

Abstract
Diabetic neuropathy is the most common and debilitating complication of diabetes mellitus with over half of all patients developing altered sensation as a result of damage to peripheral sensory neurons. Hyperglycemia results in altered nerve conduction velocities, loss of epidermal innervation, and the development of painful or painless signs and symptoms in the feet and hands. Current research has been unable to determine if a patient will develop insensate or painful neuropathy or be protected from peripheral nerve damage all together. One of the mechanisms that has been recognized to have a role in the pathogenesis of sensory neuron damage is the process of reactive dicarbonyls forming advanced glycation endproducts (AGEs) as a direct result of hyperglycemia. The glyoxalase system, composed of the enzymes glyoxalase I (GLO1) and glyoxalase II, is the main detoxification pathway involved in breaking down toxic reactive dicarbonyls before producing carbonyl stress and forming AGEs on proteins, lipids, or nucleic acids. This review discusses AGEs, GLO1, their role in diabetic neuropathy, and potential therapeutic targets of the AGE pathway.
Diabetes Mellitus
Diabetes mellitus is a chronic, multi-system metabolic disorder caused by a combination of environmental and genetic factors and characterized by hyperglycemia. The World Health Organization estimates that 220 million people worldwide currently have diabetes. Type 2 diabetes mellitus is the most prevalent form and accounts for 90-95% of these cases. According to the Center for Disease Control and Prevention, 25.8 million children and adults currently suffer from diabetes mellitus in the United States. Importantly, 35% of the adult population is estimated to have prediabetes or metabolic syndrome, a condition with higher than normal blood glucose and impaired insulin sensitivity that has yet to reach diagnostic criteria for diabetes mellitus [1]. Individuals with prediabetes are at a higher potential for developing type 2 diabetes mellitus. Hence, a staggering 79 million adults are at risk for developing type 2 diabetes mellitus. Although type 1 diabetes mellitus accounts for a far smaller percentage, 5-10% of cases, the incidence has been steadily rising in the past decades to nearly 5% annually in the United States [2]. Hence, both type 1 and type 2 diabetes mellitus remain growing problems throughout the world.
Despite the differences in etiology, clinical presentation, and disease prevalence, secondary complications, such as heart disease, stroke, retinopathy, nephropathy, and neuropathy, occur in both type 1 and 2 diabetes mellitus. Due to the continual rise in diabetes mellitus, secondary complications continue to be a large economic burden in the United States and across the world [3, 4]. Of these, diabetic neuropathy is the most common complication of long-term diabetes mellitus [3, 5, 6]. The Center for Disease Control and Prevention estimates 60-70% of diabetic patients will develop diabetic neuropathy symptoms with the prevalence increasing with the duration of diabetes mellitus [1]. Patients with diabetic neuropathy are at an increased risk for developing ulcers, recurrent foot infections, and Charcot joints, bony destruction and deformation due to repetitive, traumatic injury, often associated with reduced sensation in the feet [5, 7]. Consequently, diabetic neuropathy is the cause of 50-75% of non-traumatic amputations [5]. Diabetic neuropathy has a profound impact on patients’ quality of life and is responsible for majority of diabetes-associated morbidity and mortality.
Diabetic neuropathy is a collection of syndromes, either focal or diffuse in nature, affecting sensory, motor, and/or autonomic peripheral neurons [5, 6]. These disorders can range from clinical to subclinical and differ in their anatomical distribution, clinical course, and spectrum of symptoms. The most prevalent of the syndromes is distal symmetrical sensorimotor polyneuropathy, referred to as diabetic neuropathy in this review, that results from damage to peripheral sensory nerves and accounts for nearly 80% of diabetic neuropathy cases [3]. One hallmark of diabetic neuropathy is the symmetrical loss of distal skin innervation due to degeneration of small cutaneous nerve fibers. Diabetes-induced nerve damage causes a dying-back of distal axons that begins in the feet and progresses proximally in a stocking-and-glove distribution [8, 9]. Diabetic peripheral neuropathy has an insidious onset and is chronic and progressive in nature; therefore, diabetic neuropathy often results in severe, irreversible symptoms after longstanding diabetes mellitus.
