

Abstract
High-altitude pulmonary hypertension (HAPH) is a consequence of chronic alveolar hypoxia, leading to hypoxic vasoconstriction and remodeling of the pulmonary circulation. Brisket disease in cattle is a naturally occurring animal model of hypoxic pulmonary hypertension. Genetically susceptible cattle develop severe pulmonary hypertension and right heart failure at altitudes >7,000 ft. No information currently exists regarding the identity of the pathways and gene(s) responsible for HAPH or influencing severity. We hypothesized that initial insights into the pathogenesis of the disease could be discovered by a strategy of (1) sequencing of functional candidates revealed by single nucleotide polymorphism (SNP) analysis and (2) gene expression profiling of affected cattle compared with altitude-matched normal controls, with gene set enrichment analysis (GSEA) and Ingenuity pathway analysis (IPA). We isolated blood from a single herd of Black Angus cattle of both genders, aged 12-18 months, by jugular vein puncture. Mean pulmonary arterial pressures were 85.6±13 mmHg STD in the 10 affected and 35.3±1.2 mmHg STD in the 10 resistant cattle, P<0.001. From peripheral blood mononuclear cells, DNA was hybridized to an Affymetrix 10K Gene Chip SNP, and RNA was used to probe an Affymetrix Bovine genome array. SNP loci were remapped using the Btau 4.0 bovine genome assembly. mRNA data was analyzed by the Partek software package to identify sets of genes with an expression that was statistically different between the two groups. GSEA and IPA were conducted on the refined expression data to identify key cellular pathways and to generate networks and conduct functional analyses of the pathways and networks. Ten SNPs were identified by allelelic association and four candidate genes were sequenced in the cohort. Neither endothelial nitric oxide synthetase, NADH dehydrogenase, TG-interacting factor-2 nor BMPR2 were different among affected and resistant cattle. A 60-gene mRNA signature was identified that differentiated affected from unaffected cattle. Forty-six genes were overexpressed in the affected and 14 genes were downregulated in the affected cattle by at least 20%. GSEA and Ingenuity analysis identified respiratory diseases, inflammatory diseases and pathways as the top diseases and disorders (P<5.14×10-14), cell development and cell signaling as the top cellular functions (P<1.20×10-08), and IL6, TREM, PPAR, NFkB cell signaling (P<8.69×10-09) as the top canonical pathways associated with this gene signature. This study provides insights into differences in RNA expression in HAPH at a molecular level, and eliminates four functional gene candidates. Further studies are needed to validate and refine these preliminary findings and to determine the role of transcribed genes in the development of HAPH.
Keywords: brisket disease, microarray analysis, hypoxia
INTRODUCTION
High mountain disease (brisket disease) is right heart failure due to hypoxic pulmonary hypertension in cattle residing at high altitudes.[1–3] Hypoxia is the most potent stimulus for pulmonary hypertension, and the hypoxia of high altitude (>7,000 ft) is a well known cause.[4–8] Some cattle (Bos taurus) possess a heritable susceptibility to severe high-altitude pulmonary hypertension (HAPH).[9–12] While most cattle thrive at high altitudes, susceptible cattle develop pulmonary hypertension that is sufficient to cause right heart failure, edema of the brisket and death. Experiments conducted by Grover and Reeves in cattle susceptible and resistant to HAPH suggest an autosomal-dominant mode of inheritance, or inheritance related to a few major genes.[11–15] No information currently exists on the identity of these genes. HAPH in cattle occurs in about 20% (10–50%) of animals brought to high altitudes (>7,000 ft) to replenish herds,[8,14–16] suggesting that the gene of interest may be a relatively common polymorphism only expressed with the stimulus of altitude hypoxia. There is no known phenotype of the gene at low altitude.
We hypothesized that the insights into pathogenesis of brisket disease may be gained through two approaches: (1) allelic association analysis of single nucleotide polymorphism (SNP) differences; and (2) a comparative gene expression analysis of hypertensive versus resistant cattle. Therefore, we analyzed DNA and RNA from 20 cattle of the same herd; 10 with severe pulmonary hypertension and 10 with normal pressures. We used an Affymetrix 10K Bovine Gene SNP array and sequenced four genes of interest. Expression data were then analyzed for genes and canonical pathways that were statistically different between the two groups. Gene set enrichment and Ingenuity map analyses were performed to further refine the data.[16–19] Collectively, the results of this study provide molecular and cellular observations in possible genes of interest in HAPH leading to brisket disease.
MATERIALS AND METHODS
Selection of study animals and blood samples
We obtained blood from a single herd of black Angus cattle of both genders, aged 12–18 months, residing at 8,500 feet, by jugular vein puncture on the same day of right heart catheterization screening for HAPH by one of us (Timothy N. Holt, hereinafter TH). The project was approved by the Vanderbilt Medical Center IACUC. Right heart catheterization is standard practice at altitude to discover hypertensive animals so that they can be shipped down to a low altitude.
