

Abstract
Proliferative pulmonary vascular remodeling is the pathologic hallmark of pulmonary arterial hypertension (PAH) that ultimately leads to right heart failure and death. Highly proliferative endothelial cells known as endothelial colony-forming cells (ECFC) participate in vascular homeostasis in health as well as in pathological angiogenic remodeling in disease. ECFC are distinguished by the capacity to clonally proliferate from a single cell. The presence of ECFC in the human pulmonary arteries and their role in PAH pathogenesis is largely unknown. In this study, we established a simple technique for isolating and growing ECFC from cultured pulmonary artery endothelial cells (PAEC) to test the hypothesis that ECFC reside in human pulmonary arteries and that the proliferative vasculopathy of PAH is related to greater numbers and/or more proliferative ECFC in the pulmonary vascular wall. Flow cytometric forward and side scatter properties and aggregate correction were utilized to sort unmanipulated, single PAEC to enumerate ECFC in primary PAEC cultures derived from PAH and healthy lungs. After 2 weeks, wells were assessed for ECFC formation. ECFC derived from PAH PAEC were more proliferative than control. A greater proportion of PAH ECFC formed colonies following subculturing, demonstrating the presence of more ECFC with high proliferative potential among PAH PAEC. Human androgen receptor assay showed clonality of progeny, confirming that proliferative colonies were single cell-derived. ECFC expressed CD31, von Willebrand factor, endothelial nitric oxide synthase, caveolin-1 and CD34, consistent with an endothelial cell phenotype. We established a simple flow cytometry method that allows ECFC quantification using unmanipulated cells. We conclude that ECFC reside among PAEC and that PAH PAEC contain ECFC that are more proliferative than ECFC in control cultures, which likely contributes to the proliferative angiopathic process in PAH.
Keywords: endothelium, endothelial colony forming cell, endothelial progenitor cell, pulmonary arterial hypertension, angiogenesis
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a poorly understood disease that carries a devastating prognosis. Increased pulmonary vascular tone in PAH leads to right heart failure and death. In addition to elevated pulmonary artery pressure, PAH is characterized by increased and dysfunctional angiogenesis of the pulmonary circulation.[1,2] The pathogenesis of these vascular lesions is not well understood.
Dysregulation of angiogenesis is a notable feature of many disease processes.[3–5] Though endothelial cells are normally quiescent, injury, inflammation, hypoxia and other factors can initiate angiogenesis through mobilizing pro-angiogenic cells from the bone marrow and activating local endothelial cells to proliferate.[2,3,6,7] These mobilized pro-angiogenic cells are myeloid in origin, while in contrast a population of true endothelial cells found in the vessel wall called endothelial colony-forming cells (ECFC) are believed to contribute to angiogenesis through cell proliferation.[8] While sprouting angiogenesis can be an appropriate response to vascular injury and hypoxia, increased and dysregulated angiogenesis is a hallmark of many disease states such as cancer,[8,9] macular degeneration[10,11] and pulmonary arterial hypertension.[1,2] Identifying the key cellular players and how they interact in healthy and pathologic angiogenesis are important goals in understanding the pathogenesis of numerous diseases.
ECFC, also termed late outgrowth endothelial cells or blood outgrowth endothelial cells, are proliferative endothelial cells residing in the vessel wall and found rarely in the peripheral blood.[7,12] ECFC were identified by Ingram and colleagues using a single cell in vitro colony-forming assay.[8,12] Although identified in the search for an endothelial progenitor cell, it is unclear whether ECFC represent a unipotent or multipotent stem cell or are just fully differentiated endothelial cells with high proliferative potential. ECFC express endothelial cell-specific surface markers, such as CD34, CD146, CD31, Flk-1 and CD105, but there are currently no markers that distinguish ECFC from other endothelial cells.[8,12] ECFC meet rigorous tests of endothelial cell function, including possessing the capacity to form functional vessels in vivo when implanted in immunodeficient mice.[13] ECFC therefore demonstrate the capacity to form structural cells of mature vessels.
Although originally isolated from peripheral blood where they comprised only 1 in 108 plated human mononuclear cells, ECFC have since been identified in several different vascular beds, including the human umbilical vein and aorta, as well as the rat pulmonary artery.[8,13,14] A hallmark of ECFC is their remarkable proliferative potential, having been documented to achieve over 100 population doublings in vitro.[8,12] The role of ECFC in disease has been suggested by studies in diabetes and pulmonary arterial hypertension (PAH) that suggest that ECFC function is compromised in these illnesses. Circulating ECFC demonstrate impaired angiogenic tube formation in both diabetes and PAH.[15,16]
While there have been several studies investigating the role of circulating pro-angiogenic hematopoietic cells in PAH, the importance of ECFC in the genesis of the vascular lesions of PAH is less well studied.[16–20] Given the angiogenic and proliferative capacities of ECFC and the exuberant angiogenesis that occurs in PAH, it is possible that differences in either proliferative capacity or numbers of these cells may contribute to the vasculopathy of PAH. We hypothesized (1) ECFC reside in the human pulmonary arteries in PAH and controls and (2) that PAH PAEC will contain a greater number or more proliferative ECFC. We further sought to validate that the assay used to isolate and grow ECFC would yield clonal populations of cells. In this study we describe the characterization of ECFC derived from the cultured endothelial cells from the human pulmonary artery and differences in these cell populations between healthy control and PAH patients.
MATERIALS AND METHODS
Pulmonary artery endothelial cell isolation and culture
PAH and control PAECs were obtained from explanted PAH human lungs and donor lungs not used in transplantation, respectively. All patients were female for purposes of clonality analysis (see HUMARA assay below). Pulmonary arteries were dissected down to the distal small arterioles, longitudinally cut, and incubated with collagenase type II to detach endothelial cells. Cells were grown in MCDB-107 (Sigma, St. Louis, Mo.) on fibronectin-coated tissue culture plates. Tissue culture plates were pre-coated with 1 mL of bovine serum fibronectin (Calbiochem, La Jolla, Calif.) diluted in phosphate-buffered saline (PBS) to 50μg/mL for 20-30 minutes. PAEC were passaged at 70% to 80% confluence by dissociation with 0.25% trypsin-ethylenediaminetetraacetic acid (Invitrogen, Carlsbad, Calif.) with trypsin reaction stopped with MCDB-107 media containing serum. Endothelial cell phenotype was confirmed by immunocytochemistry for the endothelial cell-specific markers CD31 (1:30 dilution; Dako, Glostrup, Denmark) and von Willebrand factor (vWF; 1:200 dilution; Dako, Glostrup, Denmark), and fluorescence-activated cell sorting (FACS) analyses for CD31 and VEGFR2 expression (Becton Dickinson, San Jose, Calif.). Immunohistochemical analysis of cultured cells identified that >95% were CD31-positive and >99% vWF-positive. FACS analysis confirmed that >95% of the cells were CD31- and VEGFR2-positive.[17,21] Primary cultures at passage 5 were used in experiments. DNA from original PAEC cultures was analyzed for mutations and chromosomal abnormalities known to be associated with PAH. Cultured PAEC were harvested by manual scraping with a cell scraper and DNA was extracted using the Qiagen DNA Mini kit (Qiagen, Valencia, Calif.) according to the manufacturer's recommended protocol. BMPR2 mutation analysis was performed by direct sequencing and multiplex ligation-dependent probe amplification as previously described.[17] To detect genome-wide copy number changes, DNA was hybridized to single nucleotide polymorphism arrays, as previously described.[22]
Endothelial colony-forming cell assay
We adapted the ECFC assay originally described by Ingram and colleagues for the culture of PAEC.[8] The principle is to sort single endothelial cells into the wells of a 96-well plate and subsequently culture and expand dividing cells. Through the use of single-cell sorting, the range of clonogenic potentials of PAEC can be assessed using this assay. ECFC that possess high proliferative potential form secondary colonies upon replating.
