

Abstract
Proteinase inhibitor I (Inh I) and proteinase inhibitor II (Inh II) from potato tubers are effective proteinase inhibitors of chymotrypsin and trypsin. Inh I and Inh II were shown to suppress irradiation-induced transformation in mouse embryo fibroblasts suggesting that they possess anticarcinogenic characteristics. We have previously demonstrated that Inh I and Inh II could effectively block UV irradiation-induced activation of transcription activator protein 1 (AP-1) in mouse JB6 epidermal cells, which mechanistically may explain their anticarcinogenic actions. In the present study, we investigated the effects of Inh I and Inh II on the expression and composition pattern of the AP-1 complex following stimulation by UV B (UVB) irradiation in the JB6 model. We found that Inh I and Inh II specifically inhibited UVB-induced AP-1, but not NFκB, activity in JB6 cells. Both Inh I and Inh II up-regulated AP-1 constituent proteins, JunD and Fra-2, and suppressed c-Jun and c-Fos expression and composition in bound AP-1 in response to UVB stimulation. This regulation of the AP-1 protein compositional pattern in response to Inh I or Inh II may be critical for the inhibition of UVB-induced AP-1 activity by these agents found in potatoes.
Plants typically respond to environmental stress such as insect herbivory, mechanical damage, and ultraviolet (UV) irradiation by inducing defense-related proteins (1, 2). Among these proteins, proteinase inhibitors I (Inh I) and II (Inh II) isolated from potato leaves are two well-characterized chymotrypsin inhibitors. Both proteins accumulate in potato and tomato leaves and are involved in signal transduction pathways in the plant's protective response against environmental herbivores and pathogens (3–5). In addition, these inhibitors have also been reported to have an inhibitory effect on irradiation-induced cell transformation in mammalian cells (6). We reported previously that both Inh I and Inh II block UVB- or UVC-induced transcription activator protein 1 (AP-1) activity in mouse JB6 cells (7). Considering the critical role that AP-1 activation plays in malignant cellular transformation and tumorigenesis (8–15), the inhibitory effects of these compounds on AP-1 activation may explain their reported antitumor effects. The precise mechanism explaining the inhibition is, however, unclear.
AP-1 is an inducible eukaryotic transcription factor composed of products of the jun and fos oncogene families that form Jun–Jun or Jun–Fos dimers (16, 17). When stimulated, AP-1 binds to specific transactivation promoter regions or TREs (12-O-tetradecanoylphorbol 13-acetate response elements) on DNA to induce transcription of genes involved in cell proliferation, metastasis, and metabolism (18). Many positive and negative components are involved in the regulation of AP-1 activity. Among these regulators, mitogen-activated protein kinase (MAP kinase) signaling pathways are common mediators of AP-1 function (19). However, we have previously observed that both Inh I and Inh II fail to block extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs), or p38 kinase, three members of the MAP kinase family (7). This observation suggested that inhibition of AP-1 activity by Inh I and Inh II is mediated by a mechanism independent of the MAP kinase pathways.
Nuclear factor κB (NFκB) is another eukaryotic transcriptional factor that appears to be critically involved in regulating the expression of a variety of genes that participate in the inflammatory response and suppression of apoptosis, as well as cell proliferation (20, 21). Furthermore, subunits of AP-1 and NFκB are able to “cross-talk,” and thus both transcription factors may play a role in cellular transformation (22, 23). Recent reports have indicated that both AP-1 and NFκB are involved in tumor promoter-induced progression in the human keratinocyte transformation model (24, 25).
To elucidate how the proteinase inhibitors suppress AP-1 activity and whether NFκB is involved in this mechanism, we investigated changes in both AP-1 and NFκB activities and DNA-binding capacities in response to cellular treatment with Inh I and Inh II and exposure to UVB, respectively. We also determined changes in the AP-1 protein composition after treatment with the Inh I and Inh II and subsequent exposure to UVB in JB6 cells. In this paper, we report that both Inh I and Inh II block UVB-induced activities of AP-1, but not NFκB. Additionally, the regulation of the AP-1 protein compositional pattern by Inh I and Inh II appears to be involved in the inhibition of AP-1 activity.
Materials and Methods
Cell Culture and Reagents.
