

Abstract
NO synthases are widely distributed in the lung and are extensively
involved in the control of airway and vascular homeostasis. It is
recognized, however, that the O2-rich environment of the
lung may predispose NO toward toxicity. These Janus faces of NO are
manifest in recent clinical trials with inhaled NO gas, which has shown
therapeutic benefit in some patient populations but increased morbidity
in others. In the airways and circulation of humans, most NO
bioactivity is packaged in the form of S-nitrosothiols
(SNOs), which are relatively resistant to toxic reactions with
O2/O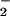 . This finding has led to the
proposition that channeling of NO into SNOs may provide a natural
defense against lung toxicity. The means to selectively manipulate the
SNO pool, however, has not been previously possible. Here we report on
a gas, O-nitrosoethanol (ENO), which does not react with
O2 or release NO and which markedly increases the
concentration of indigenous species of SNO within airway lining fluid.
Inhalation of ENO provided immediate relief from hypoxic pulmonary
vasoconstriction without affecting systemic hemodynamics. Further, in a
porcine model of lung injury, there was no rebound in cardiopulmonary
hemodynamics or fall in oxygenation on stopping the drug (as seen with
NO gas), and additionally ENO protected against a decline in cardiac
output. Our data suggest that SNOs within the lung serve in matching
ventilation to perfusion, and can be manipulated for therapeutic gain.
Thus, ENO may be of particular benefit to patients with pulmonary
hypertension, hypoxemia, and/or right heart failure, and may offer a
new therapeutic approach in disorders such as asthma and cystic
fibrosis, where the airways may be depleted of SNOs.
. This finding has led to the
proposition that channeling of NO into SNOs may provide a natural
defense against lung toxicity. The means to selectively manipulate the
SNO pool, however, has not been previously possible. Here we report on
a gas, O-nitrosoethanol (ENO), which does not react with
O2 or release NO and which markedly increases the
concentration of indigenous species of SNO within airway lining fluid.
Inhalation of ENO provided immediate relief from hypoxic pulmonary
vasoconstriction without affecting systemic hemodynamics. Further, in a
porcine model of lung injury, there was no rebound in cardiopulmonary
hemodynamics or fall in oxygenation on stopping the drug (as seen with
NO gas), and additionally ENO protected against a decline in cardiac
output. Our data suggest that SNOs within the lung serve in matching
ventilation to perfusion, and can be manipulated for therapeutic gain.
Thus, ENO may be of particular benefit to patients with pulmonary
hypertension, hypoxemia, and/or right heart failure, and may offer a
new therapeutic approach in disorders such as asthma and cystic
fibrosis, where the airways may be depleted of SNOs.
The airways and blood vessels of the lung, the nerves that innervate these structures, and the cells that reside within them are enriched in three major isoforms of NO synthase (NOS; refs. 1 and 2). A high level of NOS expression is already evident in the developing fetus and plays a key role in the transition from fetal to adult circulation (3). Vascular NOSs function to maintain normoxic pulmonary vascular tone and to counter hypoxic pulmonary vasoconstriction; NOS up-regulation by hypoxia further serves to protect against pulmonary hypertension and right heart failure (4). NOSs may have additional homeostatic roles in the airway, including control of bronchial tone, mucus secretion, sodium/water permeability, ciliary motility, and in defense against pathogens and pollutants.
There is now considerable evidence to indicate that high concentrations
of NO can be toxic (1, 5–7) and that O2-rich
environments such as the lung are particularly predisposed to its
potentially mutagenic (8), proapoptotic (9) and proinflammatory
properties (5). These findings are well rationalized by the chemical
reactions of NO with O2 and reactive oxygen
species, which yield higher oxides of nitrogen
(NOx) that can cause inflammation, hemorrhage,
and edema (1). In particular, the airways of subjects administered
inhaled NO gas reveal the same footprints of oxidative injury that are
found in animals and humans exposed to NOx or
hyperoxia (10, 11), and various NOx have been
implicated in pollutant-induced asthma (12) and in “silo fillers'
lung” (a potentially fatal disorder; ref. 13). However,
endogenous NOSs do not generate high levels of free NO in
the respiratory system. Rather, it has been shown that the NO produced
is complexed with thiols to form a major reservoir of NO-related
bioactivity in the form of S-nitrosothiol (SNO; ref. 14),
which is resistant to potentially toxic reactions with
O2/O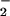 . It has not been
previously possible, however, to delineate the exact roles of SNOs in
vascular or airway homeostasis or to assign specific functions to NO
vs. SNO.