DNA and RNA preparation and analysis
DNA was isolated from whole blood using QIAmp DNA mini-kit as directed by the manufacturer's instructions (Qiagen, Valencia, CA, USA) and subsequently quantified using a spectrophotometer. Total cellular RNA was prepared from blood on silica-gel membranes with on-column DNAse treatment (RNeasy Total RNA Isolation Kit; Qiagen). RNA integrity was assessed via agarose gel electrophoresis and samples showing evidence of degradation were discarded.
Affymetrix bovine 10K SNP genotyping and bioinformatic analysis
For SNP comparison, genomic DNA was extracted as described above and hybridized to Affymetrix - GeneChip Bovine Mapping 10K SNP chip according to manufacturer's instructions (Affymetrix, Santa Clara, Calif., USA). Following bovine genotype collection, we remapped all of the SNP loci represented on the bovine 10K array using the Batch query within dbSNP online (NCBI) in conjunction with the latest bovine genome assembly (Btau 4.0). In addition, SNP loci present on the array more than once (duplications) were identified, and all duplications were removed. Likewise, SNPs with multiple chromosomal assignments were also flagged and subsequently removed. At the conclusion of our bioinformatic analyses, 8,011 unique bovine SNPs with single chromosomal assignments and updated bovine genome annotation were available for genome-wide association (GWA) analysis.
Statistical approach to SNP microarray
Because the precise mode of inheritance related to HAPH in cattle has not been fully elucidated, we used several standard case–control GWA approaches in a preliminary effort to identify new potential candidate genes. All statistical tests were performed using formulae incorporated within the software program HelixTree 6.4.2 (Golden Helix Inc., Bozeman, MT, USA). In the first approach, we performed a simple allelic association test across all unique SNP loci for 10 cattle exhibiting severe HAPH (n=20 alleles) and 10 cattle defined as “resistant” at altitude (n=20 alleles). Thereafter, we also performed standard genotypic association tests in an effort to evaluate whether SNPs implicated by a simple allelic test would systematically assemble into disparate genotypic classes for cattle with severe HAPH, as compared with altitude-matched HAPH-resistant controls. Raw P-values for all tests were corrected by either full-scan permutation and/or the Bonferroni procedure implemented within HelixTree 6.4.2.
Sequencing SNP candidates
TG-interacting factor-2 (TGIF2), endothelial nitric oxide synthetase (eNOS) and NADH coding regions were sequenced using a cDNA-based approach. Briefly, we performed first strand cDNA synthesis using the Superscript First-Strand System (Invitrogen Life Technologies, Carlsbad, CA, USA) with 3 mg of total RNA and an oligo-dT primer. We then used one-tenth volume of the first strand reaction as a template for polymerase chain reaction (PCR) amplification using the Elongase Amplification System (Invitrogen Life Technologies). The forward and reverse primers corresponded to sequences located in the 5’ and 3’ untranslated mRNA sequence of BMPR2, eNOS and TGIF-2 (primer sequences and conditions available upon request). We then carried out individual PCR reactions and visualized all amplicons on a 2% agarose gel. Thereafter, we used 2 μl ExoSAP-it (USB) per 5 ul of PCR reaction (37°C for 15 min, followed by 80°C for 15 min) to eliminate residual primer and dNTPs. Sequencing was accomplished using the Big Dye Terminator cycle sequencing technology (Applied Biosystems, Carlsbad, Calif., USA) in 10 μl reactions containing 2 μl Big Dye, 2 μl Big Dye Buffer, 50 ng template and 2.5 ul of primer. Thermal cycling parameters were as follows: 95°C for 5 min; 30 cycles at 95°C for 30 s, 55°C for 10 s and 60°C for 4 min. After ethanol precipitation, the products were resuspended in 10 μl formamide and analyzed on a 3100 Genetic Analyzer (Applied Biosystems).
Microarray and gene set enrichment analysis analyses
Pulmonary tissue was not available from these animals. It is also possible that the lung tissue expression signature might be overwhelmed by adaptive cellular responses. This would then make the interpretation of the expression data particularly susceptible to false-positive and false-negatives.[16–19] Our solution to this was to avoid disease effector cells and to use peripheral blood mononuclear cells (PBMCs) in our studies. Peripheral blood mononuclear cells are genetically the same as lung tissue intraindividuals, and are genetically different among hypertensive and resistant cattle and are likely to be free of end-stage disease effects. RNA was extracted from PBMCs. After the RNA was isolated, it was used to probe the Affymetrix GeneChip Bovine Genome Array (900561). Raw data were then analyzed by the Partek software package at the Vanderbilt Functional Genomics Shared Resource to produce expression intensities for each probe set. To study pathway-level differences between groups, GSEA was conducted on the microarray gene expression data.[1] We used the Gene Ontology Biological Process (c5; from the MSigDB database) as our gene set database.