AP-1 or NFκB luciferase reporter plasmid stably transfected mouse epidermal JB6 P+1-1 and the JB6 mouse epidermal cell line Cl 41 were constructed as previously reported (7, 26). The cells were cultured in monolayers at 37°C under a 5% CO2/95% air atmosphere in Eagle's minimal essential medium (MEM) containing 5% fetal bovine serum (FBS), 2 mM l-glutamine, and 25 μg/ml gentamicin. FBS and MEM were from BioWhittaker (Walkersville, MD); aprotinin and leupeptin were from Sigma; rabbit polyclonal IgG against c-Jun, JunB, JunD or c-Fos, FosB, Fra-1, and Fra-2 (TransCruz Gel Supershift reagent, 200 μg/0.1 ml) were from Santa Cruz Biotechnology; and Inh I and Inh II were isolated from potato tubers and characterized as described previously (3, 4).
Luciferase Assay for AP-1 or NFκB Activity.
Confluent monolayers of JB6 P+1-1 cells were trypsinized, and viable cells (8 × 103), suspended in 100 μl of 5% FBS in MEM, were added to each well of a 96-well plate. Plates were incubated for 24 h at 37°C in a humidified atmosphere of 5% CO2/95% air. The cells were then starved by culturing them in 0.1% FBS in MEM for 24 h before treatment for 30 min with or without Inh I and Inh II at the concentrations indicated. The cells were then exposed to UVB (4 kJ/m2), cultured for an additional 12 h and extracted with lysis buffer (0.1 M potassium phosphate buffer, pH 7.8/1% Triton X-100/1 mM DTT/2 mM EDTA), and the luciferase activity was measured with a luminometer (Monolight 2010). Results are expressed as relative AP-1 activity ± standard deviation.
Isolation of Nuclear Proteins and Gel-Shift and Gel-Supershift Assays.
Nuclear protein extracts were prepared from JB6 Cl 41 cells, and electrophoretic mobility-shift and supershift assays were performed essentially as reported previously (27–29). Briefly, Cl 41 cells were cultured in 10-cm dishes and starved in 0.1% FBS in MEM at 37°C in a 5% CO2 incubator as described previously. After a 24-h starvation, the cells were treated for 30 min with various concentrations of Inh I or Inh II as indicated and then exposed to UVB (4 kJ/m2) and incubated another 12 h. The cells were then harvested and disrupted in 500 μl of lysis buffer A (25 mM Hepes, pH 7.8/50 mM KCl/0.5% Nonidet P-40/100 μM DTT containing 10 μg/ml leupeptin, 25 μg/ml aprotinin, and 1 mM PMSF). After a 1-min centrifugation (16,000 × g, 4°C), the nuclei-containing pellet was washed once with 500 μl of buffer B (buffer A without Nonidet P-40), resuspended in 150 μl of extraction buffer [buffer B but with 500 mM KCl and 10% (vol/vol) glycerol], and shaken for 30 min at 4°C. The resulting nuclear extracts were stored at −70°C until analysis. The DNA-binding reaction (for electrophoretic mobility-shift assay) was carried out at room temperature for 30 min in a mixture containing 4 μg of nuclear proteins, 1 μg of poly(deoxyinosinic acid⋅deoxycytidylic acid) [poly(dI⋅dC)], and 15,000 cpm of a 32P-labeled double-stranded AP-1 oligonucleotide (5′-CGCTTGATGAGTCAGCCGGAA-3′) or NFκB oligonucleotide (5′-TAGTTGAGGGGACTTTCCCAGGCA-3′). The samples were then fractionated through a 5% polyacrylamide gel in 0.5× TBE (1× TBE is 90 mM Tris/64.6 mM boric acid/2.0 mM EDTA, pH 8.0). Electrophoretic mobility-supershift assays of AP-1 were performed by preincubating 4 μg of nuclear protein with 4 μg of the specific antibody against c-Jun, JunB, JunD, c-Fos, FosB, Fra-1, or Fra-2 at 4°C for 2 h and then processing as described above. Completed gels were then dried and analyzed by using the Storm 840 Phospho-Image System (Molecular Dynamics).
Western Immunoblotting.
Cl 41 cells were starved and treated as described for electrophoretic mobility-shift assay. The cells were lysed with 1× SDS sample buffer, heated at 95°C for 5 min, and fractionated by 8% polyacrylamide/SDS gel electrophoresis. Proteins were then transferred to Immobilon membranes (Millipore) and probed with antibodies against c-Jun, JunB, JunD, c-Fos, FosB, Fra-1, or Fra-2.
