

Abstract
The past decade has seen increased application of 18-flurodeoxyglucose positron emission tomography (18FDG-PET) imaging to help diagnose and monitor disease, particularly in oncology, vasculitis and atherosclerosis. Disordered glycolytic metabolism and infiltration of plexiform lesions by inflammatory cells has been described in idiopathic pulmonary arterial hypertension (IPAH). We hypothesized that increased 18FDG uptake may be present in the lungs, large pulmonary arteries and right ventricle of patients with pulmonary hypertension, and that this uptake would be related to markers of immune activation. We imaged the thorax of 14 patients with pulmonary hypertension (idiopathic and chronic thromboembolic) and six controls by 18FDG-PET/computed tomography (CT) and measured uptake in the lung parenchyma, large pulmonary arteries and right ventricle. 18FDG uptake in the lungs and pulmonary arteries was normalized for venous blood activity to give a target-to-background ratio (TBR). Blood was contemporaneously drawn for high-sensitivity CRP - C-reactive protein (CRP) (hsCRP), N-Terminal Probrain natriuteric peptide (NT-ProBNP) and other inflammatory cytokines. IPAH patients had significantly higher lung parenchymal TBR (P=0.034) and right ventricle FDG uptake (P=0.007) than controls. Uptake in the main pulmonary arteries was similar in chronic thromboembolic pulmonary hypertension, IPAH and controls. There were no correlations between 18FDG uptake and hsCRP or inflammatory cytokine levels. NT-ProBNP correlated with RV uptake in those with pulmonary hypertension (r=0.55, P=0.04). In this pilot study, we found increased 18FDG uptake in the lung parenchyma and right ventricle of subjects with IPAH. The lung uptake might be useful as a surrogate marker of increased cellular metabolism and immune activation as underlying mechanisms in this disease. Further evaluation of the impact of targeted therapies in treatment-naïve patients and the significance of right ventricular uptake is suggested.
Keywords: inflammation, pulmonary artery, pulmonary arterial hypertension, positron emission tomography, right ventricle
INTRODUCTION
Idiopathic pulmonary arterial hypertension (IPAH) is a condition associated with endothelial dysfunction, small pulmonary artery smooth muscle cell and fibroblast proliferation, and in situ thrombosis. These changes result in an elevated pulmonary vascular resistance (PVR), which may progress to failure of the right ventricle and death. A large contribution to the PVR comes from small precapillary “resistance” arteries.[1] The etiology of IPAH is unclear, with abnormalities found in many cellular pathways[2] as well as heritable cases being caused by mutations in the bone morphogenetic protein receptor type II (BMPR- II) gene.[3]
Pulmonary hypertension may be associated with other conditions, such as chronic thromboemboli in the pulmonary arteries (chronic thromboembolic pulmonary hypertension (CTEPH)).[4]
There is mounting evidence that immune dysregulation and possibly inflammation play a role in IPAH. Patients with IPAH have elevated serum levels of the proinflammatory cytokines interleukin (IL)-1 and IL-6[5] (5) and C-reactive protein[6] in comparison with healthy controls. Recently, elevated levels of ILs 6, 8, 10 and 12p70 have been shown to predict survival in patients with pulmonary arterial hypertension.[7] The source of production of these cytokines in pulmonary hypertension is largely unknown, but may be the lungs.[8] Infiltrates of T-cells, B-cells and macrophages are seen in the plexiform lesions often found in IPAH.[9] The role of immune dysregulation in CTEPH is less clear. Raised serum levels of the proinflammatory cytokines IL-10 and tumor necrosis factor alfa (TNFα) have been described in CTEPH.[10]
The past decade has seen the development of imaging techniques able to detect and track changes in vascular inflammation.[11,12] There has been particular interest in the use of 18-flurodeoxyglucose positron emission tomography (18FDG-PET), an imaging technique that is sensitive to cellular processes that metabolize glucose. 18FDG is taken up into cells via glucose transporter proteins, and phosphorylated to 18FDG-6-phosphate, which cannot be metabolized further along the glycolytic pathway and accumulates within cells in proportion to their glucose uptake (and hence metabolic activity). While commonly used in oncology to identify primary and metastatic tumor cells, several other cell types may also demonstrate high levels of 18FDG accumulation, including activated leucocytes,[13] lymphocytes[14] and fibroblasts.[15] Increased 18FDG uptake has been demonstrated in animal models of inflammation[16] as well as a range of human inflammatory conditions including abscesses,[17] tuberculosis,[18] sarcoidosis[19] and large vessel vasculitis.[20] 18FDG uptake in atherosclerosis of the aorta and carotid and vertebral arteries at PET imaging correlates with the presence of vascular symptoms,[21] risk factor load[22] and inflammatory biomarker levels.[23] Such imaging might have a role in monitoring the response of atherosclerosis to therapy.[24]
Several human cancer cell lines have high mitochondrial membrane potentials and low expression of the Kv1.5 potassium channel, which may lead to an apoptosis-resistant phenotype and a switch to glycolytic metabolism;[25] these energy-handling abnormalities have also been described in IPAH.[26,27] Increased glycolytic metabolism is present in endothelial cells derived from IPAH-transplant explants;[28] this study also described higher 18FDG lung uptake on PET scan than normal control patients, with the uptake ascribed to increased vascular endothelial glycolytic activity.