In silico functional analysis HPAH signature using ingenuity pathways analysis
We used IPA in the Ingenuity System to generate networks and conduct functional analyses of the HAPH signature. A data set containing gene identifiers was uploaded into the application. These genes, called focus genes, were used to query a global molecular network developed from information contained in the Ingenuity Pathways Knowledge Base. IPA generates models of gene interactions called networks that are presented graphically to show relationships between genes and the pathways they regulate. These networks are ranked according to a score calculated via a right-tailed Fisher's exact test. In network graph, proteins encoded by genes are represented as nodes and their relationships as edges (links). All edges are supported by references from the literature. The functional analysis of a network identified the biological functions and/or diseases that were most significant to the genes in the network. The network genes associated with biological functions and/or diseases in the Ingenuity Pathways Knowledge Base were considered for further analysis. Fisher's exact test was used to calculate a P-value, determining the probability that each biological function and/or disease assigned to a network is by chance alone. Canonical pathways analysis identified the pathways from the IPA library of canonical pathways that were most significant to the data set. The significance of the association between the data set and a canonical pathway was measured in two ways.[1] A ratio of the number of genes from the data set that map to the pathway divided by the total number of genes that map to the canonical pathway is displayed.[2] Fischer's exact test was used to calculate a P-value determining the probability that the association between the genes in the dataset and the canonical pathway is explained by chance alone.
RESULTS
Study animals
Pulmonary arterial (PA) pressures were measured by jugular vein puncture at the time of right heart catheterization. Mean PA pressures were 86.10±5 mmHg STD in the affected and 31.20±0.7 mmHg STD in the resistant cattle, P<0.0001, by two-tailed Upaired t test (Fig. 1). A mean PA pressure of 30–40 mmHg is normal for cattle residing at altitude for at least 12 months.[3,8] A mean PA pressure greater than 49 mmHg denotes a high risk for development of brisket disease.[8] Inclusion criteria were animals with a mean pulmonary artery pressure (mPAP) of 72–116 mmHg, thereby considered “affected,” and those with a mPAP of 32–37 mmHg, thereby considered “resistant” at altitude.
Figure 1.
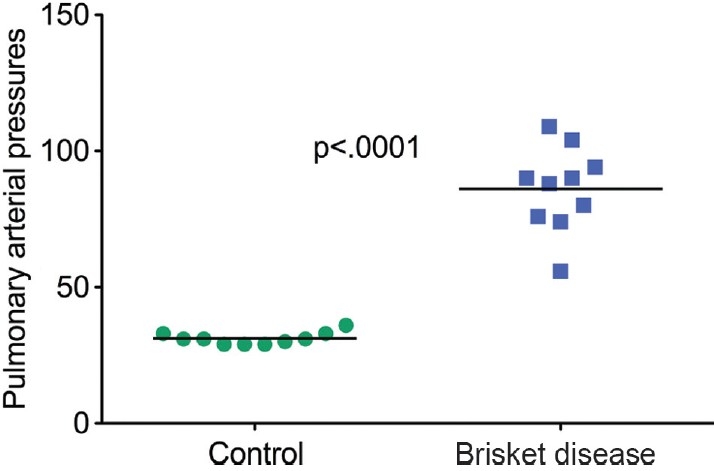
Mean pulmonary arterial pressures in the 10 cattle with pulmonary hypertension at altitude versus 10 unaffected at the same altitude.
GWA using the affymetrix geneChip bovine mapping 10K array
Among the bovine cohort investigated, 6,344 of the 8,011 unique bovine SNPs with single chromosomal assignments and updated bovine genome annotation were diallelic, thereby making them potentially informative for subsequent analyses. The most robust allelic association was detected on BTA10 (0.0000028 ≤ praw≤ 0.0000451), with weaker signals detected on additional bovine chromosomes. As expected, full-scan permutation and/or Bonferroni correction for multiple testing revealed a much weaker BTA10 signal for both approximate and exact tests (0.0476 ≤ pcorrected≤ 0.2856), respectively. Summary data for the top 10 bovine SNPs implicated by an allelic GWA test are presented in Table 1. Additionally, bovine genes proximal to each SNP are also given, with relevant human-bovine comparative data.
Table 1.