PAEC at passage 5 from frozen stock stored in liquid nitrogen in 10% DMSO in fetal bovine serum (FBS) were brought to room temperature and cultured in complete endothelial growth media-2 (EGM-2; bullet kit, Lonza, Walkersville, Md.) in a fibronectin-coated 100 mm tissue culture plate (see below). Cells were grown in 9 mL of EGM-2 supplemented with 10% FBS, 2% penicillin/streptomycin and 0.25 μg/mL amphotericin B (EGM-2 complete) and incubated in a humidified incubator at 37°C with 5% CO2. At 50-90% confluency, PAEC were subsequently rinsed twice with PBS and trypsinized with 1 mL of warm trypsin-EDTA. Trypsinization was stopped with 4 mL of EGM-2 complete and cells were harvested and spun down at 233 g for 5 minutes at 20° C. Media was aspirated and cells were resuspended with 1-3 mL of EGM-2 complete and then filtered with a 40 μm filter for single-cell sorting into 96 well plates. Suspended cells were kept on ice to reduce aggregation.
Ninety-six-well plates and all subsequent plates used for culture of ECFC were coated with 5 μg/cm2 of rat tail collagen type I (BD Biosciences, San Jose, CA) in 0.02N acetic acid prior to adding cells. Stock solution of 0.02N acetic acid was prepared from glacial acetic acid (17.4N) diluted in Milli-Q-purified water (Millipore, Billerica, Mass.) and filtered with a 0.22 μm vacuum filter under a laminar flow tissue culture hood. Prior to preparation of plates for sorting, collagen type I solution was made fresh by adding the appropriate amount of collagen type I to the acetic acid stock to make a 50 μg/mL collagen type I (collagen-I) solution. After determining the surface area of each well, enough collagen-I solution was added to cover the plating area with 5 μg/cm2 of collagen-I. Plates were coated 2 hrs. to overnight in a humidified incubator at 37°C with 5% CO2. Collagen-I solution was then aspirated and wells were rinsed twice with PBS. Two hundred μL of EGM-2 complete was added to each well of a 96-well plate. For subculturing, larger culture vessels received appropriate volumes of EGM-2 complete.
PAEC suspended in complete EGM-2 were sorted by FACS using a FACSAria II flow cytometry cell sorter (BD Biosciences, San Jose, Calif.) to place a single cell in each well of a 96-well tissue culture plate. Cells were sorted by forward scatter and side scatter (FSC/SSC) gated for the cellular fraction with aggregate correction to ensure that multiple cells were not sorted into each well. One hundred cells were sorted into the first well on each 96-well plate to assist with focusing of the microscope during visualization. PAEC were sorted into 3 96-well plates for each patient. Media was changed on Days 4 and 8. After 2 weeks, wells were examined for formation of ECFC colonies by an inverted microscope. Cell counts were performed by visual inspection where possible or otherwise with a hemacytometer. To detach cells for subculturing and counting the supernatant overlying, each well of a 96-well plate was removed and the well was rinsed once with 70 μL PBS. Twenty μL of warm trypsin was added to the well and incubated for 2.5 minutes at 37° C. Trypsinization was stopped by adding 30 μL EGM-2 complete. Plates were put on ice after this step to prevent aggregation. Where counting was achieved with a hemacytometer (>50 cells), 10 μL of the cell suspension was loaded onto a hemacytometer and the 4 corner fields were counted and averaged. A colony was defined by the presence of 2 or more cells in a well. ECFC were defined as single cells giving rise to a colony containing 50 cells or greater.[8]
Replating of ECFC
After the number of cells/colony was counted, ECFC colonies were subcultured in 600 μL of complete EGM-2 in a 24-well tissue culture plate pre-coated with 5 μg/cm2 rat tail collagen-I and examined for secondary colony formation or confluence after 1 week. Wells in which there was colony formation were subsequently subcultured in 2 mL complete EGM-2 to a 6-well tissue culture plate pre-coated with 5 μg/cm2 rat tail collagen-I in complete EGM-2 and allowed to grow to confluence. Confluent wells were then subcultured in a 100 mm tissue culture plate pre-coated with 5 μg/cm2 rat tail collagen-I in complete EGM-2.
Immunohistochemistry
ECFC which were expanded to 6-well plates and 100 mm tissue culture dishes were analyzed for endothelial cell-specific markers to confirm endothelial phenotype: vWF; endothelial nitric oxide synthase (eNOS); caveolin-1 (cav-1); CD31; and CD34. Cells were trypsinized with 1 mL of warm trypsin and transferred onto 4-well chamber slides pre-coated with rat tail collagen type I and grown in complete EGM-2 as previously described. After overnight incubation at 37° C, media was removed and cells were fixed using the following methods specific to the staining protocol for each antibody: vWF–4.0% paraformaldehyde/PBS/0.2% Triton 100 for 10 minutes at room temperature; eNOS–methanol for 10 minutes at -20° C; CD31–4.0% paraformaldehyde/PBS/0.2% Triton 100 for 10 minutes at room temperature; caveolin-1–acetone-methanol (1:1) for 10 minutes at -20° C; and CD34–acetone for 10 minutes at -20° C. After fixation, cells were washed 3 times with PBS and blocked with 3.0% normal goat serum (vWF, eNOS, CD31) or horse serum (cav-1, CD34). Following aspiration of blocking serum, cells were incubated with 200 μL of the following primary antibodies at the listed dilutions at room temperature for 90 minutes with gentle agitation: polyclonal rabbit anti-human: vWF at 1:1200 (Dako, Glostrup, Denmark); polyclonal rabbit anti-human NOS3 (eNOS) at 1:500 (Santa Cruz Biotechnology, Santa Cruz, Calif.); polyclonal rabbit anti-CD31 prediluted at 1:4 (Abcam, Cambridge, Mass.); monoclonal mouse anti-caveolin 1 at 1:200 (BD Transduction Laboratories, San Jose, Calif.); and monoclonal mouse anti-human CD34 Class II at 1:25 (Dako, Glostrup, Denmark). A negative control which was incubated with blocking serum only instead of primary antibody was used for each PAEC sample or ECFC clone. Cells were then washed with PBS/0.05% Tween 20 three times for 5 minutes and the appropriate secondary antibody was added and incubated at room temperature for 30 minutes. For vWF, eNOS, and CD31, goat anti-rabbit IgG (Vector Laboratories, Burlingame, Calif.) was used and for cav-1 and CD34, horse anti-mouse IgG (Vector Laboratories, Burlingame, Calif.) was used. Cells were then washed with PBS/0.05% Tween 20 three times for 5 minutes with gentle agitation and avidin/biotin complex was added and incubated for 30 minutes at room temperature. Cells were washed again with PBS/0.05% Tween 20 three times for 5 minutes. Following wash step, microchambers were removed and visualization was achieved with ImmPACT DAB peroxidase substrate for 2-10 minutes. Slides were rinsed with running tap water, counterstained with hematoxylin (Vector Laboratories, Burlingame, Calif.) for 2-3 minutes, and rinsed again with running tap water until rinse water was colorless. Slides were destained with one quick dip in 1.0% HCl in 70% ethanol, rinsed with running tap water, and dipped 10-20 times in bluing solution (Richard Allan Scientific, Kalamazoo, Mich.). Following a final rinse with running tap water, slides were dehydrated using an ethanol and xylene series and mounted with Permount.