18FDG-PET imaging of the heart in PAH shows excessive 18FDG uptake within the wall of the pressure-overloaded right ventricle, with the degree of uptake correlating with pulmonary hemodynamics.[29]
Given the possible pathological role of inflammation in idiopathic PAH and CTEPH, the presence of deranged endothelial cell bioenergetics and the recent development of techniques allowing the imaging of vascular inflammation, we hypothesized that increased 18FDG uptake in the large “conduit” pulmonary arteries, lung parenchyma and right ventricle would be present in subjects with pulmonary hypertension compared with controls. We also determined whether 18FDG uptake correlated with systemic inflammation as evaluated by levels of relevant inflammatory cytokines.
MATERIALS AND METHODS
Study population
Outpatients attending our national pulmonary hypertension referral service at Papworth Hospital, Cambridge, UK, with a diagnosis of IPAH or CTEPH with a distal distribution of disease were recruited. All patients met recognized diagnostic criteria.[30] Exclusion criteria were a diagnosis of connective tissue, diabetes, intercurrent infection or inflammatory lung disease (e.g., bronchiectasis, emphysema). All patients gave written informed consent and the study was approved by the Cambridgeshire Regional Ethics Committee.
The comparator group was comprised of a group of healthy ex-smokers (median smoking history of 10 pack years) without chronic obstructive pulmonary disease or pulmonary hypertension from a recent imaging study.[31] Sample sizes were based on previous vascular PET/computed tomography (CT) studies and powered to show differences in uptake between vascular beds.[31,32]
18FDG-PET/CT
Imaging was performed using a PET/CT system (GE Discovery 690). All subjects fasted for 6 h before imaging; 222-239 MBq (average 232 MBq) of 18FDG was injected intravenously after which subjects rested in a quiet room. PET/CT acquisition began 85–96 min after injection. PET acquisition was for 20 min over the mediastinum to include the pulmonary arteries and right ventricle. Low-dose CT of the thorax was performed immediately prior to the PET to allow anatomic colocalization and attenuation correction of PET data. No CT contrast was administered. Attenuation-corrected PET images were iteratively reconstructed using “Time of Flight” information.[33]
PET/CT image analysis was performed using Osirix version 3.7.1 (Osirix Imaging Software, Geneva, Switzerland). The ascending aorta, main pulmonary arteries and pulmonary trunk, lung parenchyma and right and left ventricles were analyzed, described in detail below. Regions of interest were placed by one reader blinded to the diagnosis (GH) and reviewed independently by two readers prior to the final analysis.
Mean and maximum body weight-corrected standardized uptake values (SUV) were calculated from the pixel activity within each region of interest (ROI). Where appropriate, SUVs were normalized to blood 18FDG activity by dividing the average SUV by the uptake of superior vena cava (SVC) blood to give a blood-corrected SUV, known as a target-to-background ratio (TBR), a validated technique for vascular PET imaging.[34]
Ascending aorta
The ascending aorta was identified from anatomic landmarks on CT. As previously described,[31] the maximum SUV was measured from a circular ROI placed around the lumen on each transaxial slice (approximately 10 per subject), from which TBR was derived.
Large pulmonary arteries
We were unable to find published methods for determining 18FDG uptake in the pulmonary arteries on PET/CT. We therefore adapted methods validated in atherosclerosis imaging studies.[23,24] The pulmonary trunk and right and left main pulmonary arteries were included in the analysis and identified from anatomic CT landmarks. The region bordered inferiorly by the right ventricular outflow tract to the superior limit of the pulmonary trunk was included, and ROIs drawn around the pulmonary trunk to the distal limit of the main pulmonary arteries on each transaxial slice. An average SUV max and TBR were derived.