. It has not been
previously possible, however, to delineate the exact roles of SNOs in
vascular or airway homeostasis or to assign specific functions to NO
vs. SNO.
The major therapeutic challenge in diseases complicated by right heart failure, hypoxemia, and/or pulmonary hypertension is to lower pulmomary vascular resistance (PVR) without decreasing left ventricular function or systemic arterial pressure. Inhaled NO gas is the first agent to meet these therapeutic objectives, and it has provided dramatic benefit to neonates with pulmonary hypertension (15). The gas is preferentially delivered to well-ventilated areas of the lung, thereby coupling ventilation (V) to perfusion (Q), and it acts as a selective pulmonary vasodilator (16). However, inhaled NO therapy has some drawbacks: impairment of renal function (17), methemoglobinemia (18), left heart failure (19), and perhaps intraventricular cerebral hemorrhage (20) can complicate therapy, and a “rebound” increase in pulmonary artery pressure (PAP) on discontinuing the gas is common and may result in cardiovascular collapse (21). In addition, the usage of NO gas has been tempered by a worrisome increase in morbidity and decrease in ventilator-free survival (the percentage of patients alive and off ventilatory support) in a large multicenter trial of patients with acute lung injury and by similar concerns raised in post hoc analyses of other studies (17, 22).
We reasoned that the ideal drug would more closely simulate NOS
activity by transforming NO groups into SNOs (23) (and thereby avoid
the potentially toxic reactions with
O2/O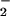 ), and that it should be
a gas (to meet the desired V/Q matching and pulmonary/systemic
activity quotients). We established a screen for potential compounds by
using the following criteria: (i) high volatility;
(ii) resistance to decomposition in the presence of
O2; (iii) reaction with glutathione to
form S-nitrosoglutathione (GSNO); (iv) limited
ability to oxidize hemoglobin; and (v) biocompatibility.
O-nitrosoethanol (ENO) fulfilled these criteria. In
particular, ENO can be synthesized as a liquid but easily partitions
into the gas phase because of its low boiling point (17°C).
Heterolytic cleavage of ENO (NO+ transfer
chemistry) is strongly favored over the homolytic mechanism of
decomposition that generates NO. Consequently, ENO is stable in 100%
O2 and does not generate
NOx. Rather ENO reacts preferentially with
nucleophiles, of which the most prevalent in the lung are the sulfurs
of glutathione and proteins. S-nitrosothiols including GSNO
are thus predicted to be the major products of ENO decomposition in
lung tissues.
), and that it should be
a gas (to meet the desired V/Q matching and pulmonary/systemic
activity quotients). We established a screen for potential compounds by
using the following criteria: (i) high volatility;
(ii) resistance to decomposition in the presence of
O2; (iii) reaction with glutathione to
form S-nitrosoglutathione (GSNO); (iv) limited
ability to oxidize hemoglobin; and (v) biocompatibility.
O-nitrosoethanol (ENO) fulfilled these criteria. In
particular, ENO can be synthesized as a liquid but easily partitions
into the gas phase because of its low boiling point (17°C).
Heterolytic cleavage of ENO (NO+ transfer
chemistry) is strongly favored over the homolytic mechanism of
decomposition that generates NO. Consequently, ENO is stable in 100%
O2 and does not generate
NOx. Rather ENO reacts preferentially with
nucleophiles, of which the most prevalent in the lung are the sulfurs
of glutathione and proteins. S-nitrosothiols including GSNO
are thus predicted to be the major products of ENO decomposition in
lung tissues.
Methods
Synthesis of ENO Gas.
ENO can be synthesized from the action of sodium nitrite on ethanol in cold sulfuric acid (24), and can be stored in ethanol at 4°C. Lower concentrations of ENO can be purchased from Aldrich. An ENO gas admixture was generated by passing either O2 or N2 through a Fisher Milligan (Springfield, NJ) gas washer containing ethyl nitrite in ethanol solution. The concentrations of ENO delivered by the system were varied by changing the solution concentrations (or flow rates) and were verified in a test lung (Michigan Instruments, Grand Rapids, MI) in line with a ventilator (see text for further details).
GCMS Analysis of ENO Gas.
Gas samples were analyzed by using a Hewlett–Packard 5890 II gas chromatography system coupled to a 5972 mass spectrometer. Ten-microliter samples were injected, via a capillary injector, onto a 30 m 0.32-mm i.d. GS-Q column (J & W Scientific, Folsom, CA). GC analyses were run at 150°C under helium. The GC column eluate was analyzed by negative ion mass spectrometry. Data were collected by using the Hewlett–Packard chemstation software package, with mass spectrometer operating in scan mode over the range of 10 to 150 m/z.