Acetylated LDL uptake
ECFC which grew to populate a 100 mm tissue culture dish were trypsinized and cultured in a 2-well chamber slide pre-coated with rat tail collagen-I as previously described. ECFC were incubated with 20 μg/mL with DiI-acetylated-low-density lipoprotein (DiI-Ac-LDL; Invitrogen, Carlsbad, Calif.) for 2 hrs. in EGM-2 complete at 37°C. Cells were washed twice with complete EGM-2 and examined for DiI-Ac-LDL uptake using a Leica DM IRB inverted microscope.
Electron microscopy
For ECFC that expanded to populate a 100 mm tissue culture dish, we confirmed endothelial phenotype by examining for caveolae and Weibel-Palade bodies with electron microscopy. Samples were trypsinized and grown in a 2-well chamber slide pre-coated with collagen-I. ECFC were trypsinized, transferred to chamber slides, and grown overnight in 1.6 mL complete EGM-2 in a 5% CO2 humidified incubator at 37° C. Media was aspirated and samples were fixed with 2.5% glutaradehyde and 4% paraformaldehyde with 0.2M cacodylate buffer and kept overnight at 4° C. Samples were washed with sodium cacodylate buffer (0.2M, pH 7.3) 3 times for 5 min. each. Cacodylate buffer was removed and 1% osmium tetroxide (in H2O) was added and incubated for 60 min. at 4° C. Samples were washed again with sodium cacodylate twice, 5 min. each, and rinsed with maleate buffer (pH 5.1) ×1 for 5 min. Staining was then performed by adding 1% uranyl acetate in maleate buffer and stained for 60 min. Uranyl acetate was removed and samples were washed with maleate buffer 3 times. Samples were subsequently dehydrated by rinsing with increasing concentrations of cold ethanol (50-95%), 5 min. each, followed by 3 rinses with 100% room temperature ethanol. Following this, samples were rinsed with propylene oxide 3 times for 15 min. each. Propylene oxide was removed and replaced with 1:1 propylene oxide/eponate 12 medium (Ted Pella Inc., Redding, Calif.) at room temperature overnight. Subsequently, media was aspirated and pure eponate 12 medium was added for 4-6 hrs at room temperature. Polymerization was allowed to progress for 24 hrs., after which ultra thin sections of 85 nm were cut with diamond knife, stained with uranyl acetate and lead citrate, and then observed with a Philips CM12 electron microscope operated at 60 kV.
HUMARA assay
We assessed the clonality of ECFC that proliferated to populate a 100 mm tissue culture dish by examining the X-inactivation distribution of ECFC and the parent PAEC culture with the human androgen receptor assay (HUMARA). HUMARA uses the methylation status of a highly polymorphic CAG repeat region flanking the human androgen receptor gene as a surrogate for X-inactivation status.[23] The inactive X chromosome is methylated in this region while the active X chromosome is not. X-inactivation is normally random in a human female; however, clonal populations of cells demonstrate inactivation of the same X-chromosome. HUMARA was performed as previously described.[17]
ECFC in 100 mm tissue culture dishes were trypsinized and resuspended in EGM-2 and DNA was extracted using the Qiagen DNA Mini kit (Qiagen, Valencia, Calif.) according to the manufacturer's recommended protocol. One hundred ng of DNA was digested 16 h with RsaI and 100 ng DNA was digested with RsaI+HhaI. NEB Buffer 4 and 1X bovine serum albumin (BSA; New England BioLabs, Ipswich, Mass.) was used for both digestions. After digestion, restriction enzymes were inactivated at 80° C for 20 min. For HUMARA PCR, 1 μL or 4 μL of the diluted digestion product (RsaI and RsaI+HhaI, respectively) was added to 2 μL of ABgene buffer (Applied Biosystems, Carlsbad, Calif.), 2 μL dNTPs (2 mM), 2 μL custom synthesized fluorescently-labeled primers (Integrated DNA Technologies, Coralville, Iowa), 0.5 μL of dimethyl sulfoxide, 0.2 μL ABgene Taq polymerase, and the remainder with distilled water to achieve a final volume of 20 μL. PCR reaction was run for 36 cycles on a thermal cycler. Ten μL of the PCR products were run on a 1.25% agarose gel to check for efficient amplification. Two μL of the remaining product was diluted 1:10-1:100 in distilled water, as judged from the intensity of the band on the agarose gel. 1 μL of diluted PCR product was loaded with 8.75 μL of HiDi formamide (Applied Biosystems, Carlsbad, Calif.) and 0.25 μL of a fluorescent ladder on an Applied Biosystems AB3730XL fluorescent sequence using the “genotyping” module. Data were analyzed with Gene Mapper v4.0 software (Applied Biosystems, Carlsbad, Calif.). The raw peak height values of the digested samples were corrected for amplification efficiency by using the average of the 2 samples digested with RsaI alone.[23] Results for ECFC were compared to HUMARA analysis of the original PAEC culture from which ECFC were derived. HUMARA PCR primer sequences were as follows: Forward—5’-GTT TCC AGA ATC TGT TCC AGA GCG TGC-3’; and Reverse-5’-6-FAM/ATG GGC TTG GGG AGA ACC ATC CTC-3′. 6-FAM is 6-carboxyfluorescein, a fluorescent PCR primer tag.
Statistical analysis
All data were analyzed using the JMP 9.0 software program. The Wilcoxon test was used for comparison of nonparametric data and the Wilcoxon signed rank test was used for comparison of paired nonparametric data, as appropriate. P values of less than 0.05 were considered significant. Mean ± standard error of mean for each group is shown.
RESULTS
Patient selection
PAEC were isolated from donor lungs not used in transplantation (n=4) and explanted lungs from PAH patients at the time of transplantation (n=11). Mean age of patients was 36.3±4.9 for control versus 48.8±3.5 for PAH, P=0.11. All patients were Caucasian females. The PAH group included patients with different subtypes of PAH and were genetically characterized for chromosomal or mutational changes known to be associated with PAH.[22] These genetic alterations included bone morphogenetic protein receptor 2 (BMPR2) mutations and a proportion of cells harboring mosaic deletion of the X-chromosome. Also included were PAH patients with no known genetic abnormalities (Table 1).
Table 1.
Patient Demographics
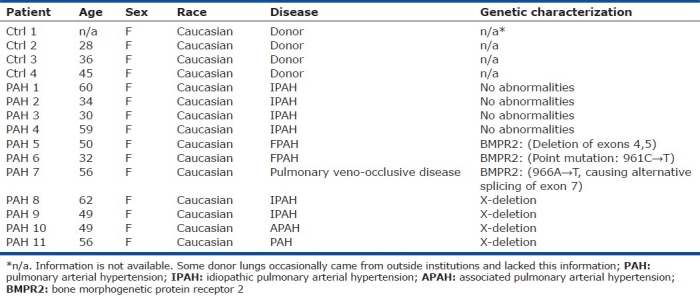
Single-cell sorting of PAEC
PAEC were isolated from control and PAH lungs as outlined in the “Methods” section (Fig. 1). Cultured PAEC at passage 5 were grown to between 50%-90% confluency and sorted using a BD FACsAria II flow cytometer to put a single cell in each well of a 96-well tissue culture plate pre-coated with rat tail collagen-I. Aggregate exclusion was performed as described in “Methods.” Visual inspection of colonies with eosin staining 24 hrs. after sorting confirmed that 97% of wells received a single cell that was able to become adherent. Adherent cells included healthy-looking cells, pyknotic cells, and cells already undergoing division. Thus flow cytometric techniques were effectively employed to achieve single-cell sorting of unstained PAEC (Fig. 2).
Figure 1.