Top six bovine SNPs implicated by allelic association tests for a small bovine cohort consisting of severe HAPH cases and altitude-matched HAPH-resistant controls
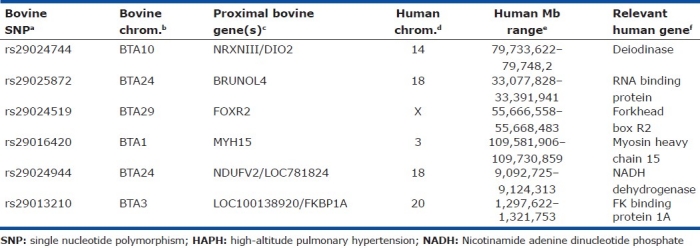
Examination of relevant genotypes determined for the top 10 SNP loci elucidated by an allelic association test revealed six corresponding loci whereby severe bovine HAPH cases were fixed for a genotype underrepresented in our altitude-matched HAPH-resistant controls (Table 1). However, genotypic association tests for all variable loci (n=6344) revealed no statistically significant associations after correction for multiple testing. Relevant bovine genotypic data and corresponding distributions are summarized in (Table 1).
Because of its role in human PAH[20–23] and its place in the TGF-b family of growth and repair receptors, the bovine BMPR2 gene was an obvious initial functional candidate for HAPH. We screened 10 severely affected and 10 unaffected cattle with a set of bovine microsatellites proximal to bovine BMPR2 to evaluate whether local genetic variation exhibited any evidence of segregation with the brisket disease phenotype (HAPH). This microsatellite analysis revealed a total of seven BMPR2 alleles, with no significant differences in the allelic distributions between the HAPH-affected and -resistant cattle, as determined by Fisher's exact test. No microsatellite allele was found uniformly among severely hypertensive cattle or resistant cattle thus providing no evidence of linkage disequilibrium between any particular allele and the HAPH phenotype. These data suggest, but do not prove, that variation within bovine BMPR2 is not responsible for modulating HAPH in cattle.
Two other high-priority functional candidates were screened by cDNA-based sequence analysis, including eNOS and TGIF2.[24,25] Low NO production has been implicated in the pathogenesis of high-altitude pulmonary edema (HAPE),[26,27] while TGIF2 is a homeobox transcriptional repressor and an important downregulator of BMPR2 and TGF-b signaling.[27,28] Sequence comparison of the eNOS and TGIF2 coding regions of HAPH-affected and -resistant cattle did not result in the identification of any statistically significant variants predictive of HAPH in our samples.
Statistical analysis of microarray data reveals altered expression in HAPH cattle
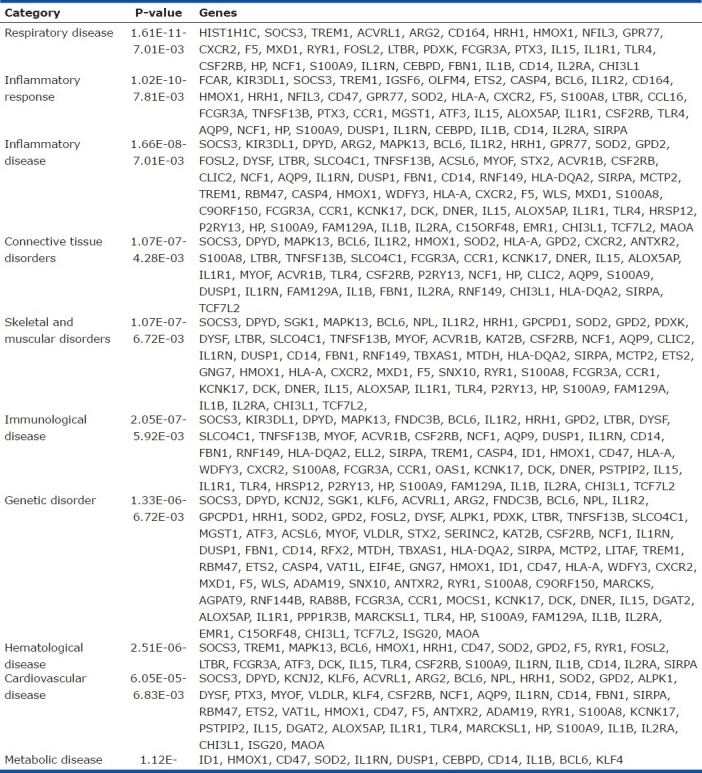
Gene expression profiles were determined by oligonucleotide microarray analyses using total RNAs from the PBMCs cells as described in the Methods section. In order to identify candidate genes involved in HAPH or protection from HAPH, we used a hierarchical clustering analysis to analyze the expression signature from the PBMCs. A combination of relative level (fold-change) of expression and statistical significance (Student's t-test) was used to distinguish these genes. This analysis identified 675 genes that were significantly upregulated and 320 genes that were downregulated (the HAPH signature) in affected animals. The top 15 candidates are listed in Table 2. Interestingly, the gene with the highest expression in affected animals is important for antigen presentation while the gene with the lowest expression is important for vessel elasticity as well as regulation of the TGF-beta pathway (Table 2). Genes related to disease process are shown in (Table 3).
Table 2.