SNO Formation in Airways.
ENO or NO was added to the inhaled gas of neonatal pigs breathing room air in three randomized doses. Airway-lining fluid was sampled by bronchoscopy from second order bronchial airways after inhalation of drug for 5 min. With each drug dose, 2 ml of 10 mM phosphate/100 μM diethylenetriaminepentaacetic acid, pH 7.4, was injected, and aspirates were then assayed immediately for SNO. SNO concentrations were normalized to protein content, as measured by the Bradford method (Pierce).
Measurement of S-nitrosthiols.
SNO formation was assessed both by the Saville method (in vitro only) and by means of the Sievers Nitric Oxide Analyzer system, using the chemical reduction chamber designed for liquid analysis. Samples were added to a reduction system containing 100 μM cysteine and 1 μM CuCl in a 6-ml volume (25); this solution was made fresh for each assay.
Porcine Model of Transient Pulmonary Hypertension.
Neonatal pigs (1–2 weeks old) were instrumented and ventilated as previously described (26). A variety of parameters were monitored, including pulmonary PAP, systemic arterial pressure, cardiac output (CO), partial pressure of oxygen in the systemic arterial circuit (PaO2), systemic vascular resistance (SVR), and PVR. Pulmonary hypertension was induced by lowering the fraction of inspired O2 (FiO2) to 0.14. Under these conditions, the PAP increased by ≈50%. Hemodynamic measurements were retaken after 5 min of ENO breathing. ENO was then stopped, and the low FiO2 was maintained for 4 min, at which point the hemodynamic measurements were repeated. The FiO2 was then increased to 0.21. This procedure was repeated three times by using different doses of ENO in randomized order.
Porcine Model of Lung Injury-Induced Pulmonary Hypertension.
Pulmonary hypertension was induced in neonatal pigs, instrumented and ventilated on 100% O2 as described above, by repeated saline lavage (to remove surfactant). Lavage was stopped once the PaO2 had fallen below 100 mmHg (1 mmHg = 133 Pa) and had remained there for 30 min. Baseline measurements were then taken, and either no gas (control), ENO (≈5–10 ppm), or NO (20 ppm followed by 5 ppm) was added to the inhaled gas for 2 hr. At this point, NO or ENO was discontinued, and hemodynamic measurements and blood gases were taken every 5 min for a total 20 min (while the animals continued to breath 100% O2).
Results and Discussion
We devised a simple method to deliver ENO as an inhaled gas admixture. ENO(l) was added to ethanol in a Fisher Milligan gas washer. Nitrogen (or oxygen; 0.6 liters/min) was passed through the solution, saturating the gas, and then blended in a ventilator to produce the inspired gas admixture (6 liters/min; see Methods for details). The concentration of ENO in the gas phase was easily adjusted by altering its solution concentration (Fig. 1A). GCMS analysis of the gas output from the ventilator revealed the absence of NOx and virtually undetectable levels of NO (Fig. 1B). When introduced into an aqueous solution, ENO decomposed to ethanol and nitrite. In the added presence of 0.5 mM glutathione—to simulate dissolution into human airway lining fluid (1)—ENO generated GSNO stoichiometrically (i.e., with 100% yield; Fig. 1C). In contrast, NO generates GSNO with lower yield (Fig. 1C). These data indicate that ENO and NO undergo alternative chemical reactions: ENO directly transnitrosates glutathione to form GSNO whereas NO generates GSNO and nitrite through the intermediacy of N2O3 (27)—a potentially toxic reaction channel.
Figure 1.

ENO is a gaseous nitrosant. (A) Nitrogen gas (0.6 liters/min) was passed through a Fisher Milligan gas washer containing selected concentrations of ENO (dissolved in ethanol) and then blended with a room air gas flow (6 liters/min) in a pediatric ventilator. Samples were collected and analyzed by GCMS for ENO content. Data are the mean ± SD of three separate experiments. (B) Mass spectrometry trace of gas chromatography eluate. A gas sample from the ventilator was loaded onto a GS-Q gas chromatography column at 60°C, and column effluent was monitored by negative ion mass spectrometry. ENO elution can be followed at 30 and 46 atomic mass units (amu; the O-nitroso bond is cleaved in the ionization process). The 30-amu trace is shown. The large peak at 3.2 min represents ENO and the very small peak at 1.7 min represents NO. Examination of the column effluent at other atomic masses revealed the complete absence of NO2. (C) The ability of ENO and NO to nitrosate 0.5 mM glutathione (equivalent to airway concentrations) was assessed in vitro. The SNO yield is 0.5 SNO per NO added—in keeping with the nitrosation via a N2O3 intermediate (27)—and ≈1.0 SNO per ENO added, indicating direct transnitrosation of glutathione.
