase. Biochemistry. 2002;41:741–751. doi: 10.1021/bi011148d. [DOI] [PubMed] [Google Scholar]
- 46.Byler DM, Susi H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers. 1986;25:469–487. doi: 10.1002/bip.360250307. [DOI] [PubMed] [Google Scholar]
- 47.D’Amico S, Gerday C, Feller G. Dual effects of an extra disulfide bond on the activity and stability of a cold-adapted alpha-amylase. J Biol Chem. 2002;277:46110–46115. doi: 10.1074/jbc.M207253200. [DOI] [PubMed] [Google Scholar]
- 48.Klink TA, Woycechowsky KJ, Taylor KM, Raines RT. Contribution of disulfide bonds to the conformational stability and catalytic activity of ribonuclease A. Eur J Biochem. 2000;267:566–572. doi: 10.1046/j.1432-1327.2000.01037.x. [DOI] [PubMed] [Google Scholar]
- 49.Shimaoka M, Springer TA. Therapeutic antagonists and the conformational regulation of the beta2 integrins. Curr Top Med Chem. 2004;4:1485–1495. doi: 10.2174/1568026043387575. [DOI] [PubMed] [Google Scholar]
- 50.Lutz KL, Siahaan TJ. Modulation of the cellular junction protein E-cadherin in bovine brain microvessel endothelial cells by cadherin peptides. Drug Delivery. 1997;4:187–193. [Google Scholar]
- 51.Sinaga E, Jois SD, Avery M, Makagiansar IT, Tambunan US, Audus KL, Siahaan TJ. Increasing paracellular porosity by E-cadherin peptides: discovery of bulge and groove regions in the EC1-domain of E-cadherin. Pharm Res. 2002;19:1170–1179. doi: 10.1023/a:1019850226631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.