The top 15 up and down genes expressed in peripheral blood mononuclear cells comparing highaltitude pulmonary hypertension with resistant cattle at 8,500 ft altitude
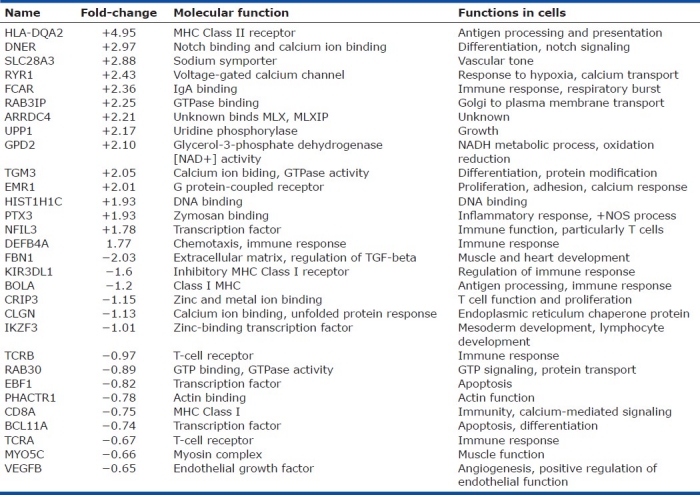
Table 3.
Top 10 disease processes associated with high-altitude pulmonary hypertension in cattle
DISCUSSION
HAPH with right heart failure was first described by Glover and Newsome in 1915 as “Brisket disease: Dropsy of high altitude.”[1] The link of altitude to hypoxic pulmonary hypertension was made after the discovery of hypoxic pulmonary vasoconstriction by von Euler and Liljestrand in 1946.[28] The exact mechanism causing acute hypoxic vasoconstriction remains elusive, although much is known about modifying influences.[29–31] Chronic hypoxia is a powerful stimulus for pulmonary hypertension, which involves not only vasoconstriction but also remodeling[32] of the pulmonary arteries. K-channel function has a central role, as does oxidant stress,[33,34] with Rho–Rho kinases involved in transduction of the hypoxic pressor response.[35–39]
To date, no major genetic variants have been identified that modify the strength of the hypoxic pressor response.[37] Some observations have been made regarding genetic associations with pulmonary responses to altitude in humans.[30–40] For example, eNOS and tyrosine kinase gene variants were overrepresented in a Japanese cohort of individuals susceptible to HAPE as compared with healthy controls at altitude.[27,40] Eddahibi and colleagues detected an association between the LL genotype of the human SLC6A4 (formerly SERT) with pulmonary artery pressure in a cohort of patients with chronic obstructive pulmonary disease as compared with controls.[41] Recent studies implicate HIF2a polymorphism in Tibetans versus Han as a possible difference in successful high-altitude adaptation.[42] Many other signaling pathways possess potential candidate genes.[43]
Relevant to the present study, the risk of acquiring brisket disease in low-altitude cattle brought to very high altitude (10,000 ft) is about 10–50%.[31,44] Notably, the prevalence of brisket disease among herds chronically residing and bred above 2100 m (c. 7,000 ft) has been reduced to about 1%, presumably because of selective loss of susceptible cattle to heart failure and to removal from herds after HAPH has been identified.[3] Previously, Reeves, Weir and Grover bred a cohort of cattle with HAPH whose mean PA pressure was 50mmHg (n=8) and a group of resistant animals whose mean PA was 29mmHg (n=11), all residing at 10,000 ft.[11,14] The 19 cattle were initially taken down to an altitude of 4,916 ft, where the cattle with HAPH recovered, and then the cattle were bred within each phenotypic class. Bulls and cows were equally represented. First-generation offspring were studied at both low and high altitude. Mean PA pressures were 27±4 (STD) at low altitude. After residing at 10,000 ft for 2 months, the offspring of cattle with HAPH (brisket disease) had a mean PA pressure of 87±7 (SE), and those from resistant stock had a mean PA of 44±3 mmHg. Second-generation breeding (breeding experiments not overtly clear) within these two cohorts of calves yielded the same pattern of susceptibility and resistance. Pressures continued to diverge over time at altitude. Proof that the stimulus to pulmonary hypertension was hypoxia and not “altitude” was subsequently demonstrated by exposures in a hypoxic chamber at low altitude, whereby similar pulmonary hypertensive responses were recorded. Arterial blood gas measurements in susceptible and resistant cattle revealed similar severity of hypoxemia, similar alveolar ventilation (defined by the arterial carbon dioxide partial pressure, PC02) and only small differences in hematocrit, not considered sufficient to cause viscosity effects. The mode of inheritance of HPAH currently remains unclear.[8] Nevertheless, what does remain clear among several studies is that HAPH in cattle is heritable, which indicates that cattle may be selected for resistance or susceptibility, and that the major gene(s) involved are likely to be discovered using large-scale genomic approaches followed by fine mapping.