Antibody-bound proteins were detected by chemiluminescence (ECL, Amersham Pharmacia Biotech) and analyzed by using the Storm 840 Phospho-Image System (Molecular Dynamics).
Statistical Analysis.
Significant differences in AP-1 activity were determined by using Student's t test. The results are expressed as means ± standard deviation (SD).
Results and Discussion
Inh I and Inh II Block UVB-Induced AP-1 Activity but Increase UVB-Induced NFκB Activity.
Inh I and Inh II are effective proteinase inhibitors of chymotrypsin and trypsin (3, 4) and have been reported to display anticarcinogenic properties by suppressing irradiation-induced transformation of mouse embryo fibroblasts (6). Although the mechanisms underlying these effects are poorly understood, the inhibition of chymotrypsin-like enzymes is believed to play an important role in the reported anticarcinogenic effect (30). We have previously demonstrated that blocking AP-1 activity inhibits tumor transformation, suggesting that AP-1 activity is necessary for tumor promoter-induced transformation (10). To investigate whether Inh I or Inh II suppresses UVB-induced AP-1 activity, we used the mouse epidermal JB6 cell model, which is a well-established cell line used extensively to study tumor promotion (31–34), and a stable transfectant with the AP-1 luciferase reporter (33, 34). Our data showed that Inh I or Inh II had no significant effect on AP-1 activity in JB6 cells (Fig. 1A). However, both Inh I and Inh II markedly blocked UVB-induced AP-1 activity (P < 0.05; Fig. 1B). Neither of the inhibitors, at the tested concentrations, was toxic to the cells as indicated by the incorporation of [3H]thymidine (data not shown). These findings are also consistent with our previous work (7). The data indicated that the compounds suppressed AP-1 activity effectively with little effect on cellular metabolism.
Figure 1.

Inh I or Inh II suppresses UVB-induced AP-1 activity in a concentration-dependent manner. Stably transfected JB6 P+1-1 AP-1 luciferase reporter cells were cultured and starved as indicated in the text. After a 30-min treatment of the cells with Inh I or Inh II at the concentrations indicated, the cells were or were not exposed to UVB irradiation (4 kJ/m2) and cultured for another 12 h before harvest and determination of AP-1 activity. AP-1 activity is expressed as relative luciferase units as assessed by a luminometer. (A) Inh I or Inh II treatment resulted in slight, but insignificant (P > 0.05; mean ± SD of triplicate experiments, six wells each), increase of basal AP-1 activity. (B) UVB stimulation alone induced a 13.8- to 14.3-fold increase in AP-1 activity. Inh I (40–80 μM) or Inh II (20–80 μM) significantly inhibited AP-1 activity in response to UVB. ∗, P < 0.05; mean ± SD for triplicate experiments, six wells each.
NFκB is a dimer composed of two DNA-binding subunits, NFκB p50 and p65, which belong to the c-rel protooncogene family (20). Communication or cross-talk between AP-1 and NFκB has been reported before (23). In that report, the bZIP regions of c-Fos and c-Jun interacted with NFκB p65 through the Rel homology domain. The complex of NFκB p65 and Jun or Fos increased DNA-binding activity and biological function by both the NFκB and AP-1 response elements. In yet another study, the activation of NFκB and AP-1 was shown to be crucially involved in UV-induced expression of FasL, an apoptosis-related ligand. However, in the current experiments, both Inh I and Inh II significantly enhanced NFκB activity induced by UVB (P < 0.05; Fig. 2B). The enhancement of Inh I and Inh II on UVB-induced NFκB activity did not appear to be dose-dependent (Fig. 2B). Neither Inh I nor Inh II, at the concentrations indicated, induced a significant effect on NFκB activity in JB6 cells (Fig. 2A). The markedly different effects of Inh I and Inh II on UVB-induced AP-1 and NFκB activation suggest that UVB-activated AP-1 and NFκB transactivation occur by different mechanisms.
Figure 2.