Sural nerve biopsies from diabetic patients also demonstrate loss of small unmyelinated C-fibers and small myelinated Aδ fibers in early stages of diabetic neuropathy with progressive involvement of large myelinated Aβ fibers with duration of disease [10, 11]. Despite histological and ultrastructural findings of axonal regeneration, collateral sprouting, and remyelination within peripheral nerves [12], impaired nerve regeneration has been documented [13, 14]. However, regeneration is ultimately unable to compensate for the continued vicious cycle of damage and neurodegeneration of sensory neurons [13, 15].
Diabetes-induced damage results in peripheral nerve pathology that correlates with clinical signs and symptoms. Nerve injury that results in structural changes can be measured through clinical neurological assessment, quantitative sensory testing, nerve conduction studies, and peripheral nerve biopsies that are often used in combination to diagnose diabetic neuropathy [16-20]. The clinical features of diabetic neuropathy can be divided based on the type of fibers that are damaged and lost. Impairment of small, myelinated Aδ fibers and unmyelinated C-fibers results in altered mechanical, thermal, and pain sensation. Deficits in vibration, proprioception, and balance are often a consequence of large, myelinated Aβ fiber damage. Sensory symptoms usually predominate in diabetic peripheral neuropathy [21], although with progression of the disease, motor dysfunction may also be present [22].
The majority of diabetic patients experience insensate neuropathy characterized by painless symptoms including reduced vibratory perception, numbness, and insensitivity to touch and pain [23, 24]. However, others have painful diabetic neuropathy that manifests as positive symptoms of hyperalgesia, tactile allodynia, paresthesias, abnormal sensitivity to temperature, and unremitting pain [23, 25]. Data on the prevalence of painful diabetic has varied widely with some reporting a prevalence rate of 7-20% [26] and others reporting 40-50% of those with diabetic neuropathy having neuropathic complications. The European Diabetes (EURODIAB) Prospective Complications Study found approximately 25% of type 1 diabetic patients developed painful symptoms throughout the course of the 7-year investigation [26]. Although there is a considerable understanding of the molecular and pathological process responsible for damage to the peripheral nervous, the mechanisms that produce painful versus insensate signs and symptoms are not known. Furthermore, neurophysiological and histological findings do not distinguish between patients suffering with positive and negative symptoms [27]. The mechanisms underlying the development of these dichotomous symptoms remain a significant question that must be addressed to enable the development of improved and targeted therapies for diabetic neuropathy.
Hyperglycemia as a Risk Factor for Diabetic Neuropathy
A number of large studies have clearly identified hyperglycemia as a key feature involved in the pathogenesis of diabetic neuropathy [9, 28-30]. Similarly, poor glycemic control has also been recognized as an important risk factor for the development and progression of neurological complications. The Diabetes Control and Complications Trial (DCCT) followed 1,441 patients with type 1 diabetes mellitus and no history of neuropathy that were randomly assigned to either conventional therapy (one or two insulin injections daily) or intensive therapy (three or four daily insulin injections or continuous subcutaneous insulin infusion) [29]. The DCCT study found that intensive therapy reduced the risk of developing confirmed clinical neuropathy by 64% compared to conventional therapy after an average follow-up of 6.5 years [31]. The Epidemiology of Diabetes Interventions and Complications (EDIC) study, the follow up study to the DCCT, enrolled 93% of the former intensive therapy group and 91% of former conventional therapy group to assess peripheral neuropathy for 13-14 years after the DCCT study commenced; however, due to the clear beneficial effects, the conventional treatment group was encouraged to begin and maintain intensive treatment [29]. Throughout the study period, the prevalence of clinical neuropathy and nerve conduction abnormalities continued to increase in both former treatment groups; however, the group assigned to the former intensive therapy during the DCCT study had significantly lower prevalence of indices of neuropathy despite similar glycemic control between the treatment groups [29, 32]. Similarly, the EURODIAB study found both duration of diabetes and quality of glycemic control as risk factors for the development of diabetic neuropathy [30]. Thus, prior exposure to hyperglycemia predisposed to greater risk of developing diabetic peripheral neuropathy.