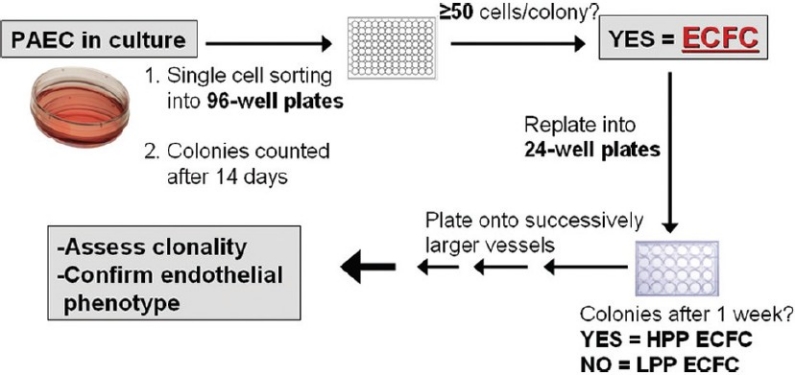
Schematic of ECFC assay. Cultured control and PAH PAEC were single-cell sorted into 96-well plates by FACS based on forward and side scatter properties and allowed to grow for two weeks after which wells were scored for the formation of colonies. ECFC are defined as single cells that gave rise to a colony containing ≥50 cells. ECFC were subsequently subcultured into progressively larger culture vessels. Those colonies that retain the ability to give rise to colonies upon secondary culturing were considered high proliferative potential (HPP) ECFC. ECFC which lacked this capacity are considered low proliferative potential (LPP) ECFC. The most proliferative ECFC were assessed for endothelial cell phenotype and for clonal origin.
Figure 2.
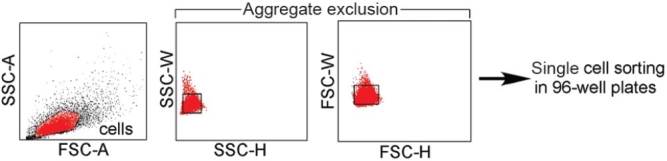
Flow cytometric single cell sorting. PAEC suspended in EGM-2 were sorted into 96-well plates by FACS. FSC-A/SSC-A dot plot was used to define gate for endothelial cells. In order to prevent the sorting of >1 endothelial cell in each well aggregate correction was performed. Aggregates lead to increased SSC-W and FSC-W due to prolonged scatter of the laser due to their increased size. Use of aggregate exclusion led to 97% of wells receiving a single endothelial cell that was able to adhere to the collagen matrix when assessed 24 hrs. after sorting.
ECFC among control and PAH PAEC
A modified version of the ECFC assay described by Ingram and colleagues was used to isolate and culture ECFC as described.[8] We determined the quantity of ECFC contained in cultured PAEC, the numbers of cells contained in each ECFC colony, and the capacity for ECFC to form secondary colonies following subculture into larger culture vessels.
Our findings indicate that altogether, 216/1140 (18.9%) cells sorted from control PAEC formed colonies, while 820/3040 (27.0%) cells from sorted PAH PAEC formed colonies. The remainder of the cells did not divide and remained as single cells. Treating data from each patient as independent events, 18.0±4.6% of control versus 25.7± 2.4% of PAH PAEC gave rise to colonies (P=0.17, Wilcoxon test). In several cases, colonies containing greater than 50 cells when seen under light microscopy were trypsinized for counting with a hemacytometer, but no cells were subsequently visualized on the hemacytometer. We imputed a value of 50 cells/colony for these cases since the colonies were seen to be there. There was a broad range of colony sizes in both groups, ranging from cells that divided only once to cells that formed colonies containing over 3,000 and 10,000 cells in the control and PAH groups, respectively. The median number of cells per colony in controls was 55±5 cells versus 209±57 cells in the PAH group (P=0.14). Eight out of 11 patients from the PAH PAEC demonstrated a median number of cells per colony greater than the highest value in the control group. Although not statistically significant, PAH PAEC tended to give rise to colonies with larger numbers of cells.
We found that dividing cells from both control and PAH PAEC contained similar percentages of cells that gave rise to colonies of ≥50 cells (ECFC); (73.0±5.3% control vs. 75.0±5.3% PAH, P=0.40). However, PAH PAEC contained a larger proportion of highly proliferative ECFC and contained the more proliferative ECFC (Fig. 3). The median number of cells in each ECFC colony was 144±36 cells in control versus 355±64 cells PAH, P=0.03 (Fig. 4a). Thus, PAH PAEC give rise to larger ECFC colonies. There was not a statistically significant difference in the number of ECFC between the 2 groups. In each 96-well plate sorted, there were 14±5 ECFC in control versus 20±3 ECFC in PAH PAEC (P=0.17). This number ranged between 7 and 27 ECFC per 96-well plate in controls versus 9 and 34 ECFC/96-well plate in PAH. Both control and PAH PAEC contain ECFC supporting the hypothesis that ECFC are resident cells of the pulmonary artery endothelium. Together the data indicate that PAH pulmonary artery endothelium harbors more proliferative ECFC.
Figure 3.
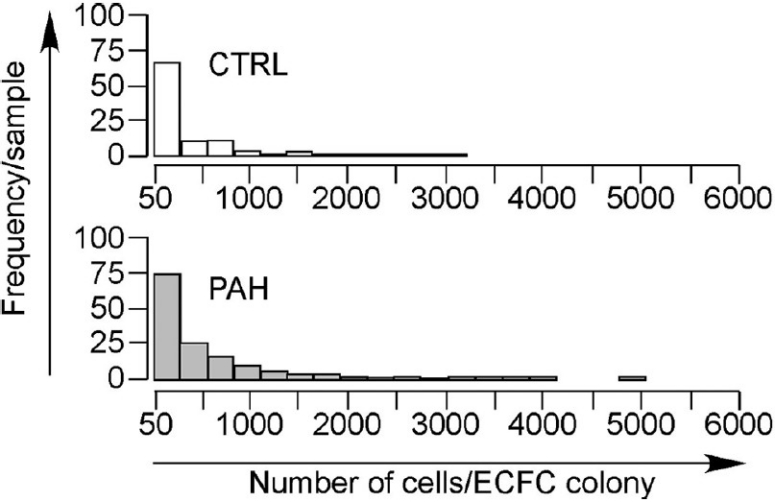
Distribution of number of cells per ECFC colony in PAH and control PAEC. Horizontal axis represents number of cells per ECFC colony. Vertical axis represents frequency per sample. ECFC derived from PAH PAEC gave rise to the most proliferative colonies arising from a single cell.
Figure 4.
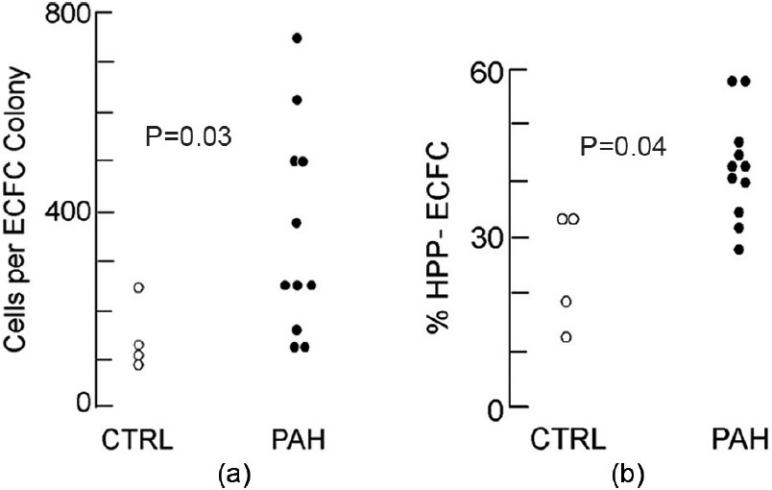
PAH PAEC gave rise to ECFC that generate larger colonies and a higher proportion of HPP ECFC: (a) Median number of cells per ECFC colony from control and PAH PAEC. Vertical axis represents cells per ECFC colony. Each circle represents the median number of cells per ECFC colony from an individual patient. Open circles represent control and filled circles represent PAH. (b) ECFC were subcultured into 24-well plates to assess for HPP ECFC based on ability to form colonies. Vertical axis is percentage of HPP ECFC as a fraction of total ECFC.