Inh I or Inh II enhances UVB-induced NFκB activity in a concentration-independent manner. Stably transfected JB6 P+1-1 NFκB luciferase reporter cells were cultured and starved as described in the text. After a 30-min treatment of the cells with Inh I or Inh II at the concentrations indicated, the cells were or were not exposed to UVB irradiation (4 kJ/m2) and cultured for another 12 h before harvest and determination of NFκB activity. NFκB activity is expressed as relative luciferase units as assessed by a luminometer. (A) Inh I or Inh II caused a slight, but insignificant (P > 0.05; mean ± SD of triplicate experiments, six wells of each), increase in basal NFκB activity. (B) UVB stimulation induced a 15.8- to 16.8-fold increase in NFκB activity. Inh I (10–40 μM) or Inh II (10–40 μM) significantly increased UVB-induced NFκB activity. ∗, P < 0.05; mean ± SD of triplicate experiments, six wells each. Higher concentrations of Inh I or Inh II did not further enhance UVB-induced NFκB activity.
Neither Inh I nor Inh II Had an Effect on UVB-Induced AP-1 or NFκB DNA Binding.
To determine whether Inh I or Inh II has an effect on UVB-induced AP-1 or NFκB binding activity, electrophoretic mobility gel-shift assays were performed. UVB (4 kJ/m2) induced a significant increase in both AP-1 and NFκB DNA-binding activities (lane 3, Figs. 3 and 4). In both cases, the binding of the probe was effectively eliminated by adding a 10-fold excess of unlabeled AP-1 or NFκB oligonucleotide (lane 1, Figs. 3 and 4), confirming that the band was only either AP-1 or NFκB. The results also showed that Inh I and Inh II had no effect on the UVB-induced increase in AP-1 or NFκB DNA binding (Figs. 3 and 4, lanes 3, 4, and 5). These results indicate that the observed effects of Inh I and Inh II on UVB-induced AP-1 or NFκB transactivation did not occur through changes in DNA binding by AP-1 or NFκB.
Figure 3.

Inh I and Inh II do not affect UVB-induced AP-1 DNA binding. JB6 Cl 41 cells were treated, nuclear proteins were extracted, and electrophoretic mobility-shift assays were carried out as described in the text. (A) UVB strongly induces AP-1 DNA binding (lane 3), and the binding is specific as indicated by the effective competition of a 10-fold excess of the unlabeled AP-1 probe (lane 1). Inh I or Inh II has little effect on UVB-induced AP-1 DNA binding (lanes 4 and 5). (B) Densitometry analysis of A from three independent experiments.
Figure 4.

Inh I and Inh II do not affect UVB-induced NFκB DNA binding. JB6 Cl 41 cells were treated, nuclear proteins were extracted, and electrophoretic mobility-shift assays were carried out as described in the text. (A) UVB strongly induces NFκB DNA binding (lane 3), and the binding is specific as indicated by the effective competition of a 10-fold excess of the unlabeled NFκB probe (lane 1). Inh I or Inh II has little effect on UVB-induced NFκB DNA binding (lanes 4 and 5). (B) Densitometry analysis of A from three independent experiments.
The DNA transactivation potential of AP-1 and NFκB depends not only on the quantities of particular proteins present but also on their distribution patterns (22, 35, 36). Any change in the level of expression of these components leads to changes in their transactivational activity. Results of the present study suggested that Inh I and Inh II differentially affected the response of UVB-induced AP-1 transactivation by regulating the dimeric composition of AP-1 rather than the DNA-binding activity.
The Composition of UVB-Induced AP-1 Was Changed by Inh I and Inh II.