We began by assessing the effect of ENO inhalation on resting pulmonary and systemic hemodynamics and on PaO2, in piglets breathing room air (21% O2). The results in Table 1 show that ENO had no significant effect on PaO2, mean blood pressure (MnBP), or SVR. There was, however, a reduction in PAP at the highest dose (0.125%). In other words, ENO shows a selective, albeit modest, effect on pulmonary vascular tone in normal animals—a response suggestive of potent vasodilator activity because the pulmonary vascular bed is already close to maximally dilated under resting conditions and, therefore, relatively insensitive to vasodilators.
Table 1.
ENO administration in neonatal pigs (21% O2)
| Parameter | ENO % | Pre-compound | Compound | Post-compound |
|---|---|---|---|---|
| PAP, mmHg | 0.0025 | 12.4 (1.1) | 11.7 (1.2) | 13.3 (1.3) |
| 0.025 | 12 (1.1) | 10 (1.2) | 12.2 (1.6) | |
| 0.125 | 12.1 (1) | 9.8 (0.9)* | 13.7 (1.3) | |
| PVR, dynes/s/10−5 cm | 0.0025 | 3,004.1 (340.8) | 2,961.3 (295.9) | 3,500.4 (376.8) |
| 0.025 | 2,893.9 (322.7) | 2,628.4 (286.2) | 3,301.2 (440.8) | |
| 0.125 | 2,638.9 (314.6) | 2,409.1 (288.5) | 3,354.2 (384.2) | |
| PaO2, mmHg | 0.0025 | 74.2 (2.3) | 75.7 (3.4) | 74.5 (2.7) |
| 0.025 | 71 (1.9) | 73.7 (2.7) | 74.7 (0.8) | |
| 0.125 | 70 (2.4) | 72.5 (2.1) | 70 (5.5) | |
| MnBP, mmHg | 0.0025 | 48.5 (2.4) | 47.5 (3.4) | 47.5 (3.7) |
| 0.025 | 48.2 (3) | 48.7 (4.3) | 45.5 (4.1) | |
| 0.125 | 51.7 (4.3) | 47.5 (3.4) | 50.5 (4.4) | |
| SVR, dynes/s/10−5 cm | 0.0025 | 12,122.3 (1451) | 12,579.4 (1447.2) | 12,944.4 (1540.2) |
| 0.025 | 12,213.7 (1508.5) | 13,084.4 (1365.4) | 12,643.5 (1353.6) | |
| 0.125 | 11,342.8 (1442.6) | 11,676.4 (1268.6) | 12,316.9 (1323.3) | |
| CO, ml/min | 0.0025 | 438.8 (139.4) | 412.9 (115.2) | 410.4 (105.6) |
| 0.025 | 428.6 (128.3) | 397.7 (103.6) | 402.5 (103.4) | |
| 0.125 | 459.5 (140.6) | 429.7 (137.2) | 442.6 (140.4) |
Values are shown as mean (standard error). *, P < 0.05, as compared with pre-compound value.
Next we examined the effect of ENO in a piglet model of hypoxic pulmonary hypertension with two objectives in mind. First, to explore whether it was capable of reversing the increases in PAP and PVR and, second, to determine whether the ENO effect on PAP/PVR was selective or, alternatively, also exerted on systemic vessels. Vascular tone is exquisitely sensitive to O2 tension. Reduction of the fraction of inspired O2 (FiO2) to ≈0.14 increases PAP (hypoxic vasoconstriction) and lowers SVR and MnBP (hypoxic vasodilation; Table 2). Once pressures had stabilized (≈5 min), ENO was delivered in three randomized doses at ≈5-min intervals (Fig. 2).
Table 2.
Induction of pulmonary hypertension (14% O2)
| Normoxia | Hypoxia | |
|---|---|---|
| PAP | 14.8 (3.32) | 23.4 (4.97) |
| PVR | 3,697.2 (1062) | 5,197.8 (2352) |
| MnBP | 55 (10.11) | 44.7 (9.82) |
| SVR | 14,147 (4776) | 9,715.8 (3756) |
| CO | 352.6 (31.1) | 412.9 (36.87) |
Values are shown as mean (standard error). Units are as in Table 1.
Figure 2.