ECFC are classified as possessing either high proliferative potential (HPP) or low proliferative potential (LPP) based on ability to form secondary colonies upon subculturing (Fig. 5).[8] We investigated whether there was a greater proportion of HPP ECFC in PAH PAEC compared to control. We found that 24.9±5.6% of control ECFC versus 41.2±2.9% PAH ECFC were HPP ECFC, P=0.04 (Fig. 4b). HPP ECFC formed larger colonies in 96-well plates than LPP ECFC. The median number of cells per colony for LPP ECFC was 732±79 cells fewer than HPP ECFC at the 96-well stage (869 cells per colony in PAH vs. 137 in controls, P<0.01), suggesting that the capacity for ECFC to form secondary colonies is directly linked to their proliferative potential. Furthermore, the most proliferative ECFC were also derived from PAH PAEC. Out of 4,180 single cells plated into 96-well plates, 15 grew to form subsequent colonies greater than 100,000 cells. All of these most proliferative ECFC were derived from PAH ECFC. Thus PAH PAEC give rise to a greater percentage of HPP ECFC and yield the more proliferative ECFC compared to control. This corroborates previous evidence demonstrating a hyperproliferative/apoptosis resistant phenotype among PAH PAEC and suggests more proliferative ECFC may contribute to this phenotype.
Figure 5.

HPP ECFC. ECFC colonies from 96-well plates were subcultured into 24-well plates and assessed for colony formation or confluence after 1 week. Representative images are shown. Secondary ECFC colonies ranged in size from small (A), medium (B), or large/confluent (C). Scale bar is 200 μm.
ECFC express endothelial cell markers
ECFC isolated from the circulation as well as other vascular beds have been demonstrated to be of endothelial origin as assessed by cell surface markers, functional assays, and gene expression profile.[8,12,13,24] 16ECFC express several endothelial cell-specific markers and do not express the leukocyte common antigen CD45, consistent with a putative role as the source of dividing endothelial cells during angiogenesis.[6–8]
We chose to assess for the expression of CD31, vWF, cav-1, eNOS, and CD34. CD31 is expressed by platelets, endothelial cells, and macrophages and is used frequently used to stain for endothelial cells in immunohistochemical staining.[25] Von Willebrand factor, a protein that binds Factor VIII and is important for platelet adhesion, is highly expressed by endothelial cells.[26] eNOS is expressed in endothelial cells, and reduction of nitric oxide levels is implicated in the increased pulmonary vascular tone.[27] PAH PAEC express more eNOS than endothelial cells from the systemic circulation. Cav-1 is a protein that localizes to caveolae, invaginations of the plasma membrane which are found in most cells and is particularly important for vesicular trafficking in endothelial cells.[28] Cav-1 also regulates eNOS activity and may play a role in PAH pathogenesis.[27] CD34 is a cell surface protein implicated in cell migration and halting of differentiation that has been documented to be expressed in ECFC and PAEC, and which has served most commonly as a hematopoietic stem cell marker.[8,29]
Our findings indicate that ECFC are endothelial cells. They express CD31, vWF, cav-1, eNOS, and CD34, consistent with the expression pattern expected of endothelial cells and corroborating previous reports on ECFC surface marker expression.[8,9] Staining of PAEC cultures prior to ECFC sorting also demonstrated expression of these markers (Fig. 6). The staining pattern was similar in both PAEC and ECFC groups, suggesting expression of CD31, vWF, Cav-1, eNOS, and CD34 is maintained in highly proliferative as well as less proliferative endothelial cells. ECFC also demonstrated uptake of acetylated LDL, a functional property of endothelial cells (Fig. 7). Endothelial cell ultrastructure is typified by the presence of caveloae invaginations of the plasma membrane, as well as by Weibel-Palade bodies. ECFC demonstrated the presence of both these elements as visualized by electron microscopy (Fig. 8). Thus we found strong evidence that ECFC exhibit endothelial cell phenotype and are not colonies of any potential contaminating cells in the primary PAEC cultures.
Figure 6.
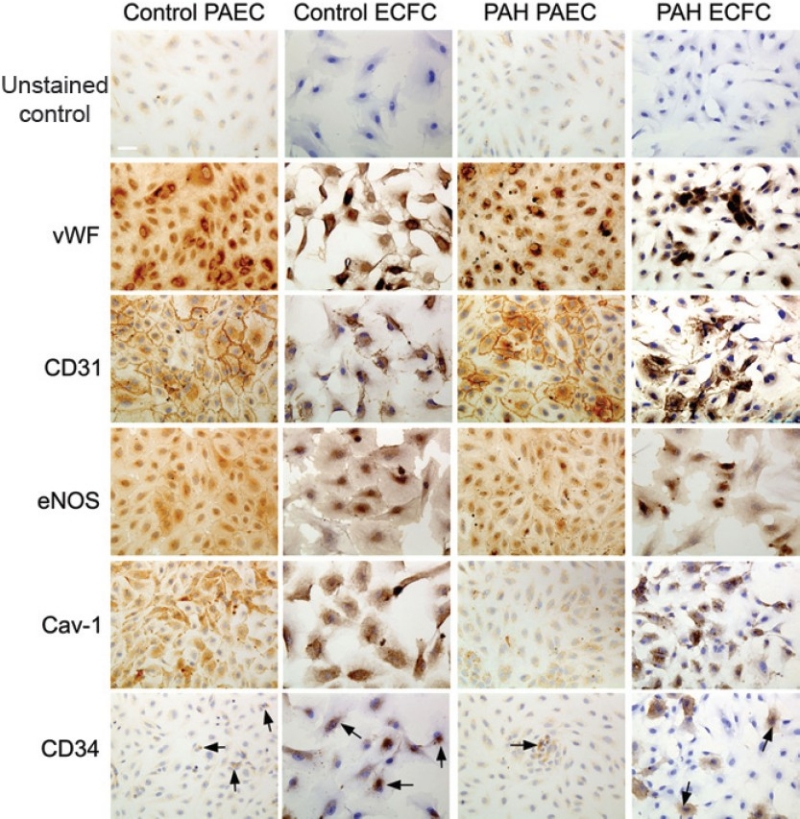
ECFC express endothelial cell-specific markers. Representative images are shown. Both PAEC and ECFC demonstrate positive staining for CD31, vWF, cav-1, eNOS, and CD34. vWF stains intracellularly in both PAEC and ECFC, as expected for endothelial cells. CD31 exhibits staining localized to the membrane, particularly at sites of cell-cell contact, and in the cytosol. The cell-junctional staining for CD31 staining in ECFC is not as pronounced owing to cells not being confluent. eNOS staining is present in both PAEC and ECFC, particularly in the perinuclear region. Cav-1 stains both PAEC and ECFC. There is also CD34 staining in both PAEC and ECFC in both control and PAH groups (arrows). There are no significant differences in staining pattern between PAH and control groups. Scale bar is 50 μm.
Figure 7.
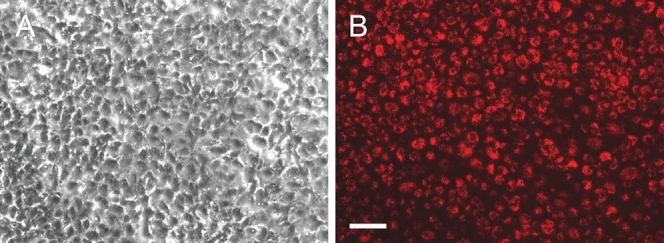
ECFC take up acetylated LDL. ECFC were incubated with 20 μg/mL DiI-acetylated LDL for 2 hrs. at 37°C and assessed for uptake with fluorescence microscopy. (A) Phase contrast ECFC image taken with inverted microscope (B) Strong uptake of the DiI-labeled acetylated LDL was demonstrated by ECFC. Scale bar is 100 μm.