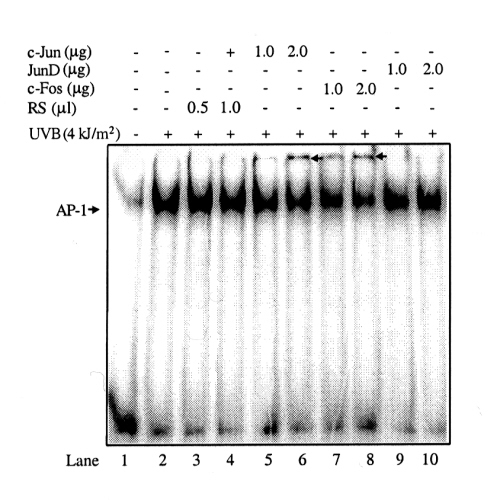
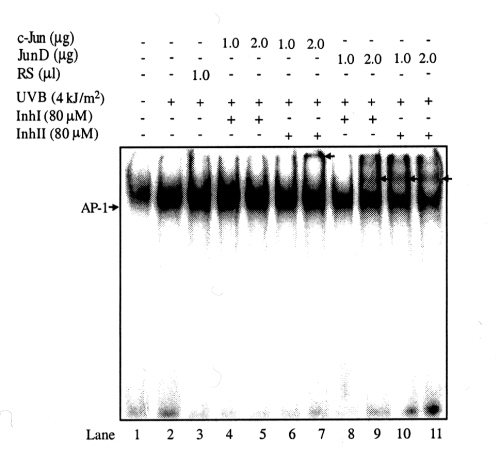
Because Inh I and Inh II inhibited UVB-induced AP-1 activity, but did not affect UVB-induced AP-1 binding to its DNA recognition site, we propose that the inhibitory effect of Inh I and Inh II on UVB-induced AP-1 transcriptional activity happens as a result of alterations in the composition of the AP-1 complex proteins. To test this hypothesis, we used electrophoretic mobility-supershift assays to identify AP-1-associated proteins that were induced by Inh I and Inh II. In this assay, the respective antibody (c-Jun, JunB, JunD, c-Fos, FosB, Fra-1, or Fra-1) deactivates its target protein, thereby preventing that protein from interacting with the AP-1 DNA-binding site. This interaction would result in a decreased signal or an extra shifted band (i.e., “supershift”), suggesting that the specific protein is an important component of AP-1. Results indicated that, in unstimulated JB6 cells, the AP-1 is composed mainly of JunD and Fra-2 proteins (Fig. 5A, lanes 5 and 9). UVB stimulation induced a significant increase in c-Jun, c-Fos (Fig. 5B, lanes 4 and 7 and Fig. 7, which is published as supplemental data on the PNAS web site, www.pnas.org), and a mild increase in JunB (Fig. 5B, lane 5) in the composition of AP-1 but a decrease in JunD and Fra-2 (Fig. 5B, lanes 6 and 10) compared with the unstimulated AP-1 composition (Fig. 5A, lanes 5 and 9). When the cells were treated for 30 min with Inh I or Inh II before UVB irradiation, the AP-1 composition pattern changed and JunD (Fig. 8, which is published as supplemental data) and Fra-2 components again were increased (Fig. 5 C and D, lanes 5 and 9) and c-Jun or c-Fos were markedly decreased (Fig. 5 C and D, lanes 3 and 6).
Figure 5.

Different AP-1 compositional patterns after UVB stimulation and Inh I or Inh II treatment as assessed by gel supershift assays. Cl 41 cells were cultured and treated, nuclear proteins were isolated, and electrophoretic mobility-supershift assays were carried out as described in the text. Negative control (lane 1) indicates untreated cells. Rabbit serum (RS) was used as an internal control because the antibodies used for the supershift assay were dissolved in rabbit serum. Arrows indicate supershifted bands. (A) Cl 41 cells without any treatment. Incubation of extracted nuclear proteins with JunD or Fra-2 antibodies induced a clearing of the AP-1 band or a supershifted band (lanes 5 and 9). (B) Cl 41 cells were treated with UVB. Incubation of extracted nuclear proteins with c-Jun or c-Fos antibody induced a strong supershifted band (lanes 4 and 7). Incubation of extracted nuclear proteins with JunB antibody also induced a slight supershifted band (lane 5). In contrast to unstimulated control cells (A), incubation of nuclear extracts with JunD or Fra-2 antibodies failed to induce clearing of the AP-1 band or a supershifted band (lanes 6 and 10). (C) Cells were treated with Inh I followed by exposure to UVB. Incubation of extracted nuclear proteins with JunD or Fra-2 antibodies once again induced a clearing and supershifted band (lanes 5 and 9). The bands induced by c-Jun or JunB antibodies can still be seen but with attenuation (lanes 3 and 4). (D) Treatment with Inh II followed by exposure to UVB shows results similar to those seen with Inh I. Rabbit serum did not induce any supershifted bands in these experiments (lane 2 of A and lane 3 of B).