Lung parenchyma
ROIs of radius 1 cm were drawn in peripheral lung locations, away from obvious pulmonary vessels in the anterior, middle and posterior sections of each lung (to minimize any effects of gravitational pooling of the blood pool and lung heterogeneity) on a transaxial slice taken at the level of the carina. ROI placement was validated by a thoracic radiologist blinded to the patient group (NS). SUV max was measured from which the TBR was derived as per published methods.[35,36]
Right ventricle
As previously described,[29] we placed a number (five to 14) of circular ROIs on each of the RV and LV free walls of a transaxial image to obtain an averaged mean SUV. We compared 18FDG uptake in the right and left ventricles using the ratio method of Kluge.[37]
Biomarkers
Immediately prior to PET/CT imaging, venous blood was drawn for high-sensitivity C-reactive protein (hsCRP), N-Terminal Probrain natriuteric peptide (NT-ProBNP) and inflammatory cytokine analyses. NT-ProBNP was analyzed using a Siemens Dimension Clinical chemistry system and hsCRP using a Dade Behring immunonephelometry assay. Blood for inflammatory cytokines was centrifuged for 10 min at 3,000 revolutions per minute, and the serum supernatant removed and stored at -85°C for analysis.
Serum levels of inflammatory cytokines (IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, IL-13, interferon [IFN]-γ and TNFα) were measured using a high-sensitivity cytokine multiplex assay and a multiplex analyzer following the manufacturer's protocols.
Statistics
One-way ANOVA was used for comparisons among the three groups. When a P-value under 0.05 was found, Tukey's post test comparison was performed between individual groups. For comparison between two groups, an unpaired Student's t test was used if the data were normally distributed; otherwise, the Mann-Whitney test was used. Associations between variables were tested using Pearson's correlation for normally distributed values and Spearman's rank correlation for nonnormally distributed data. The intraclass correlation coefficient (ICC) was used to ascertain intrareader reproducibility. Analysis was performed using GraphPad Prism software (Version 5.02). Statistical significance was set at the 5% level.
Intrareader reproducibility
Six scans were reread after 3 months to estimate the intrareader variability. There was no significant difference between results, and the TBR values were highly reproducible (intraclass correlation coefficient for aorta: 0.95, large pulmonary arteries: 0.99).
RESULTS
Demographic, imaging and biochemical data for all three groups are summarized in (Table 1). Eight IPAH patients, six CTEPH patients and six comparator subjects were enrolled: Seven/eight idiopathic PAH and five/six distal CTEPH patients were receiving PAH-targeted therapy (phosphodiesterase-5 inhibitor, endothelin antagonist or prostanoid). There was no difference in the body mass index or 18FDG circulation time among the three groups. No IPAH patients carried a BMPR2 mutation.
Table 1.
Demographics, blood and positron emission tomography results for the three groups
PET and fused PET/CT images of the right ventricle and the pulmonary trunk of an IPAH subject are shown in (Fig. 1).
Figure 1.

Positron emission tomography (PET) and fused PET/computed tomography of the right ventricle and pulmonary trunk of an idiopathic pulmonary arterial hypertension subject.
Large vessel uptake
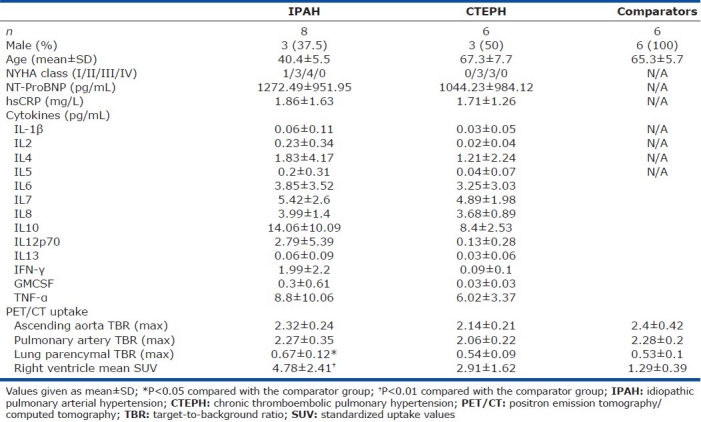
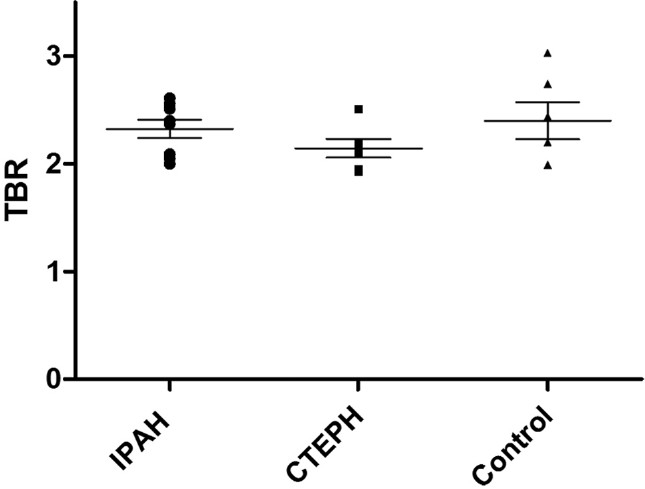
There were no significant differences in 18FDG uptake (expressed as TBR) in the ascending aorta (Fig. 2) or in the main pulmonary arteries between the IPAH, CTEPH and comparator groups (Fig. 3).