Figure 8.
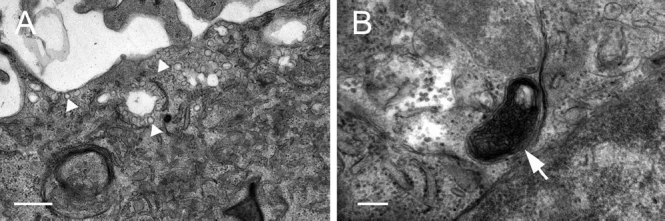
ECFC show ultrastructural properties typical of endothelial cells. (A) ECFC demonstrate the presence of numerous caveolae along the plasma membrane (arrowheads), consistent with endothelial cell morphology. Scale bar is 500 nm. (B) ECFC also demonstrate the presence of Weibel-Palade bodies, which manufacture vWF (arrow). Scale bar is 200 nm.
Clonality of expanded ECFC confirmed by HUMARA
The ECFC assay allows the clonogenic potential of individual endothelial cells to be assessed. In order to further verify that this methodology could isolate cells that could undergo significant clonal expansion, we assessed the clonality of 3 of the most proliferative clones using HUMARA. Our data suggest that the expanded ECFC were clonal in origin (Fig. 9). For PAH 3, PAEC demonstrated random X-inactivation. After methylation-specific digestion, both a 226 bp and 223 bp peak were amplified in roughly equal proportions. However, in 2 ECFC clones derived from PAH 3, there was a loss of the 223 bp peak following methylation-specific digestion, suggesting that the same X chromosome is inactivated in all cells, implying a common clonal origin. Similarly, in PAH 11, which contains a mosaic X-chromosome deletion, the ECFC clone contained a single X-chromosome in all cells, indicating that it derived from a single cell with the X-deletion. Furthermore, the single X-chromosome was active in all cells. This verifies the ability of the ECFC assay to isolate clones and potentially allow for studies of the most proliferative endothelial cells contained in culture.
Figure 9.
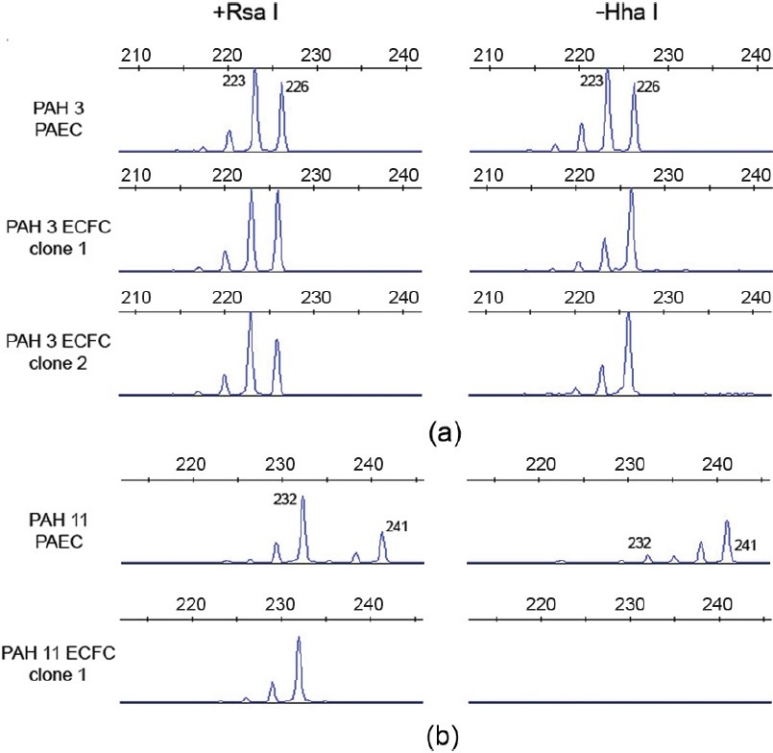
X-inactivation analysis. Allele patterns at the androgen receptor locus are shown before (left panel) and after digestion with the methylationsensitive restriction enzyme HhaI (right panel). Alleles are labeled with their size in basepairs. (a) Patient PAH 3. PAEC DNA shows an identical pattern before and after HhaI digestion, indicating random X-inactivation, whereas the two ECFC show a single peak after digestion, consistent with clones derived from a single cell. (b) Patient PAH 11 has a known mosaic X-chromosome deletion, with loss of the 241bp allele in approximately 50% of cells. HhaI digestion reveals significant skewing of X-inactivation in the “parent” PAEC culture. The ECFC clone contains a single 232bp allele, indicating that it is derived from a cell carrying the X-chromosome deletion. Furthermore, there was no amplification following HhaI digestion, suggesting that the single X-chromosome is active in all cells.
DISCUSSION
ECFC are proliferative endothelial cells that demonstrate in vitro and in vivo angiogenic capacity, are structural cells of the endothelium, and are present but rare in the circulation.[8,12,30,31] They have been isolated from cultured endothelial cells from the human umbilical vein and aorta, as well as the rat pulmonary arteries and microvessels.[12,14] ECFC possess significant clonal proliferative potential and are implicated in both physiologic and pathologic angiogenesis. For example, ECFC demonstrate the ability to participate in vascular repair in mouse models of hind limb ischemia.[32,33] Conversely, circulating ECFC dysfunction has been demonstrated in diseases of dysregulated angiogenesis, such as diabetes mellitus and PAH.[15,16] This combination of proliferative capacity, in vivo angiogenic function, and evidence of dysfunction in diseases of aberrant angiogenesis all argue for a crucial role of ECFC in angiogenesis, perhaps as the source of endothelial cells required for new vessel formation.
A role for ECFC in PAH pathogenesis has been suggested from work done by Toshner and colleagues, who documented that circulating human ECFC in PAH with BMPR2 mutation display a hyperproliferative phenotype and have impaired in vitro tube-forming capacity.[16] However this work did not establish whether circulating ECFC were derived from the pulmonary arteries. Thus, it is not known whether these findings represent widespread endothelial dysfunction or a PAEC-specific pathology. ECFC isolated from the pulmonary vasculature itself has previously only been well-characterized in the rat lung, where ECFC were found in rat PAEC and were enriched in the pulmonary microvascular endothelial cells.[14] While this study offered important insight into the presence, distribution, and biology of ECFC in the pulmonary circulation, it did not establish a role for these cell populations in human disease. In the human pulmonary artery, the presence, quantity, and potentials of ECFC are largely unknown. The preseny study represents the first attempt to address these questions.
In this study, we demonstrated a simple method to isolate ECFC using flow cytometry to sort single endothelial cells into 96-well plates based on forward and side scatter with aggregate exclusion. We confirmed single-cell sorting by visual inspection with eosin staining and by indirect assessment of clonality (HUMARA). Using this method, we identified the presence of ECFC in cultured human PAEC derived from control and PAH populations. Our methodology notably avoided the use of retroviral vectors to introduce GFP into endothelial cells as described in the literature, which has the potential to introduce unforeseen changes to cell growth as well as adding to the biohazard for the manipulation of these cells.[8,12] We observed that ECFC among PAEC are not rare, representing between 5-30% of all sorted endothelial cells in the healthy and PAH subject samples that we studied. There was a wide range of proliferative capacities among ECFC, with some giving rise to over a million cells while others proliferated to 50 cells and were unable to form colonies after subculturing. We found that PAH PAEC tend to give rise to larger ECFC colonies than controls and also contained more HPP ECFC, suggesting PAH ECFC proliferate more or are more apoptosis-resistant than control. Although there was not a statistically significant difference in the number of ECFC between control and PAH groups, the number of cells per ECFC colony was higher in PAH and the most proliferative cells after subculturing of ECFC were also found in the PAH group. Sorted PAH PAEC demonstrated a skew towards a more proliferative phenotype compared to control. Overall these observations corroborate a previous report by our group demonstrating PAH PAEC in culture exhibit a hyperproliferative/apoptosis-resistant phenotype,[21] as well as findings by Toshner and colleagues that circulating ECFC are hyper-proliferative in PAH.[16,21] Our findings indicate that more proliferative ECFC are present among PAH PAEC and may be linked to the hyper-proliferative phenotype of these cells in relation to control.