Western blot analysis showed that UVB stimulation resulted in decreased expression of JunD and Fra-2 proteins compared with unstimulated control and increased expression of c-Jun, JunB, c-Fos, FosB, and Fra-1 proteins (Fig. 6). Pretreatment of cells for 30 min with Inh I or Inh II reversed the UVB-induced decrease of JunD and Fra-2. Inh I and Inh II also inhibited UVB-induced expression of c-Jun and c-Fos proteins (Fig. 6). The results indicated that the inhibitory effect of Inh I and Inh II on AP-1 transcriptional activity appears to be mediated through an alteration in the protein composition of the AP-1 complex.
Figure 6.

Various AP-1 protein compositional patterns after UVB stimulation and Inh I or Inh II treatment, assessed by Western blotting. Cl 41 cells were cultured and treated and Western blotting was carried out as indicated in the text. JunD and Fra-2 proteins were attenuated after UVB treatment and increased again when cells were treated with Inh I or Inh II before UVB. c-Jun, JunB, c-Fos, FosB, and Fra-1 proteins were increased after UVB exposure and inhibited when cells were pretreated with either Inh I or Inh II before UVB treatment. Western immunoblotting was performed three times with samples from various cell preparations. Similar results were obtained each time and a representative result is shown. β-Actin was used as internal control to ensure equal protein loading.
Inh I and Inh II have been shown to prevent x-ray irradiation-induced mammalian cell transformation (6). Our previous work suggested that the compounds suppressed tumor promoter UV-induced AP-1 activity (7). However, the mechanism explaining the inhibition of Inh I and II on AP-1 activity is poorly understood, probably because AP-1 DNA-binding affinity and transactivation potential are affected by many complicated factors.
AP-1 can be modified at the transcriptional, posttranscriptional, and posttranslational levels. These modifications alter the DNA-binding affinity and transactivation potential of AP-1 (19). Evidence showed that the MAP kinase superfamily, including ERK1/2, JNKs, and p38 kinase, mediates the expression of “immediate-early” genes, c-jun and c-fos, and the phosphorylation of the gene products, c-Jun and c-Fos, thus influencing transcriptional and posttranslational regulation of AP-1 activity, respectively (19). Our previous observation, however, did not demonstrate that Inh I and Inh II suppressed UVB-induced phosphorylation or activity of MAP kinases, including ERKs, p38 kinase, or JNKs (7), thus indicating that the inhibition of UV-induced AP-1 activity by Inh I and II might be through MAP kinase-independent pathways. Our present data show that UVB induced increases in c-Jun and c-Fos expression, but decreased JunD and Fra-2 expression. Inh I or Inh II reversed these changes in AP-1 composition. Thus, the inhibition of AP-1 activity and cell transformation by Inh I and II is probably not connected to MAP kinases or kinases upstream (e.g., protein kinase C, Ras/Raf, or MAP kinase kinases). The results suggested that the inhibition of cell transformation by the compounds might be targeted directly at the transcriptional factor AP-1, but not the MAP kinase cascades.
Several proteins can form complexes that bind to AP-1 sites. These proteins differ considerably in their ability to activate transcription of target genes. For instance, both c-Fos and Fra-1 form stable heterodimers with any of the Jun proteins, with similar DNA-binding activities and specificities, yet c-Fos has a much stronger transactivational activity (37). It was reported that elevated expression of JunD decreased cell transformation and AP-1 activity (38). In our study, AP-1 activity induced by UVB stimulation plus Inh I or Inh II had a different pattern of protein composition mainly because of the expression of JunD and Fra-2, which may be responsible for the changes observed in the AP-1 transactivation capacity.
The three Jun proteins are closely related in amino acid sequence, particularly in the region (HR-2) containing the DNA-binding domain (38). This may explain why in our experiment, when inhibited by Inh I or Inh II, the UVB-induced AP-1 complex had a high proportion of JunD or Fra-2 proteins and did not show a decrease in its DNA-binding ability. These results are in consensus with a significant report in which JunD partially suppressed cell transformation by an activated ras gene, whereas c-Jun cooperated with ras to transform cells (35). Others have reported that the inhibition of proliferation by vitamin D is accompanied by an increase of JunD in the AP-1 complex (36). Sonoda et al. (39) showed that reversion of the malignant tumor phenotype to the normal one by oxamflatin is by means of induction of JunD. These data indicated that two closely related transcription factor proteins could function in an opposing manner. Although the Jun family members can bind to the same DNA sequences, they have been shown to do so with different affinities and thus may elicit different responses with regard to expression of target genes (40–42).