Figure 2.
Ascending aorta target-to-background ratio. No significant difference is present among the three groups.
Figure 3.
Large pulmonary artery target-to-background ratio. No significant difference is present among the three groups.
Lung parenchymal uptake
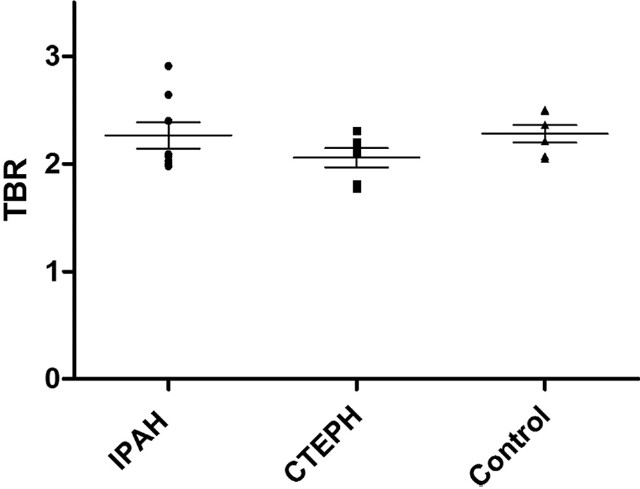
Idiopathic PAH patients had a significantly higher lung parenchymal TBR max than the comparator group (P=0.034). There were no overall differences between the CTEPH group and either the comparator or the idiopathic PAH group (Fig. 4).
Figure 4.
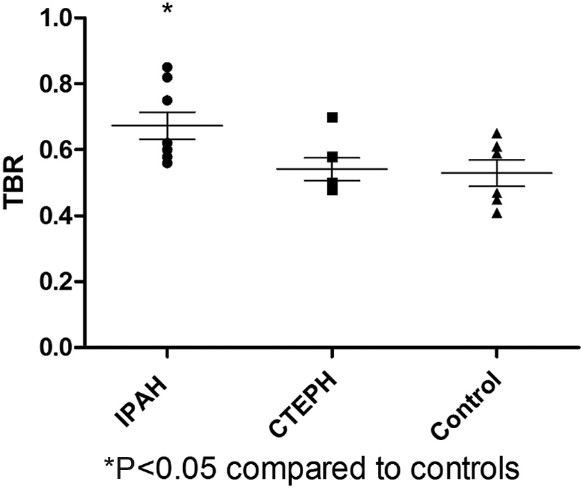
Lung parenchymal max target-to-background ratio. Idiopathic pulmonary arterial hypertension lung had a significantly higher uptake compared with controls.
LV uptake, RV uptake and RV/LV ratio
There was no difference in LV uptake among the three groups. Both RV uptake (Fig. 5) and the RV/LV ratio were significantly higher in idiopathic PAH patients than in the comparator group (P=0.007 for RV, P=0.017 for RV/LV). There were no differences between the CTEPH group and the other two groups.
Figure 5.
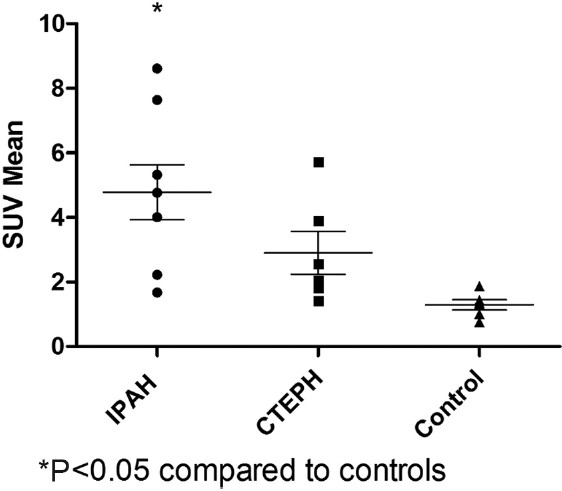
Right ventricle standardized uptake values. Idiopathic pulmonary arterial hypertension patients had significantly higher right ventricular uptake than controls.