ENO mitigates hypoxia-induced pulmonary hypertension. Neonatal pigs were instrumented and ventilated as previously described (26). Pulmonary hypertension was induced by lowering the FiO2 to 14% (Hypoxia). (A) PAP; (B) MnBP; (C) PVR; (D) SVR; (E) PaO2; and (F) CO. Changes in PAP, PVR, and PaO2 are significant, whereas systemic effects are not evident. ●, 0.0025% ENO; ○, 0.025% ENO; and ▾, 0.125% ENO. *, Significantly different from hypoxia (P < 0.05).
Inhalation of ENO produced rapid, dose-dependent reductions in PVR and PAP, whereas the drug had no effect on SVR, MnBP, or CO (Fig. 2). At the highest dose tested, PAP and PVR were not different from baseline in room air; that is, ENO fully reversed the hypoxic vasoconstriction. ENO also produced a slight increase in PaO2 that was independent of the fall in PVR, whereas the alveolar-arterial (A-a) O2 ratio was unchanged (data not shown). In this respect, it is important to understand that pulmonary vasoconstriction is a physiological adaptation to hypoxia, and that lowering of PVR could well have worsened oxygenation. Taken as a whole, the effects of ENO are best rationalized by matched changes in V/Q ratio, perhaps in part secondary to bronchodilation, and establish that the gas is a selective vascular pulmonary smooth muscle relaxant.
One of the most frequent complications of inhaled NO therapy is “rebound” (21). On discontinuing the drug, PAP may rebound to a level higher than baseline, perhaps because of inhibition of endothelial NOS. Complete hemodynamic collapse is seen in as many as 25% of patients (21). We reasoned that ENO therapy would be more resistant to rebound if it were to fortify the endogenous SNO reservoir within the lung (as SNOs are longer lived than NO). As a first assessment of the metabolic fate of ENO and NO gases, we measured the in vivo production of SNOs in the lungs of animals treated by inhalation. Airway lining aspirates were obtained by bronchoscopy, and samples were analyzed for SNO content as previously described (28). Fig. 3 illustrates that (i) ENO produced marked increases in airway SNO content at all doses tested, whereas NO was far less effective [e.g., ≈1 ppm of ENO yielded a greater increase in SNO (5-fold) than 75 ppm of NO (≈2-fold)]; (ii) maximal increases in steady-state SNO levels produced by ENO were ≈8-fold; and (iii) SNO concentrations in vivo were maintained within a relatively narrow range. These data are consistent with results of recent studies showing that SNOs are metabolized and their levels tightly regulated in vivo (29–31).
Figure 3.

ENO increases the SNO concentration in the lung. ENO or NO was added to the inhaled gas of neonatal pigs breathing room air in three randomized doses. Lung aspirates were collected and assayed immediately for SNO and for protein content. SNO concentrations normalized to protein content are expressed as fold-increase over endogenous levels (0.145 ± 0.03 nM/μg for NO group, 0.22 ± 0.04 nM/μg for ENO group). *, P < 0.05, significantly different from precompound.
Further evidence for a role of SNOs in the action of ENO was provided by comparing the effects of NO and ENO in a lung washout (injury) model of pulmonary hypertension (32). ENO and NO were each administered for 2 hr, and both were effective at reducing PVR and improving oxygenation (Fig. 4 A and B). PAP was lower in animals treated with NO than with ENO (Fig. 4C), but this result is largely a reflection of lower CO in the NO group (Fig. 4D). Removal of NO from the inhaled gas admixture resulted in a rapid rebound in pulmonary pressure and fall in PaO2, whereas these parameters (PAP, PVR, and A-a gradient) remained completely unchanged on cessation of ENO (Fig. 4 A–D). Specifically, PAP/PVR returned to baseline in all animals within 3 min of stopping NO, but was unchanged for the entire 20-min period of observation after removal of ENO. Furthermore, ENO seemed to protect against a decline in CO (which fell by 40% in untreated animals during the 2-hr study period), whereas NO did not protect (Fig. 4D), suggesting that ENO has a positive inotropic effect, including the possibility that it prevents myocardial ischemia (33). These results thus strongly suggest that flux through the SNO pool functions to dispense NO bioactivity in the lung and that the response is physiological. In other words, SNO throughput is adapted to serve in matching ventilation to perfusion and to sustain CO. The mechanisms by which lung SNOs exert these bioactivities, however, are not known.
Figure 4.