Taken together, the results presented in this study demonstrate that Inh I and Inh II specifically inhibit UVB-induced AP-1 activity by regulating its protein compositional pattern. The up-regulation of JunD and Fra-2 by these compounds was critical for the down-regulation of UVB-induced AP-1 activity to occur.
Supplementary Material
Acknowledgments
We thank Ms. Andria Hansen for secretarial assistance. This work was supported by The Hormel Foundation and grants from the National Institutes of Health (CA77646, CA81064, and CA74916) and the American Institute for Cancer Research (99A062).
Abbreviations
- Inh I
proteinase inhibitor I
- Inh II
proteinase inhibitor II
- AP-1
transcription activator protein 1
- MAP kinase
mitogen-activated protein kinase
- NFκB
nuclear factor κB
- UVB
ultraviolet B
References
- 1.Bergey D R, Howe G A, Ryan C A. Proc Natl Acad Sci USA. 1996;93:12053–12058. doi: 10.1073/pnas.93.22.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conconi A, Smerdon M J, Howe G A, Ryan C A. Nature (London) 1996;383:826–829. doi: 10.1038/383826a0. [DOI] [PubMed] [Google Scholar]
- 3.Melville J C, Ryan C A. J Biol Chem. 1972;247:3445–3453. [PubMed] [Google Scholar]
- 4.Bryant J, Green T R, Gurusaddaiah T, Ryan C A. Biochemistry. 1976;15:3418–3424. doi: 10.1021/bi00661a004. [DOI] [PubMed] [Google Scholar]
- 5.Stratmann J W, Ryan C A. Proc Natl Acad Sci USA. 1997;94:11085–11089. doi: 10.1073/pnas.94.20.11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Billings P C, Morrow A R, Ryan C A, Kennedy A R. Carcinogenesis. 1989;10:687–691. doi: 10.1093/carcin/10.4.687. [DOI] [PubMed] [Google Scholar]
- 7.Huang C, Ma W-Y, Ryan C A, Dong Z. Proc Natl Acad Sci USA. 1997;94:11957–11962. doi: 10.1073/pnas.94.22.11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adler V, Pincus M R, Polotskaya A, Montano X, Friedman F K, Ronai Z. J Biol Chem. 1996;271:23304–23309. doi: 10.1074/jbc.271.38.23304. [DOI] [PubMed] [Google Scholar]
- 9.Lamb R F, Hennigan R F, Turnbull K, Katsanakis K D, MacKenzie E D, Birnie G D, Ozanne B W. Mol Cell Biol. 1997;17:963–976. doi: 10.1128/mcb.17.2.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong Z, Birrer M J, Watts R G, Matrisian L M, Colburn N H. Proc Natl Acad Sci USA. 1994;91:609–613. doi: 10.1073/pnas.91.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang C, Ma W-Y, Dawson M I, Rincon M, Flavell R A, Dong Z. Proc Natl Acad Sci USA. 1997;94:5826–5830. doi: 10.1073/pnas.94.11.5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong Z, Huang C, Brown R E, Ma W-Y. J Biol Chem. 1997;272:9962–9970. doi: 10.1074/jbc.272.15.9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Domann F E, Levy J P, Birrer M J, Bowden G T. Cell Growth Differ. 1994;5:9–16. [PubMed] [Google Scholar]
- 14.Huang C, Ma W-Y, Hanenberger D, Cleary M P, Bowden G T, Dong Z. J Biol Chem. 1997;272:26325–26331. doi: 10.1074/jbc.272.42.26325. [DOI] [PubMed] [Google Scholar]
- 15.Agadir A, Chen G, Bost F, Li Y, Mercola D, Zhang X. J Biol Chem. 1999;274:29779–29785. doi: 10.1074/jbc.274.42.29779. [DOI] [PubMed] [Google Scholar]
- 16.Angel P, Karin M. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 17.Curran T, Franza B R., Jr Cell. 1988;55:395–397. doi: 10.1016/0092-8674(88)90024-4. [DOI] [PubMed] [Google Scholar]