SVC venous blood uptake
There was no significant difference between venous blood uptake among the three groups.
Correlation between uptake in RV, pulmonary arteries and lung parenchyma
The RV uptake did not correlate with lung parenchymal uptake or pulmonary artery uptake in any group. Pulmonary artery uptake correlated with lung parenchymal uptake for both the IPAH group (r=0.81, P=0.0153) and the combined IPAH and CTEPH groups (r=0.71, P=0.0039) and also in the comparator group(r=0.91, P=0.012).
Correlation with inflammatory cytokines and NT-ProBNP
Cytokine measurements were not available for the comparator group. The levels of hsCRP, cytokines or NT-ProBNP did not differ significantly between the IPAH and the CTEPH groups. There was an association (r=0.55, P=0.04) between NT-ProBNP and RV uptake for the combined IPAH and CTEPH groups. NT-ProBNP levels did not correlate with lung parenchymal or large vessel (aorta or pulmonary) uptake in either group. There was no correlation between large pulmonary artery TBR, lung parenchymal TBR or RV uptake and hsCRP or any of the inflammatory cytokines in either group.
DISCUSSION
To our knowledge, this is the first study to attempt to measure endothelial metabolism, inflammatory cell activity or vascular remodeling using the surrogate of 18FDG uptake in the pulmonary arteries in pulmonary hypertension. In this pilot prospective imaging study, we demonstrated that lung 18FDG uptake, expressed as TBR, is higher in patients with idiopathic PAH than the comparator or CTEPH group. RV uptake was also higher in our IPAH patients than comparators. Correlations between lung parenchymal uptake and pulmonary artery uptake were present. There was no difference in the uptake in the large pulmonary arteries or aorta among the three groups. The absence of significantly increased uptake in the large pulmonary arteries suggests that these processes are confined to the distal vessels in this disease.
Our results are concordant with previously published studies. Increased lung parenchymal 18FDG uptake in IPAH was reported in a smaller study (n=4).[28] A positive correlation between adverse pulmonary hemodynamics and RV 18FDG uptake has been described.[29] The RV uptake in our pulmonary hypertension group (IPAH and CTEPH) correlated with NT-ProBNP. We did not examine correlations between right heart catheter or echocardiographic indices and 18FDG uptake as there was a time interval of a few months between the hemodynamic investigations and the PET/CT scan for most patients. We found no correlation between 18FDG uptake in any region and inflammatory cytokine or hsCRP levels. As RV uptake in IPAH correlates with hemodynamics,[29] and little or no correlation exists between inflammatory cytokines or hsCRP and hemodynamics,[5,6,7] a correlation between RV uptake and inflammatory cytokines may not be expected.
The reasons for the increased lung parenchymal TBR in IPAH are unclear. Increased glycolytic metabolism of pulmonary artery endothelial cells has been proposed.[28] Rapidly dividing cells, including leucocytes[13] and fibroblasts,[15] have also increased glucose uptake and, therefore, lung TBR may be a surrogate marker of pulmonary vascular remodeling. Inflammatory cells in plexiform lesions may also represent a source of 18FDG uptake. The correlation between lung parenchymal TBR and pulmonary artery TBR may be due to the same underlying changes in glycolytic metabolism, or those patients with a more severe small resistance pulmonary artery vasculopathy having more inflammatory or fibroblast cells present in the larger conduit pulmonary arteries. The correlation was also seen in the comparator group, although numbers were small.