Our initial screen using four functional candidate genes (selected because they are involved in human PAH) was negative and suggested that a candidate gene approach may not be efficient. We then employed a 10K bovine SNP array for a GWA aimed at unearthing new potential candidate genes. This analysis revealed six or more genes of interest proximal to bovine SNPs exhibiting the most disparate distributions between severe HAPH and resistant cattle. Of the genes located near the SNPs of interest, NADH dehydrogenase (ubiquinone) flavoprotein 2 (NDUFV), myosin heavy chain 15 (MYH15) and the myocardial signaling protein (FKBP1A) were candidates possibly involved in pulmonary hypertension. NADH dehydrogenase is no different between affected and resistant cattle, and additional studies are needed to further evaluate candidate genes. Furthermore, the recent availability of higher density bovine SNP arrays[44,45] provides a natural progression to more thorough GWA, which are likely to identify additional candidate genes due to greatly enhanced genomic coverage. The use of these new higher density bovine SNP arrays with larger cohorts of severe HAPH and resistant cattle has the potential to maximize the probability of detecting SNPs that display a nonrandom relationship with either severe HAPH or putative resistance. It is likely that the major gene(s) responsible for bovine HAPH will have identifiable human ortholog(s) that may also modify human pulmonary hypertension related to hypoxia (altitude, emphysema, hypoventilation syndromes), and possibly in other conditions that lead to pulmonary hypertension.
Biological significance of HAPH signature
To gain biological insights into resistance or susceptibility to HAPH, we analyzed the expression data using the Gene Ontology and Ingenuity Map database. Our goal was to identify the disease processes, physiological, cellular and molecular functions and the canonical pathways that are enriched in HAPH. These analyses showed that the top disease process reflected in our data is respiratory disease (Table 2); the top three physiological processes are organism survival, hematological system and immune cell trafficking; the top three cellular and molecular pathways are cellular movement, cell signaling and small molecule biochemistry; and the top canonical pathways enriched in HAPH are IL-10 signaling, RXR activation,and, interestingly, immune and endothelial cell function (data not shown but available on request).
We then used the IPA application (IPA version 8.7) to examine the biological context and interactions of our signature. The IPA network analysis identified 10 networks with high P-values (1.0×10-21) (data not shown but available upon request). The top three networks had many genes (MAP13, STAT5a/b, ERK1/2, Cyclins, VEGF, PDGF, IL1B, ID1, CREB, p38MAPK, IL15, etc.) with a documented role in endothelial and immune cell function through their effects on cellular pathways such as TGF-beta, BMP and MAPK (important in human PAH) and molecular functions such as cell death, survival, inflammatory response and cell morphology. Function annotation of all the networks showed that the top canonical functions associated with these 10 networks are BMP, TGF-beta, PPAR, IL6, endothelial cell function, NOS, TREM1, VEGF, glucocorticoid, ERK/MAPK, NF-kB and HIF1a signaling (data not shown but available upon request). Not surprisingly, many of these pathways are also known to be important in human pulmonary hypertension.
In summary, herein, we have provided the results of the first molecular interrogation of HPAH, including a low-density SNP search, sequencing of four candidate genes and analysis of expression arrays to seek clusters of genes that may be a signature of hereditary bovine HAPH. Our hope is that this initial study of HPAH will spur further discovery and investigation of the molecular mechanisms behind HPAH. Our next challenge will be to attempt whole genome sequencing on the hypothesis that a polymorphism responsible for an autosomal-dominant phenotype at altitude will emerge from the similar background genomes of the Angus herds.
Footnotes
Source of Support: Pulmonary Hypertension Association Scientific Award, Newman PI. Elsa S. Hanigan Chair, Dr. Newman. Dr. Hamid is supported by 1R01HL102020-01
Conflict of Interest: None declared.