Advanced Glycation Endproducts
As glucose levels rise within sensory neurons due to hyperglycemia, normal metabolic pathways become overwhelmed and excess glucose is shunted into other ancillary pathways that, under these conditions, become damaging. One consequence of hyperglycemia is the increased and accelerated production of advanced glycation endproducts (AGEs) in tissues where damage results in secondary complications, including peripheral nerves. Importantly, AGEs have been shown to have a role in the pathogenesis of diabetic neuropathy. AGEs are a heterogeneous group of molecules that form from the non-enzymatic addition of sugar moieties onto arginine and lysine residues of proteins, free amino groups on lipids, or guanine nucleic acids [33]. The process of non-enzymatic glycation was first described by L.C. Maillard in the early 1900's and even at that time, he speculated it may be an important process in diabetes [34]. It has subsequently become apparent that non-enzymatic glycation and AGEs have a role in many disease processes such as aging, neurological disorders, and diabetic complications.
The classical AGE pathway involves the rearrangement of glucose or another reducing sugar, such as fructose, galactose, mannose, or ribose, that reacts with a free amino group of a protein, which forms a Schiff base (Figure 1) [35]. The Schiff base is highly unstable and degrades into the Amadori product or fructosamine [35]. Fructosamine is relatively stable, although levels tend to fluctuate with glucose concentrations [36]. The most well-known example of an Amadori product is hemoglobin A1c (HbA1c), a naturally occurring modification to the N-terminal valine amino group of the β chain of hemoglobin [34]. HbA1c is elevated in diabetic patients and gives an indication of glucose levels over the previous 2-3 months [37]. It is often used to monitor glucose control and has value at predicting risk of complications [9, 38]. With further rearrangement, oxidation, and elimination, fructosamine produces an AGE. Considerable progress has been made in understanding that AGEs form from specific metabolites despite the complexity of the glycation process. While intermediate steps in the glycation pathway are reversible, AGE formation is irreversible and causes modifications that result in both protease-resistant cross-linked and noncross-linked proteins [39, 40].
Figure 1.
A schematic illustrating the pathways that lead to the production of reactive dicarbonyls and AGEs and the mechanisms by which AGEs cause to sensory neuron dysfunction.
Besides monosaccharides, reactive dicarbonyls or α-oxoaldehydes contribute to the production of AGEs. Reactive dicarbonyls, such as 3-deoxyglucose, glyoxal, and methylglyoxal, are highly potent and reactive species that can also modify proteins, lipids, and nucleic acids and may contribute more significantly in the glycation process than the classical pathway described above (Figure 1). In fact, reactive dicarbonyls are 20,000-fold more reactive than glucose [41]. Consequently, reactive dicarbonyls have gained increasing acceptance as one of the main mechanisms that drive the production of AGEs, produce carbonyl stress, and underlie the development of diabetic complications.
Due to a combination of increased flux through glycolysis and reduced activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), both glyceradehyde 3-phosphate and dihydroxyacetone phosphate (DHAP) build up in the neuron. Under normal conditions, low levels of these metabolites are converted to methylglyoxal. However, under hyperglycemic conditions, concentrations of methylglyoxal increase within the neuron due to the nonezymatic breakdown of these two glycolytic intermediates [42, 43]. Elevated levels of methylglyoxal as well as other sugars, such as fructose, lead to the formation of advanced glycation endproducts (AGEs). AGEs modify cellular components, signal through the receptor for advanced glycation endproducts (RAGE), and compromise normal neuronal function.
The longstanding view of glycation as a relatively long process that only resulted in AGE accumulation on long-lived extracellular proteins was revolutionized with the discovery of dicarbonyl metabolites [36]. Methylglyoxal and other α-oxoaldehydes form inside cells over a relatively short time period as a byproduct of many metabolic pathways [44]. Methylglyoxal is formed from spontaneous decomposition of triosephosphates, DHAP and glyceraldehyde-3-phosphate, fragmentation of other sugars, and amino acid and ketone body degradation [45-47]. Glyoxal is also a consequence of degradation of saccharides, but also of lipid peroxidation and degradation of glycated proteins [45]. Cellular concentrations of methylglyoxal and glyoxal range from 1-5 μM and 0.1-1 μM, respectively [48]. The biogenesis of 3-deoxyglucose results from the breakdown of fructose-3-phosphate from the polyol pathway [41]. It is important to note that all of these metabolic processes are enhanced in diabetes mellitus, which leads to a significant increase in the production of reactive dicarbonyls and AGEs.