Effect of discontinuation of ENO and NO in a piglet model of lung injury. Pulmonary hypertension was induced in intubated neonatal pigs breathing 100% oxygen by repeated saline lavage. Either NO (20 ppm followed by 5 ppm), or ENO (≈5–10 ppm), or nothing (Control, i.e., values after lung injury) was then added to the inhaled gas for 2 hr. (Dosing was designed to achieve comparable reductions in PVR.) Control animals showed a progressive rise in PVR (61%) and fall in CO (40%) over the 2-hr period that followed injury; and accordingly, PAP remained essentially unchanged (±8%). (A) A-a O2 ratio; the mean PaO2 is also shown; (B) PVR; (C) PAP; (D) fall in CO (compared with baseline, i.e., values before injury). Hemodynamics were measured every 5 min for 20 min after abrupt discontinuation of inhaled gases (post). *, P < 0.05 and #, P = 0.06, compared with control.
NO can react directly with either oxygenated or deoxygenated hemes of hemogloblin to produce methemoglobin, and it is also readily converted into nitrite, which can oxidize hemoglobin (34–36). Inhaled NO is predisposed to all of the above methemoglobin-forming reactions, producing, for example, extremely high concentrations of nitrite within the airways (14). We therefore assayed for methemoglobinemia in our studies and found that the levels were below 2%. We also attempted to measure blood ethanol, which is a byproduct of ENO decomposition and is present in trace amounts in the carrier gas admixture, but these levels were below the limits of detection.
It is important to recognize that free NO is virtually undetectable in human airway lining fluid and that the concentrations in expired air are well below those that dilate blood vessels or relax airways (14, 37). The paucity of NO can be understood by appreciating that endogenous NO-related molecules undergo oxidative reactions with glutathione and proteins in human airways to produce relatively high concentrations of GSNO and SNO-proteins (14). It has been speculated that these S-nitrosylation reactions are adapted to provide NO bioactivity in the lung, and, in support of this proposition, we have shown that human airways are relaxed in vitro by endogenous concentrations of GSNO (14). The case for physiological relevance is further strengthened by studies demonstrating that airway GSNO may be depleted in human conditions associated with impairments in V/Q matching, including asthma (28), cystic fibrosis (38), and hypoxemic respiratory failure (14, 39), and by the finding that aerosolized GSNO can reverse the V/Q matching impairment associated with SNO deficiency (40). Here, we report that selective repletion of indigenous species of SNO is linked to improvements in lung function. Together, these studies strongly suggest that endogenous SNOs play a physiological role in V/Q matching.
Inhaled NO is a relatively selective pulmonary vasodilator (16, 33, 35). It has been proposed that NO gas is eliminated in reactions with hemoglobin that mitigate its toxicity. In reality, lung tissues take up most of the NO. By using positron emission tomography and N13-labeled NO, McCarthy et al. (41) showed that only a minor fraction of inhaled NO gas accesses the plasma in the form of innocuous nitrate. Moreover, NO clearance from lung tissues was much slower than N2 gas, in keeping with our earlier studies showing that inhaled NO had reacted with airway constituents including glutathione (14, 42). But, whereas reactions with glutathione preserve NOS bioactivity and mitigate endogenous NO toxicity, we find that glutathione does not efficiently sequester inhaled NO gas, thus providing a plausible explanation for NO toxicity in clinical trials (17) and hemodynamic collapse on discontinuing the drug (21). By contrast, the resistance of ENO to rebound increases in PVR would appear to result from its facile reaction with thiols to form longer-lived SNOs, and we speculate accordingly that ENO may be less toxic than NO. This viewpoint notwithstanding, the ability to manipulate the endogenous SNO pool and to thereby improve V/Q matching suggests a significant therapeutic approach to a wide-spectrum of cardiopulmonary diseases.
Acknowledgments
We thank George Quick, Damian Craig, and Michael Gentile for expert technical assistance and help with data analysis. A.G. is the recipient of a Scientist Development grant from the American Heart Association.