REFERENCES
- 1.Glover GH, Newsom IE. Brisket Disease (Dropsy of high altitude) Colo Agric Exp Station. 1915;204:3–24. [Google Scholar]
- 2.Grover RF. In: Failing hearts at high altitude. In: Attitudes on Altitude. Reeves JT, Grover RF, editors. Boulder CO: University Press of Colorado; 2001. pp. 1–24. [Google Scholar]
- 3.Rhodes J. Comparative physiology of hypoxic pulmonary hypertension: Historical clues from Brisket Disease. J Appl Physiol. 2005;98:1092–100. doi: 10.1152/japplphysiol.01017.2004. [DOI] [PubMed] [Google Scholar]
- 4.Richards DW. The J. Burns Amberson lecture: The right heart and the lung. Am Rev Respir Dis. 1966;94:691–702. doi: 10.1164/arrd.1966.94.5.691. [DOI] [PubMed] [Google Scholar]
- 5.Heath D, William DR. Man at High Altitude. Vol. 892. Edinburgh and New York: Churchill Livingstone; 1981. pp. 1158–63. [Google Scholar]
- 6.Maggiorini M, Leon-Velarde F. High-altitude pulmonary hypertension: pathophysiological entity to different diseases. Eur Resp J. 2003;22:1019–25. doi: 10.1183/09031936.03.00052403. [DOI] [PubMed] [Google Scholar]
- 7.Holt TN, Callan RJ. Pulmonary arterial pressure testing for high mountain disease in cattle. Vet Clin North Am Food Anim Pract. 2007;23:575–96. doi: 10.1016/j.cvfa.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Grover RF, Reeves JT, Will DH, Blount SG., Jr Pulmonary vasoconstriction in steers at high altitude. J Appl Physiol. 1963;18:567–74. doi: 10.1152/jappl.1963.18.3.567. [DOI] [PubMed] [Google Scholar]
- 9.Will DH, Hicks JL, Card CS, Reeves JT, Alexander AF. Correlation of acute with chronic hypoxic pulmonary hypertension in cattle. J Appl Physiol. 1975;38:495–8. doi: 10.1152/jappl.1975.38.3.495. [DOI] [PubMed] [Google Scholar]
- 10.Anand IS, Harris E, Ferrari R, Pearce P, Harris P. Pulmonary haemodynamics of the yak, cattle, and cross breeds at high altitude. Thorax. 1986;41:696–700. doi: 10.1136/thx.41.9.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weir EK, Tucker A, Reeves JT, Will DH, Grover RF. The genetic factor influencing pulmonary hypertension in cattle at high altitude. Cardiovasc Res. 1974;8:745–9. doi: 10.1093/cvr/8.6.745. [DOI] [PubMed] [Google Scholar]
- 12.Shirley LK, Beckman DW, Garrick DJ. Inheritance pulmonary arterial pressure in angus cattle and its correlation with growth. J Anim Sci. 2008;86:815–9. doi: 10.2527/jas.2007-0270. [DOI] [PubMed] [Google Scholar]
- 13.Will DH, Hicks JL, Card CS, Alexander AF. Inherited susceptibility of cattle to high-altitude pulmonary hypertension. J Appl Physiol. 1975;38:491–4. doi: 10.1152/jappl.1975.38.3.491. [DOI] [PubMed] [Google Scholar]
- 14.Grover RF. Pulmonary circulation in animals and man at high altitude. Ann NY Acad Sci. 1965;27:632–9. doi: 10.1111/j.1749-6632.1965.tb49429.x. [DOI] [PubMed] [Google Scholar]
- 15.Gjermundson CK. Danger at 5,000 feet. Angus J. 2000:47–50. [Google Scholar]
- 16.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang HC, Zheng S, VanBuren V, Zhao Z. Discovering disease-specific biomarker genes for cancer diagnosis and prognosis. Technol Cancer Res Treat. 2010;9:219–30. doi: 10.1177/153303461000900301. [DOI] [PubMed] [Google Scholar]
- 18.Lamb J. The Connectivity Map: A new tool for biomedical research. Nat Rev Cancer. 2007;7:54–60. doi: 10.1038/nrc2044. [DOI] [PubMed] [Google Scholar]
- 19.Hollander M., D.A.W . Wiley: 1999. Nonparametric Statistical Methods. [Google Scholar]
- 20.Lane KB, Machado RD, Pauciulo MW, Phillips JA, Newman JH, Loyd JE, et al. Mutations in BMPR2 encoding a TGF-beta receptor cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 21.Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, et al. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol. 2003;162:1089–98. doi: 10.1083/jcb.200212060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman JH, Wheeler L, Lane KB, Loyd E, Gaddipati R, Phillips JA III, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med. 2001;345:319–24. doi: 10.1056/NEJM200108023450502. [DOI] [PubMed] [Google Scholar]
- 23.Yuan JX, Rubin LS. Pathogenesis of pulmonary arterial hypertension: The need for multiple hits. Circ. 2005;111:534–8. doi: 10.1161/01.CIR.0000156326.48823.55. [DOI] [PubMed] [Google Scholar]
- 24.Melhuish TA, Gallo CM, Wotton D. TGIF2 interacts with histone deacetylase 1 and represses transcription. J Biol Chem. 2001;276:32109–14. doi: 10.1074/jbc.M103377200. [DOI] [PubMed] [Google Scholar]