The absence of significantly increased uptake in the large pulmonary arteries is also of interest. While some earlier reports suggest the presence of atherosclerotic-likelesions in the pulmonary arteries,[38] these reports predate the modern treatment era of pulmonary hypertension. The majority of our IPAH patients were clinically stable patients under long-term follow-up at our center. Therefore, treatment-naïve incident patients or unstable patients were underrepresented. Markers of inflammation (hsCRP and inflammatory cytokines) in our IPAH pulmonary hypertension cohort were lower than those in the published literature. Quarck et al.[6] described a mean hsCRP of 4.4 mg/L (confidence interval 3.5–5.4) in treatment-naïve idiopathic PAH patients, which was significantly higher than their control group who had a mean hsCRP of 2.3 mg/L (confidence interval 1.9–2.7), whereas our idiopathic PAH cohort had a mean hsCRP of only 1.86. Our cytokine values were also lower than that of the study of Soon et al.[7] This data suggest that our pulmonary hypertensive patients had well-controlled stable disease. Even though no differences in large pulmonary artery uptake were found between the IPAH group and the comparator group, the IPAH group had a few “outliers” that could be representative of a different IPAH phenotype. The two patients with the highest large pulmonary artery TBR values included a 33-year-old male with rapidly progressive IPAH listed for lung transplantation and a 45-year-old female with relatively stable disease. They both also had hsCRP values above the mean for the IPAH group.
There were some limitations to this study. The number of patients was small, although we did show significant differences between the groups. Lack of simultaneous hemodynamic measurements precluded direct correlation with 18FDG uptake, although NT-ProBNP levels were measured. Our imaging protocol was designed for maximizing uptake in vascular structures, not the right ventricle; therefore, caution is advised in the interpretation of the RV uptake results. A dedicated PET/CT study of the right ventricle would require ECG gating and clamping of glucose levels.
Similar to 18FDG PET, there are other vascular imaging techniques that may have a role in answering the questions raised in this study. Animal models suggest that macrophage-targeted CT contrast agents may measure inflammation in atherosclerosis.[39] Ultra-small superparamagnetic iron oxide particles accumulate in macrophages in carotid artery plaques, which then can be quantified using high-resolution magnetic resonance imaging.[40] It would be of interest to apply these novel methods of disease assessment in pulmonary arterial hypertension, particularly for assessing the impact of targeted therapy on treatment-naïve patients and identification of novel drugs at early stages of development. Further studies may also clarify the significance of RV 18FDG uptake in pulmonary hypertension of different etiologies with measurement of contemporaneous hemodynamics.
CONCLUSION
In this pilot study, we demonstrated increased 18FDG uptake in the lungs of patients with IPAH, and higher RV uptake in pulmonary hypertensive subjects compared with controls. Increased lung uptake may be due to changes in glycolytic metabolism in endothelial cells in IPAH or an increased amount of cells involved in inflammation. Further imaging studies in treatment-naïve patients, to measure the impact of targeted therapies on the lung, 18FDG signal and dedicated imaging of the right ventricle uptake may provide validation of this approach.
ACKNOWLEDGMENTS
This research was partly supported by the NIHR Cambridge Biomedical Research Centre. M. Southwood holds an NIHR Healthcare Scientist Fellowship. We are grateful to Addenbrooke's radiologist HK Cheow for governance reading of PET/CT images.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared.
REFERENCES