Several studies have shown levels of reactive dicarbonyls are higher in patients with diabetes due to hyperglycemia [49-51]. Consequently, reactive dicarbonyl-derived AGEs are elevated in plasma and accumulate in tissues prone to secondary complications including the lens, retina, kidney, and endothelial vessels of both diabetic humans and rodents [52-56]. Indeed, the concentration of AGEs in the sciatic nerve of diabetic rats is higher than nondiabetic rodents [53, 57]. Extensive accumulation of AGEs also occurs in the skin and peripheral nerves of diabetic patients particularly in the axoplasm of myelinated and unmyelinated neurons, Schwann cells, endoneurial and epineurial microvessels, perineurial basal lamina, and perineurium, suggesting AGEs have a role in the development and/or progression of neuropathies [58-63]. A recent study investigated skin autofluorescence as a measure of AGE deposition in nondiabetic and diabetic subjects with or without neuropathy and found a correlation between slowing of sensory nerve conduction velocity (SNCV) and increased autofluorescence [64]. Similarly, levels of serum carboxymethyl lysine (CML) or skin autofluorescence were significantly higher in type 1 diabetic patients with microvascular complications, like neuropathy, compared to those without complications [65, 66].
RAGE
AGEs produce neuronal damage and dysfunction by a variety of mechanisms. AGEs interact with cell surface receptors, particularly the receptor for advanced glycation endproducts or RAGE, to induce a cascade of intracellular signaling (Figure 1). RAGE is a multi-ligand member of the immunoglobulin superfamily of cell surface receptors that signals through the phosphatidylinositol-3 kinase (PI-3K), Ki-Ras, and mitogen-activated protein kinase (MAPK) pathways [59]. RAGE is present in the DRG, peripheral nerves, Schwann cells, and epidermal fibers in rodents [67]. Transient activation of PI-3K/AKT and MAPK pathways leads to nuclear translocation of NF-κB [59, 68]. NF-κB is responsible for the expression of different classes of genes including pro-inflammatory cytokines. IL-6 and TNF-α are particularly potent cytokines that have been shown to be elevated in the sciatic nerve of diabetic mice and contribute to the pro-inflammatory state of diabetic neuropathy [69]. Continual activation of NF-κB leads to altered gene expression and upregulation of RAGE creating a positive feedback loop that enhances sensory neuron damage [70]. RAGE also stimulates NAD(P)H oxidase, a potent producer of reactive oxygen species (ROS) (Figure 1) [68]. Like glycating agents, excessive ROS alter proteins, lipids, and DNA causing damage to peripheral neurons [68].
Rodent models of diabetes mellitus have demonstrated a role for RAGE in peripheral sensory nerve damage and neuropathy symptoms. Diabetic RAGE-/- mice were protected from both electrophysiological and morphological deficits of the peripheral nervous system demonstrated by diabetic wildtype mice [67]. Similarly, the diabetes-induced loss of thermal pain perception and increased nociceptive thresholds were reduced in diabetic RAGE-/- mice [69]. Patients with diabetes also exhibit increased immunoreactivity for RAGE and AGEs in sural nerve biopsies [69], which suggests AGE-RAGE interaction may also have a role clinically in neuronal dysfunction that leads to neuropathy.
Protein Modification by AGEs
The pathophysiological consequences of AGE accumulation have been investigated in normal aging and in disease states such as Alzheimer's disease, renal failure, inflammation, and some diabetic complications [71-74]. Several mechanisms are thought to mediate AGE-damage in disease. In tissues, AGE modification of structural and cellular proteins, lipids, and nucleic acids results in dysfunction of vital cellular processes with limited proteasomal degradation, increased aggregation, and enhanced half-life of glycated proteins [35, 68]. While a number of proteins, which differ in structure and function, are known targets of the glycation process, many more likely exist that have yet to be discovered [75, 76]. However, of those proteins that have been discovered and reported, a number likely have a role in the direct dysfunction of neurons. GAPDH activity was significantly reduced following methylglyoxal treatment [77], which causes a compensatory increase of the toxic metabolite creating a pervasive, damaging cycle that leads to elevated levels of methylglyoxal and further reduction in GAPDH activity [78]. Insulin and other key insulin-signaling molecules, such as insulin receptor substrate 1, were also susceptible to dicarbonyl glycation, which may alter the neurotrophic support for sensory neurons [79, 80]. Methyglyoxal-modification of extracellular matrix reduced neurite outgrowth of sensory neurons, suggesting reactive dicarbonyls could impair the regenerative capacity of DRG neurons in diabetic neuropathy. Methylglyoxal has also been shown to alter the activity and expression of the 26S proteasome, as well as other chaperones involved with protein control [81-83]. While a number of proteins have been identified as targets for reactive dicarbonyls and explain various aspects of cellular dysfunction in diabetic neuropathy, proteins that are modified and accumulate in diabetic sensory neurons have yet to be determined.