- 25.Imato I, Pinkhaskham A, Watanabe T, Saito-Ohra F, Solda E, Inazawa J. Amplifcation and overexpression of TGIF2, a novel homeobox gene of the TALE superclass in ovarian cells. Biochem Biophys Res Commun. 2000;276:264–70. doi: 10.1006/bbrc.2000.3449. [DOI] [PubMed] [Google Scholar]
- 26.Humbert MJ. Update on Pulmonary Arterial Hypertension 2007. Am J Resp Crit Care Med. 2008;117:574–9. doi: 10.1164/rccm.200801-029UP. [DOI] [PubMed] [Google Scholar]
- 27.Droma Y, Hanaoka M, Ota M, Katsuyama Y, Koizumi T, Fujimoto K, et al. Positive association of eNOS gene polymorphisms with high-altitude pulmonary edema. Circ. 2002;106:826–30. doi: 10.1161/01.cir.0000024409.30143.70. [DOI] [PubMed] [Google Scholar]
- 28.von Euler US, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand. 1946;12:301–320. [Google Scholar]
- 29.Moudgil R, Michelakis ED, Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol. 2005;98:390–403. doi: 10.1152/japplphysiol.00733.2004. [DOI] [PubMed] [Google Scholar]
- 30.Sylvester JT. Hypoxic pulmonary vasoconstriction: A radical view. Circ Res. 2001;88:1228–9. doi: 10.1161/hh1201.093167. [DOI] [PubMed] [Google Scholar]
- 31.Weissmann N, Grimminger F, Olschewski A, Seeger W. Hypoxic pulmonary vasoconstriction: A multifactorial response? Am J Physiol Lung Cell Mol Physiol. 2001;281:L314–7. doi: 10.1152/ajplung.2001.281.2.L314. [DOI] [PubMed] [Google Scholar]
- 32.Stenmark KR, McMurtry IF. Vascular remodeling versus vasoconstriction in chronic hypoxic pulmonary hypertension: A time for reappraisal? Circ Res. 2005;22:95–8. doi: 10.1161/01.RES.00000175934.68087.29. [DOI] [PubMed] [Google Scholar]
- 33.Mauban JR, Remillard CV, Yuan JX. Hypoxic pulmonary vasoconstriction: Role of ion channels. J Appl Physiol. 2005;98:415–20. doi: 10.1152/japplphysiol.00732.2004. [DOI] [PubMed] [Google Scholar]
- 34.Remillard CV, Yuan JX. High altitude pulmonary hypertension: role of k+ and ca2+ channels. High Alt J Med Biol. 2005;56:133–46. doi: 10.1089/ham.2005.6.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fagan KA, Oka M, Bauer NR, Gebb SA, Ivy DD, Morris KG, et al. Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L656–64. doi: 10.1152/ajplung.00090.2003. [DOI] [PubMed] [Google Scholar]
- 36.Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, et al. Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol. 2004;287:L665–72. doi: 10.1152/ajplung.00050.2003. [DOI] [PubMed] [Google Scholar]
- 37.Pasha MA, Newman JH. High-altitude disorders: pulmonary hypertension: Pulmonary vascular disease: The global perspective. Chest. 2010;6:13S–9S. doi: 10.1378/chest.09-2445. [DOI] [PubMed] [Google Scholar]
- 38.Rupert JL, Koehle MS. Evidence for a genetic basis for altitude related illness. High Alt Med Biol. 2006;7:150–69. doi: 10.1089/ham.2006.7.150. [DOI] [PubMed] [Google Scholar]
- 39.Mortimer H, Patel S, Peacock AJ. The genetic basis of high-altitude pulmonary edema. Pharmacol Ther. 2004;101:183–92. doi: 10.1016/j.pharmthera.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Hanaoka M, Droma Y, Hotta J, Matsuzawa Y, Kobayashi T, Kubo K, et al. Polymorphisms of the tyrosine hydroxylase gene in subjects susceptible to high altitude pulmonary edema. Chest. 2003;123:54–8. doi: 10.1378/chest.123.1.54. [DOI] [PubMed] [Google Scholar]
- 41.Eddahibi W, Chaouat A, Morrell N, Fadel E, Fuhrman C, Bugnet AS, et al. Polymorphism of the serotonin transporter gene and pulmonary hypertension in chronic obstructive pulmonary disease. Circulation. 2003;108:1839–44. doi: 10.1161/01.CIR.0000091409.53101.E8. [DOI] [PubMed] [Google Scholar]
- 42.Beall CM, Cavalleri GL, Deng L, Elston RC, Gao Y, Knight J, et al. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci U SA. 2010;25:11459–64. doi: 10.1073/pnas.1002443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rabinovitch M. Cellular and molecular pathogenesis of pulmonary hypertension. Chest. 2005;128:641–6. doi: 10.1378/chest.128.6_suppl.642S. [DOI] [PubMed] [Google Scholar]
- 44.Van Tassell CP, Smith TP, Matukumalli LK, Taylor JF, Schnabel RD, Lawley CT, et al. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods. 2008;5:247–52. doi: 10.1038/nmeth.1185. [DOI] [PubMed] [Google Scholar]
- 45.Gibbs RA, Taylor JF, Van Tassell CP, Barendse W, Eversole KA, Gill CA, et al. Genome-wide survey of snp variation uncovers the genetic structure of cattle breeds. Science. 2009;324:528–32. doi: 10.1126/science.1167936. [DOI] [PMC free article] [PubMed] [Google Scholar]