- 1.Brody JS, Stemmler EJ, DuBois AB. Longitudinal distribution of vascular resistance in the pulmonary arteries, capillaries and veins. J Clin Invest. 1968;47:783–99. doi: 10.1172/JCI105773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng Z, Morse JH, Sleger SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–44. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoeper MM, Barberà JA, Channick RN, Hassoun PM, Lang IM, Manes A, et al. Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J Am Coll Cardiol. 2009;54:S85–96. doi: 10.1016/j.jacc.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151:1628–31. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 6.Quarck R, Nawrot T, Mayns B, Delcroix M. C-reactive protein: A new predictor of adverse outcome in pulmonary arterial hypertension. J Am Coll Cardiol. 2009;53:1211–8. doi: 10.1016/j.jacc.2008.12.038. [DOI] [PubMed] [Google Scholar]
- 7.Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–7. doi: 10.1161/CIRCULATIONAHA.109.933762. [DOI] [PubMed] [Google Scholar]
- 8.Selimovic N, Bergh CH, Andersson B, Sakiniene E, Carlsten H, Rundqvist B. Growth factors and interleukin-6 across the lung circulation in pulmonary hypertension. Eur Respir J. 2009;34:662–8. doi: 10.1183/09031936.00174908. [DOI] [PubMed] [Google Scholar]
- 9.Tuder RM, Voelkel NF. Pulmonary hypertension and inflammation. J Lab Clin Med. 1998;132:16–24. doi: 10.1016/s0022-2143(98)90020-8. [DOI] [PubMed] [Google Scholar]
- 10.von Haehling S, von Bardeleben RS, Kramm T, Thiermann Y, Niethammer M, Doehner W, et al. Inflammation in right ventricular dysfunction due to thromboembolic pulmonary hypertension. Int J Cardiol. 2010;144:206–11. doi: 10.1016/j.ijcard.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 11.Rudd JH, Narula J, Strauss HW, Virmani R, Machac J, Kilmas M, et al. Imaging atherosclerotic plaque inflammation by fluorodeoxyglucose with positron emission tomography: Ready for prime time? J Am Coll Cardiol. 2010;55:2527–35. doi: 10.1016/j.jacc.2009.12.061. [DOI] [PubMed] [Google Scholar]
- 12.Owen DR, Lindsay AC, Choudhury RP, Fayad ZA. Imaging of atherosclerosis. Annu Rev Med. 2011;62:25–40. doi: 10.1146/annurev-med-041709-133809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fantone JC, Ward PA. Role of oxygen-derived free radicals and metabolites in leukocyte-dependent inflammatory reactions. Am J Pathol. 1982;107:395–418. [PMC free article] [PubMed] [Google Scholar]
- 14.Wang T, Marquardt C, Foker J. Aerobic glycolysis during lymphocyte proliferation. Nature. 1976;261:702–5. doi: 10.1038/261702a0. [DOI] [PubMed] [Google Scholar]
- 15.Munyon WH, Merchant DJ. The relation between glucose utilization, lactic acid production and utilization and the growth cycle of L strain fibroblasts. Exp Cell Res. 1959;17:490–8. doi: 10.1016/0014-4827(59)90069-2. [DOI] [PubMed] [Google Scholar]
- 16.Yamada S, Kubota K, Kubota R, Ido T, Tamahashi N. High accumulation of fluorine-18 fluorodeoxyglucose in turpentine-induced inflammatory tissue. J Nucl Med. 1995;36:1301–6. [PubMed] [Google Scholar]
- 17.Tahara T, Ichiya Y, Kuwabara Y, Otsuka M, Miyake Y, Gunasekera R, et al. High [18F]-fluorodeoxyglucose uptake in abdominal abscesses: A PET study. J Comput Assist Tomogr. 1989;13:829–31. doi: 10.1097/00004728-198909000-00014. [DOI] [PubMed] [Google Scholar]
- 18.Demura Y, Tsuchida T, Uesaka D, Umeda Y, Morikawa M, Ameshima S, et al. Usefulness of 18F-fluorodeoxyglucose positron emission tomography for diagnosing disease activity and monitoring therapeutic response in patients with pulmonary mycobacteriosis. Eur J Nucl Med Mol Imaging. 2009;36:362–9. doi: 10.1007/s00259-008-1009-5. [DOI] [PubMed] [Google Scholar]
- 19.Keijsers RG, Grutters JC, van Velzen-Blad H, ven den Bosch JM, Oyen WJ, Verzijibergen FJ. (18)F-FDG PET patterns and BAL cell profiles in pulmonary sarcoidosis. Eur J Nucl Med Mol Imaging. 2010;37:1181–8. doi: 10.1007/s00259-009-1376-6. [DOI] [PubMed] [Google Scholar]
- 20.Pipitone N, Versari A, Salvarani C. Role of imaging studies in the diagnosis and follow up of large vessel vasculitis: An update. Rheumatology (Oxford) 2008;47:403–8. doi: 10.1093/rheumatology/kem379. [DOI] [PubMed] [Google Scholar]
- 21.Rudd JH, Warburton EA, Fryer TD, Jones HA, Clark JC, Antoun N, et al. Imaging atherosclerotic plaque inflammation with [18F]-fluorodeoxyglucose positron emission tomography. Circulation. 2002;105:2708–11. doi: 10.1161/01.cir.0000020548.60110.76. [DOI] [PubMed] [Google Scholar]