- 18.Angel P, Imagawa M, Chiu R, Stein B, Imbra R J, Rahmsdorf H J, Jonat C, Herrlich P, Karin M. Cell. 1987;49:729–739. doi: 10.1016/0092-8674(87)90611-8. [DOI] [PubMed] [Google Scholar]
- 19.Karin M. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh S, May M J, Kopp E B. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 21.Baldwin A. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 22.Denhardt D T. Crit Rev Oncog. 1996;7:261–291. doi: 10.1615/critrevoncog.v7.i3-4.70. [DOI] [PubMed] [Google Scholar]
- 23.Stein B, Baldwin A S, Jr, Ballard D W, Greene W C, Angel P, Herrlich P. EMBO J. 1993;12:3879–3891. doi: 10.1002/j.1460-2075.1993.tb06066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J-J, Westergaard C, Ghosh P, Colburn N H. Cancer Res. 1997;57:3569–3576. [PubMed] [Google Scholar]
- 25.Li J-J, Rhim J S, Schlegel R, Vousden K H, Colburn N H. Oncogene. 1998;16:2711–2721. doi: 10.1038/sj.onc.1201798. [DOI] [PubMed] [Google Scholar]
- 26.Huang C, Schmid P C, Ma W-Y, Schmid H H O, Dong Z. J Biol Chem. 1997;272:4187–4194. doi: 10.1074/jbc.272.7.4187. [DOI] [PubMed] [Google Scholar]
- 27.Chen W, Borchers A H, Dong Z, Powell M B, Bowden G T. J Biol Chem. 1998;273:32176–32181. doi: 10.1074/jbc.273.48.32176. [DOI] [PubMed] [Google Scholar]
- 28.Rosenberger S F, Bowden G T. Oncogene. 1996;12:2301–2308. [PubMed] [Google Scholar]
- 29.Huang C, Ma W-Y, Maxiner A, Sun Y, Dong Z. J Biol Chem. 1999;274:12229–12235. doi: 10.1074/jbc.274.18.12229. [DOI] [PubMed] [Google Scholar]
- 30.Kennedy A R. Pharmacol Ther. 1998;78:167–209. doi: 10.1016/s0163-7258(98)00010-2. [DOI] [PubMed] [Google Scholar]
- 31.Dong Z, Lavorsky V, Colburn N H. Carcinogenesis. 1995;16:749–756. doi: 10.1093/carcin/16.4.749. [DOI] [PubMed] [Google Scholar]
- 32.Sun Y, Dong Z, Nakamura K, Colburn N H. FASEB J. 1993;7:944–950. doi: 10.1096/fasebj.7.10.8344492. [DOI] [PubMed] [Google Scholar]
- 33.Huang C, Ma W-Y, Bowden G T, Dong Z. J Biol Chem. 1996;271:31262–32168. doi: 10.1074/jbc.271.49.31262. [DOI] [PubMed] [Google Scholar]
- 34.Huang C, Ma W-Y, Dong Z. Oncogene. 1997;14:1945–1954. doi: 10.1038/sj.onc.1201056. [DOI] [PubMed] [Google Scholar]
- 35.Pfarr C M, Mechta F, Spyrou G, Lallemand D, Carillo S, Yaniv M. Cell. 1994;76:747–760. doi: 10.1016/0092-8674(94)90513-4. [DOI] [PubMed] [Google Scholar]
- 36.Lasky S R, Iwata K, Rosmarin A G, Caprio D G, Maizel A L. J Biol Chem. 1995;270:19676–19679. doi: 10.1074/jbc.270.34.19676. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki T, Okuno H, Yoshida T, Endo T, Nishina H, Iba H. Nucleic Acids Res. 1992;19:5537–5542. doi: 10.1093/nar/19.20.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryder K, Lanahan A, Perez-Alburene E, Nathans D. Proc Natl Acad Sci USA. 1989;86:1500–1503. doi: 10.1073/pnas.86.5.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonoda H, Nishida K, Yokayuki T, Ohtani M, Sugita K. Oncogene. 1996;13:143–149. [PubMed] [Google Scholar]
- 40.Doucas V, Spyrou G, Yaniv M. EMBO J. 1991;10:2237–2245. doi: 10.1002/j.1460-2075.1991.tb07760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng T, Karin M. Gene Dev. 1993;7:479–490. doi: 10.1101/gad.7.3.479. [DOI] [PubMed] [Google Scholar]
- 42.Ryseck R P, Kovary K, Bravo R. Oncogene. 1990;5:1091–1093. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}