Abbreviations
- SNO
S-nitrosothiol
- GSNO
S-nitrosoglutathione
- ENO
O-nitrosoethanol
- NOS
NO synthase
- PAP
pulmonary arterial pressure
- CO
cardiac output
- SVR
systemic vascular resistance
- PVR
pulmonary vascular resistance
- NOx
oxides of nitrogen
- MnBP
mean blood pressure
- PaO2
partial pressure of oxygen in the systemic arterial circuit
- V
ventilation
- Q
blood flow
- FiO2
fraction of inspired O2
- A-a
alveolar-arterial
References
- 1.Gaston B, Drazen J M, Loscalzo J, Stamler J S. Am J Respir Crit Care Med. 1994;149:538–551. doi: 10.1164/ajrccm.149.2.7508323. [DOI] [PubMed] [Google Scholar]
- 2.Barnes P J. Ann Med. 1995;27:389–393. doi: 10.3109/07853899509002592. [DOI] [PubMed] [Google Scholar]
- 3.North A J, Star R A, Brannon T S, Ujiie K, Wells L B, Lowenstein C J, Snyder S H, Shaul P W. Am J Physiol. 1994;266:L635–L641. doi: 10.1152/ajplung.1994.266.6.L635. [DOI] [PubMed] [Google Scholar]
- 4.Steudel W, Scherrer-Crosbie M, Bloch K D, Weimann J, Huang P L, Jones R C, Picard M H, Zapol W M. J Clin Invest. 1998;101:2468–2477. doi: 10.1172/JCI2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barnes P J, Belvisi M G. Thorax. 1993;48:1034–1043. doi: 10.1136/thx.48.10.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horvath E P, doPico G A, Barbee R A, Dickie H A. J Occup Med. 1978;20:103–110. [PubMed] [Google Scholar]
- 7.Gow A J, Thom S R, Ischiropoulos H. Am J Physiol. 1998;274:L112–L118. doi: 10.1152/ajplung.1998.274.1.L112. [DOI] [PubMed] [Google Scholar]
- 8.Zhuang J C, Lin C, Lin D, Wogan G N. Proc Natl Acad Sci USA. 1998;95:8286–8291. doi: 10.1073/pnas.95.14.8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brune B, von Knethen A, Sandau K B. Cell Death Differ. 1999;6:969–975. doi: 10.1038/sj.cdd.4400582. [DOI] [PubMed] [Google Scholar]
- 10.Lamb N J, Quinlan G J, Westerman S T, Gutteridge J M, Evans T W. Am J Respir Crit Care Med. 1999;160:1031–1034. doi: 10.1164/ajrccm.160.3.9810048. [DOI] [PubMed] [Google Scholar]
- 11.Lorch S A, Foust R, III, Gow A, Arkovitz M, Salzman A L, Szabo C, Vayert B, Geffard M, Ischiropoulos H. Pediatr Res. 2000;47:798–805. doi: 10.1203/00006450-200006000-00020. [DOI] [PubMed] [Google Scholar]
- 12.Anderson H R, Ponce de Leon A, Bland J M, Bower J S, Emberlin J, Strachan D P. Thorax. 1998;53:842–848. doi: 10.1136/thx.53.10.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pavelchak N, Church L, Roerig S, London M, Welles W, Casey G. Appl Occup Environ Hyg. 1999;14:34–38. doi: 10.1080/104732299303395. [DOI] [PubMed] [Google Scholar]
- 14.Gaston B, Reilly J, Drazen J M, Fackler J, Ramdev P, Arnelle D, Mullins M E, Sugarbaker D J, Chee C, Singel D J. Proc Natl Acad Sci USA. 1993;90:10957–10961. doi: 10.1073/pnas.90.23.10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark R H, Kueser T J, Walker M W, Southgate W M, Huckaby J L, Perez J A, Roy B J, Keszler M, Kinsella J P. N Engl J Med. 2000;342:469–474. doi: 10.1056/NEJM200002173420704. [DOI] [PubMed] [Google Scholar]
- 16.Lonnqvist P A. Intensive Care Med. 1997;23:773–779. doi: 10.1007/s001340050408. [DOI] [PubMed] [Google Scholar]
- 17.Lundin S, Mang H, Smithies M, Stenqvist O, Frostell C. Intensive Care Med. 1999;25:911–919. doi: 10.1007/s001340050982. [DOI] [PubMed] [Google Scholar]
- 18.Young J D, Dyar O, Xiong L, Howell S. Intensive Care Med. 1994;20:581–584. doi: 10.1007/BF01705726. [DOI] [PubMed] [Google Scholar]
- 19.Loh E, Stamler J S, Hare J M, Loscalzo J, Colucci W S. Circulation. 1994;90:2780–2785. doi: 10.1161/01.cir.90.6.2780. [DOI] [PubMed] [Google Scholar]