Our findings differ somewhat from previous studies on ECFC in the systemic circulation. We overall observed less ECFC proliferation than was reported in previous studies of ECFC. For example, Ingram and colleagues noted that human cord blood-derived ECFC could achieve over 100 population doublings and human umbilical vein endothelial cells (HUVEC) and human aortic endothelial cells (HAEC) could be passaged to 40 or more population doublings.[8,12] In our study 2 ECFC clones isolated from a single PAH patient (PAH 9) each achieved at least 20 population doublings. A smaller percentage of sorted PAEC divided (18.0±4.6% of control and. 25.7± 2.4% of PAH) compared to previous reports by Ingram et al. demonstrating >50% of cells divided among HUVEC and HAEC. Reasons that a sorted, adherent cell would not divide include it undergoing apoptosis or exiting the cell cycle and becoming quiescent. Reflecting this, the discrepancy between our findings and those of other groups may be multifactorial. Ingram and colleagues studied commercially available HAEC and HUVEC at passage 3, while this study included PAEC from passage 5.[12] Cells from later passages might be expected to possess either diminished proliferation, increased apoptosis, or contain a greater proportion of quiescent cells. We were unable to assess what proportion of non-dividing cells exhibited each of these traits. The amount of time cells are frozen and GFP overexpression may also affect the number of cell divisions they can undergo. Furthermore, as has been reported in rats, it is possible that in humans more proliferative ECFC reside in the pulmonary microvasculature compared to the pulmonary arteries.[14] Nonetheless, the identification of ECFC with significant proliferative potential in the pulmonary circulation mirrors what has been reported in the systemic circulation. Taken with the fact that ECFC have also been isolated from HUVEC and rat pulmonary microvasculature, it suggests that ECFC are general structural cells of the endothelium.
The ability to use the ECFC assay to clonally isolate and characterize pulmonary artery-derived ECFC can add to the growing body of knowledge regarding endothelial cell heterogeneity in the lung. Endothelial cell heterogeneity in the systemic circulation is well established, and there is increasing understanding of differences in pulmonary endothelial cell phenotype,[34] including segmentally dependent variations in nitric oxide production, barrier properties, surface antigen expression, and proliferative potential.[14,34,35] The majority of this knowledge is based on rat studies and thus much work remains to be done in humans.[35,36] Elucidating PAEC heterogeneity could be important to broadly understanding how the pulmonary vasculature responds to stress, and specifically PAH pathogenesis. PAH demonstrates segment-specific pathology.[37] For example, plexiform lesions arise in the distal precapillary circulation and are characterized by minimal muscularization while more proximally, lesions are luminal and concentric in nature, with exaggerated smooth muscle hyperplasia and invasion.[37] The ECFC assay can allow for expansion of distinct clonal populations of cells permitting a detailed study of endothelial heterogeneity that cannot be achieved simply through analysis of cells in bulk culture. It is possible, for example, that even within the same segment of the pulmonary artery only a small subset of cells produce factors that mobilize pro-angiogenic progenitors, or stimulate other endothelial cells to divide. These cells could be isolated and expanded by the ECFC assay. Additionally, in light of recent work by Aldred and colleagues demonstrating chromosomal disturbances in PAH PAEC, the ECFC assay also allows for detailed genetic analyses of whether multiple genetic hits identified in culture arise from separate hits in different cells or multiple hits within the same cell.[22] Thus the ECFC assay could be used to identify clonal genetic changes in PAEC that may confer a selective growth advantage.
More broadly, the ECFC assay allows for the characterization of the endothelial cells with the most proliferative potential. In the setting of PAH, there is widespread PAEC dysfunction, including a reduction in nitric oxide production, metabolic switch to glycolysis, and upregulation of HIF and HIF-inducible factors.[19,38] While these characteristics are noted of PAEC in culture, it is not known whether they are present in the most proliferative cells. The clonal expansion and subsequent characterization of cells in the ECFC assay could allow for determination of whether there is correlation between endothelial dysfunction and proliferative potential of a cell. This knowledge may help elucidate whether increased proliferation and other types of endothelial dysfunction are intrinsically linked.
Many additional questions remain unanswered. The in vivo functionality of the ECFC was not assessed, and thus, their relevance to angiogenesis could not be directly ascertained. Furthermore, where in the pulmonary tree the most proliferative ECFC were derived is not known, since the preparation process does not distinguish between segments of the pulmonary arteries. This hampers a segmental-analysis of PAEC heterogeneity. It is possible that, given plexiform lesions in PAH are comprised of monoclonal cell proliferations,[39] ECFC may give rise to these lesions seen in end-stage disease. Our study was also limited in sample size, and did not allow for statistically meaningful comparisons between different subclasses of PAH.
Lastly, an existing challenge in current ECFC understanding is that it is not known what distinguishes the more proliferative from less proliferative ECFC or whether they are truly phenotypically distinct from other endothelial cells. The differences in proliferation potential can currently only be assessed by the ECFC assay. No markers distinguish ECFC from endothelial cells or predict their proliferative capacity.[8] Whether ECFC represent a true progenitor cell is also not known, as no studies have shown whether they exhibit asymmetric division or have the ability to differentiate into non-endothelial cells.
The ECFC assay allows for the proliferative capacity of individual endothelial cells to be interrogated and may be a potential tool to help elucidate functional and genetic heterogeneity among endothelial cells. In this study we reported development of a simplified method for ECFC assay using non-manipulated endothelial cells. Using this method we showed that numerous ECFC are present in the pulmonary artery endothelium. PAH pulmonary artery endothelium exhibit high proliferative ECFC compared to healthy controls. The methods and findings described herein represent another step towards unraveling the mechanisms by which PAEC contribute to this devastating disease.
ACKNOWLEDGMENTS
The authors thank Denise Hatala (immunohistochemistry), Mei Yin (electron microscopy) and Dr. John Peterson (light microscopy) in the Lerner Research Institute Digital Imaging Core. In the Flow Cytometry core, we thank Moneen Morgan, Sage O’Bryant and Cathy Shemo for technical assistance with instrument operation. We also thank Dr. Amy Nowacki in the Quantitative Health Sciences Department for advice on statistical analysis.
Footnotes
Source of Support: American Heart Association 11SDG4990003, American Thoracic Society/Pulmonary Hypertension Association grant (PH-07-003), and National Institutes of Health grants RC37 HL60917 and R01 HL098199. KA is a scholar of the International Society for Advancement of Cytometry. HD is a Howard Hughes Medical Institute Medical Research Fellow (Grant ID 57006973)
Conflict of Interest: None declared.