- 22.Tahara N, Kai H, Yamagishi S, Mizogucki M, Nakaura H, Ishibashi M, et al. Vascular inflammation evaluated by [18F]-fluorodeoxyglucose positron emission tomography is associated with the metabolic syndrome. Am Coll Cardiol. 2007;49:1533–9. doi: 10.1016/j.jacc.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 23.Rudd JH, Myers KS, Bansilal S, Machac J, Woodward M, Fuster V, et al. Relationships among regional arterial inflammation, calcification, risk factors, and biomarkers: A prospective fluorodeoxygluclose positron-emission tomography/computed tomography imaging study. Circ Cardiovasc Imaging. 2009;2:107–15. doi: 10.1161/CIRCIMAGING.108.811752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tahara N, Kai H, Ishibashi M, Nakaura H, Kaida H, Baba K, et al. Simvastatin attenuates plaque inflammation: Evaluation by fluorodeoxyglucose positron emission tomography. J Am Coll Cardiol. 2006;48:1825–31. doi: 10.1016/j.jacc.2006.03.069. [DOI] [PubMed] [Google Scholar]
- 25.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 26.Yuan XJ, Wang J, Juhaszova M, Gaine SP, Rubin LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet. 1998;351:726–7. doi: 10.1016/S0140-6736(05)78495-6. [DOI] [PubMed] [Google Scholar]
- 27.McMurthy MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, et al. Gene therapy targeting surviving selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. Clin Invest. 2005;115:1479–91. doi: 10.1172/JCI23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. PNAS. 2007;104:1342–437. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, et al. Increased [18F]Fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol. 2005;45:1849–55. doi: 10.1016/j.jacc.2005.02.065. [DOI] [PubMed] [Google Scholar]
- 30.National Pulmonary Hypertension Centers of UK and Ireland, Consensus statement on the management of pulmonary hypertension in clinical practice in the UK and Ireland. Thorax. 2008;63(Suppl 2):ii1–41. doi: 10.1136/thx.2007.090480. [DOI] [PubMed] [Google Scholar]
- 31.Coulson JM, Rudd JH, Duckers JM, Rees JI, Shale DJ, Bolton CE, et al. Excessive aortic inflammation in chronic obstructive pulmonary disease: An 18F-FDG PET pilot study. J Nucl Med. 2010;51:1357–60. doi: 10.2967/jnumed.110.075903. [DOI] [PubMed] [Google Scholar]
- 32.Rudd JH, Myers KS, Bansilal S, Machac J, Rafique A, Farkouh M, et al. 18Fluorodeoxyglucose positron emission tomography imaging of atherosclerotic plaque inflammatorion is highly reproducible:Implications for atherosclerosis therapy trials. J Am Coll Cardiol. 2007;50:892–6. doi: 10.1016/j.jacc.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 33.Conti M. State of the art and challenges of time-of-flight PET. Phys Med. 2009;25:1–11. doi: 10.1016/j.ejmp.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Tawakol A, Migrino RQ, Bashian GG, Bedri S, Vermylen D, Cury RC, et al. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a non-invasive measure of carotid plaque inflammation in patients. J Am Coll Cardiol. 2006;48:1818–24. doi: 10.1016/j.jacc.2006.05.076. [DOI] [PubMed] [Google Scholar]
- 35.Chen DL, Mintun MA, Schuster DP. Comparison of methods to quantitate 18F-FDG uptake with PET during experimental acute lung injury. J Nucl Med. 2004;45:1583–90. [PubMed] [Google Scholar]
- 36.Groves AM, Win T, Screaton NJ, Berovic M, Endozo R, Booth H, et al. Idiopathic pulmonary fibrosis and diffuse parenchymal lung disease:implications from initial experience with 18F-FDG PET/CT. J Nucl Med. 2009;50:538–45. doi: 10.2967/jnumed.108.057901. [DOI] [PubMed] [Google Scholar]
- 37.Kluge R, Barthel H, Pankau H, Seese A, Schauer J, Wirtz H, et al. Different mechanisms for changes in glucose uptake of the right and left ventricular myocardium in pulmonary hypertension. J Nucl Med. 2005;46:25–31. [PubMed] [Google Scholar]
- 38.Moore GW, Smith RL, Hutchins GM. Pulmonary artery atherosclerosis: Correlation with systemic atherosclerosis and hypertensive pulmonary vascular disease. Arch Pathol Lab Med. 1982;106:378–80. [PubMed] [Google Scholar]
- 39.Hyafil F, Cornily J, Rudd JH, Machac J, Feldman LJ, Fayad ZA. Quantification of inflammation within rabbit atherosclerotic plaques using the macrophage-specific CT contrast agent N1177: A comparison with 18F-FDG PET/CT and histology. J Nucl Med. 2009;50:959–65. doi: 10.2967/jnumed.108.060749. [DOI] [PubMed] [Google Scholar]
- 40.Kooi ME, Cappendijk VC, Cleutjens KB, Kessels AG, Kitslaar PJ, Borgers M, et al. Accumulation of ultrasmall superparamagnetic particles of iron oxide in human atherosclerotic plaques can be detected by in vivo magnetic resonance imaging. Circulation. 2003;107:2453–8. doi: 10.1161/01.CIR.0000068315.98705.CC. [DOI] [PubMed] [Google Scholar]