- 20.Hogman M, Frostell C, Arnberg H, Hedenstierna G. Lancet. 1993;341:1664–1665. doi: 10.1016/0140-6736(93)90802-n. [DOI] [PubMed] [Google Scholar]
- 21.Christenson J, Lavoie A, O'connor M, Bhorade S, Pohlman A, Hall J B. Am J Respir Crit Care Med. 2000;161:1443–1449. doi: 10.1164/ajrccm.161.5.9806138. [DOI] [PubMed] [Google Scholar]
- 22.Matthay M A, Pittet J F, Jayr C. Crit Care Med. 1998;26:1–2. doi: 10.1097/00003246-199801000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Mayer B, Pfeiffer S, Schrammel A, Koesling D, Schmidt K, Brunner F. J Biol Chem. 1998;273:3264–3270. doi: 10.1074/jbc.273.6.3264. [DOI] [PubMed] [Google Scholar]
- 24.Vogel A I. In: Vogel's Textbook of Practical Organic Chemistry. Furniss B S, Hannaford A J, Smith F W G, Tatcher A R, editors. London: Longman; 1989. p. 414. [Google Scholar]
- 25.Fang K, Ragsdale N V, Carey R M, MacDonald T, Gaston B. Biochem Biophys Res Commun. 1998;252:535–540. doi: 10.1006/bbrc.1998.9688. [DOI] [PubMed] [Google Scholar]
- 26.Hillman N D, Cheifetz I M, Craig D M, Smith P K, Ungerleider R M, Meliones J N. J Thorac Cardiovasc Surg. 1997;113:1006–1013. doi: 10.1016/S0022-5223(97)70285-X. [DOI] [PubMed] [Google Scholar]
- 27.Wink D A, Nims R W, Darbyshire J F, Christodoulou D, Hanbauer I, Cox G W, Laval F, Laval J, Cook J A, Krishna M C. Chem Res Toxicol. 1994;7:519–525. doi: 10.1021/tx00040a007. [DOI] [PubMed] [Google Scholar]
- 28.Gaston B, Sears S, Woods J, Hunt J, Ponaman M, McMahon T, Stamler J S. Lancet. 1998;351:1317–1319. doi: 10.1016/S0140-6736(97)07485-0. [DOI] [PubMed] [Google Scholar]
- 29.Hausladen A, Privalle C T, Keng T, DeAngelo J, Stamler J S. Cell. 1996;86:719–729. doi: 10.1016/s0092-8674(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 30.Eu J P, Liu L, Zeng M, Stamler J S. Biochemistry. 2000;39:1040–1047. doi: 10.1021/bi992046e. [DOI] [PubMed] [Google Scholar]
- 31.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler J S. Nature (London) 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 32.Hillman N D, Meliones J N, Black D R, Craig D M, Cheifetz I M, Smith P K. J Thorac Cardiovasc Surg. 1995;110:593–599. doi: 10.1016/S0022-5223(95)70089-7. [DOI] [PubMed] [Google Scholar]
- 33.Hare J M, Stamler J S. Nat Med. 1999;5:273–274. doi: 10.1038/6486. [DOI] [PubMed] [Google Scholar]
- 34.Gow A J, Stamler J S. Nature (London) 1998;391:169–173. doi: 10.1038/34402. [DOI] [PubMed] [Google Scholar]
- 35.Gow A J, Luchsinger B P, Pawloski J R, Singel D J, Stamler J S. Proc Natl Acad Sci USA. 1999;96:9027–9032. doi: 10.1073/pnas.96.16.9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doyle M P, Herman J G, Dykstra R L. J Free Radic Biol Med. 1985;1:145–153. doi: 10.1016/0748-5514(85)90019-4. [DOI] [PubMed] [Google Scholar]
- 37.Marshall H E, Stamler J S. Am J Respir Crit Care Med. 2000;161:685–687. doi: 10.1164/ajrccm.161.3.16134. [DOI] [PubMed] [Google Scholar]
- 38.Grasemann H, Gaston B, Fang K, Paul K, Ratjen F. J Pediatr. 1999;135:770–772. doi: 10.1016/s0022-3476(99)70101-0. [DOI] [PubMed] [Google Scholar]
- 39.Gaston B, Fry E, Sears S, Heroman W M, Ignarro L, Stamler J S. Biochem Biophys Res Commun. 1998;253:899–901. doi: 10.1006/bbrc.1998.9865. [DOI] [PubMed] [Google Scholar]
- 40.Snyder A, Gaston B, Hunt J, Robbins M. Pediatr Pulmonol. 2000;20(S):P244. [Google Scholar]
- 41.McCarthy T J, Dence C S, Holmberg S W, Markham J, Schuster D P, Welch M J. Nucl Med Biol. 1996;23:773–777. doi: 10.1016/0969-8051(96)00072-8. [DOI] [PubMed] [Google Scholar]
- 42.Simon D I, Mullins M E, Jia L, Gaston B, Singel D J, Stamler J S. Proc Natl Acad Sci USA. 1996;93:4736–4741. doi: 10.1073/pnas.93.10.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]