REFERENCES
- 1.Heath D, Edwards JE. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation. 1958;18:533–47. doi: 10.1161/01.cir.18.4.533. [DOI] [PubMed] [Google Scholar]
- 2.Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28:23–42. doi: 10.1016/j.ccm.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asosingh K, Erzurum SC. Angioplasticity in asthma. Biochem Soc Trans. 2009;37:805–10. doi: 10.1042/BST0370805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 5.Morrell NW, Adnot S, Archer SL, Dupuis J, Jones PL, MacLean MR, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chao H, Hirschi KK. Hemato-vascular origins of endothelial progenitor cells? Microvasc Res. 2010;79:169–73. doi: 10.1016/j.mvr.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson MR, Yoder MC. Endothelial progenitor cells: quo vadis? J Mol Cell Cardiol. 2011;50:266–72. doi: 10.1016/j.yjmcc.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104:2752–60. doi: 10.1182/blood-2004-04-1396. [DOI] [PubMed] [Google Scholar]
- 9.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–74. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 10.Sheridan CM, Rice D, Hiscott PS, Wong D, Kent DL. The presence of AC133-positive cells suggests a possible role of endothelial progenitor cells in the formation of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2006;47:1642–5. doi: 10.1167/iovs.05-0779. [DOI] [PubMed] [Google Scholar]
- 11.Thill M, Strunnikova NV, Berna MJ, Gordiyenko N, Schmid K, Cousins SW, et al. Late outgrowth endothelial progenitor cells in patients with agerelated macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:2696–708. doi: 10.1167/iovs.07-0955. [DOI] [PubMed] [Google Scholar]
- 12.Ingram DA, Mead LE, Moore DB, Woodard W, Fenoglio A, Yoder MC. Vessel wall-derived endothelial cells rapidly proliferate because they contain a complete hierarchy of endothelial progenitor cells. Blood. 2005;105:2783–6. doi: 10.1182/blood-2004-08-3057. [DOI] [PubMed] [Google Scholar]
- 13.Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/ progenitor cell principals. Blood. 2007;109:1801–9. doi: 10.1182/blood-2006-08-043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alvarez DF, Huang L, King JA, ElZarrad MK, Yoder MC, Stevens T. Lung microvascular endothelium is enriched with progenitor cells that exhibit vasculogenic capacity. Am J Physiol Lung Cell Mol Physiol. 2008;294:L419–30. doi: 10.1152/ajplung.00314.2007. [DOI] [PubMed] [Google Scholar]
- 15.Tan K, Lessieur E, Cutler A, Nerone P, Vasanji A, Asosingh K, et al. Impaired function of circulating CD34(+) CD45(-) cells in patients with proliferative diabetic retinopathy. Exp Eye Res. 2010;91:229–37. doi: 10.1016/j.exer.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toshner M, Voswinckel R, Southwood M, Al-Lamki R, Howard LS, Marchesan D, et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2009;180:780–7. doi: 10.1164/rccm.200810-1662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asosingh K, Aldred MA, Vasanji A, Drazba J, Sharp J, Farver C, et al. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol. 2008;172:615–27. doi: 10.2353/ajpath.2008.070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montani D, Perros F, Gambaryan N, Girerd B, Dorfmuller P, Price LC, et al. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2011;184:116–23. doi: 10.1164/rccm.201006-0905OC. [DOI] [PubMed] [Google Scholar]
- 19.Farha S, Asosingh K, Xu W, Sharp J, George D, Comhair S, et al. Hypoxiainducible factors in human pulmonary arterial hypertension: A link to the intrinsic myeloid abnormalities. Blood. 2011;117:3485–93. doi: 10.1182/blood-2010-09-306357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diller GP, van Eijl S, Okonko DO, Howard LS, Ali O, Thum T, et al. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation. 2008;117:3020–30. doi: 10.1161/CIRCULATIONAHA.108.769646. [DOI] [PubMed] [Google Scholar]
- 21.Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, Vasanji A, et al. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L548–54. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 22.Aldred MA, Comhair SA, Varella-Garcia M, Asosingh K, Xu W, Noon GP, et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182:1153–60. doi: 10.1164/rccm.201003-0491OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amos-Landgraf JM, Cottle A, Plenge RM, Friez M, Schwartz CE, Longshore J, et al. X chromosome-inactivation patterns of 1,005 phenotypically unaffected females. Am J Hum Genet. 2006;79:493–9. doi: 10.1086/507565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1584–95. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woodfin A, Voisin MB, Nourshargh S. PECAM-1: A multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol. 2007;27:2514–23. doi: 10.1161/ATVBAHA.107.151456. [DOI] [PubMed] [Google Scholar]
- 26.Wang JW, Eikenboom J. Von Willebrand disease and Weibel-Palade bodies. Hamostaseologie. 2010;30:150–5. [PubMed] [Google Scholar]
- 27.Mathew R, Huang J, Gewitz MH. Pulmonary artery hypertension: Caveolin-1 and eNOS interrelationship: A new perspective. Cardiol Rev. 2007;15:143–9. doi: 10.1097/01.crd.0000249381.49138.b9. [DOI] [PubMed] [Google Scholar]
- 28.Majkova Z, Toborek M, Hennig B. The role of caveolae in endothelial cell dysfunction with a focus on nutrition and environmental toxicants. J Cell Mol Med. 2010;14:2359–70. doi: 10.1111/j.1582-4934.2010.01064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nielsen JS, McNagny KM. CD34 is a key regulator of hematopoietic stem cell trafficking to bone marrow and mast cell progenitor trafficking in the periphery. Microcirculation. 2009;16:487–96. doi: 10.1080/10739680902941737. [DOI] [PubMed] [Google Scholar]
- 30.Yoder MC, Ingram DA. The definition of EPCs and other bone marrow cells contributing to neoangiogenesis and tumor growth: Is there common ground for understanding the roles of numerous marrow-derived cells in the neoangiogenic process? Biochim Biophys Acta. 2009;1796:50–4. doi: 10.1016/j.bbcan.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duong HT, Erzurum SC, Asosingh K. Pro-angiogenic hematopoietic progenitor cells and endothelial colony-forming cells in pathological angiogenesis of bronchial and pulmonary circulation. Angiogenesis. 2011;14:411–22. doi: 10.1007/s10456-011-9228-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hur J, Yoon CH, Kim HS, Choi JH, Kang HJ, Hwang KK, et al. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol. 2004;24:288–93. doi: 10.1161/01.ATV.0000114236.77009.06. [DOI] [PubMed] [Google Scholar]
- 33.Yoon CH, Hur J, Park KW, Kim JH, Lee CS, Oh IY, et al. Synergistic neovascularization by mixed transplantation of early endothelial progenitor cells and late outgrowth endothelial cells: the role of angiogenic cytokines and matrix metalloproteinases. Circulation. 2005;112:1618–27. doi: 10.1161/CIRCULATIONAHA.104.503433. [DOI] [PubMed] [Google Scholar]
- 34.Erzurum S, Rounds SI, Stevens T, Aldred M, Aliotta J, Archer SL, et al. Strategic plan for lung vascular research: An NHLBI-ORDR Workshop Report. Am J Respir Crit Care Med. 2010;182:1554–62. doi: 10.1164/rccm.201006-0869WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevens T. Molecular and cellular determinants of lung endothelial cell heterogeneity. Chest. 2005;128:558S–64S. doi: 10.1378/chest.128.6_suppl.558S. [DOI] [PubMed] [Google Scholar]
- 36.King J, Hamil T, Creighton J, Wu S, Bhat P, McDonald F, et al. Structural and functional characteristics of lung macro- and microvascular endothelial cell phenotypes. Microvasc Res. 2004;67:139–51. doi: 10.1016/j.mvr.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 37.Jonigk D, Golpon H, Bockmeyer CL, Maegel L, Hoeper MM, Gottlieb J, et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am J Pathol. 2011;179:167–79. doi: 10.1016/j.ajpath.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A. 2007;104:1342–7. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest. 1998;101:927–34. doi: 10.1172/JCI1910. [DOI] [PMC free article] [PubMed] [Google Scholar]