Reactive Dicarbonyls and Mitochondrial Dysfunction
While mitochondrial dysfunction has been suggested to be one of the main pathogenic mechanisms in diabetic neuropathy, little is known about the nature and extent of mitochondrial damage resulting from chronic hyperglycemia. Mitochondria function in calcium homeostasis and a wide range of biochemical reactions, including fatty acid oxidation, nutrient production in the citric acid cycle, oxidative phosphorylation, and ATP production. Mitochondria are responsible for producing energy that cells harness for all cellular processes such as protein production, cellular transport, and cell growth and maintenance. Oxidation of NADH and FADH2 from glycolysis, beta oxidation, and citric acid cycle releases electrons that are passed through a coordinated series of enzyme complexes located in the mitochondrial inner membrane and are finally transferred to oxygen. Complexes from the oxidative phosphorylation pathway use the redox energy released during electron transfer to pump protons from the mitochondrial matrix into the intermembrane space which creates an electrochemical gradient across the inner mitochondrial membrane [84]. ATP Synthase or Complex V utilizes the electrochemical gradient to produce ATP [84]. This process is particularly important for neurons given their relatively high reliance on energy production due to their increased metabolic demands [85]. Consequently, mitochondrial damage and dysfunction have been linked to common neurological disorders including diabetic neuropathy [86-88].
One mechanism by which elevated intracellular reactive dicarbonyls clearly alters mitochondrial function is through glycation of mitochondrial proteins. Multiple studies have shown that reactive dicarbonyls form AGEs on mitochondrial oxidative phosphorylation proteins and produce changes in mitochondrial respiration, activity of oxidative phosphorylation proteins, and leakage of electrons from these complexes in tissues that develop secondary complications of diabetes mellitus [73, 89-91]. However, some dispute remains if mitochondrial dysfunction results in the production of reactive oxygen species and oxidative stress in sensory neurons. A large body of evidence supporting the idea that reactive dicarbonyl damage to mitochondria results in oxidative stress comes from studies in tissues other than the peripheral nervous system [73, 89, 91-93]. While the axons of DRG neurons from diabetic rats exhibited increased ROS and oxidative stress [94], impaired respiratory function and reduced expression of certain mitochondrial oxidative phosphorylation proteins resulted in reduced production of superoxide [95, 96].
The Glyoxalase System and Protection From AGEs
The glyoxalase system, which is composed of the enzymes glyoxalase I (GLO1) and glyoxalase II (GLO2) is one mechanism that protects against AGE production. The glyoxalase system is responsible for detoxifying reactive dicarbonyls prior to the formation of an AGE (Figure 1 and 2) and was discovered independently by Dakin, Dudley, and Neuberg in 1913. At that time, its function of catalyzing the conversion of methylglyoxal to lactate was also described [35, 44]. Future studies revealed that reactive dicarbonyls, like methylglyoxal, react with reduced glutathione forming a hemithioacetal [97, 98] (Figure 2). GLO1 converts the hemithioacetal to S-2-hydroxyacetylglutathione. GLO2 then catalyzes this intermediate to the corresponding α-hydroxyacid and releases reduced glutathione [99].
Figure 2.

The glyoxalase system is composed of two enzymes, glyoxalase I (GLO1) and glyoxalase II. Reactive dicarbonyls, like methylglyoxal, are effectively detoxified via this metabolic pathway. The glyoxalase enzyme pathway catalyzes the conversion of reactive α-oxoaldehydes into the corresponding α-hydroxyacids. In this schematic, methylglyoxal reacts with glutathione and is converted to S-d-Lactoylglutathione by GLO1. This intermediate is then broken down into d-lactate by glyoxalase II and reduced glutathione is recycled.
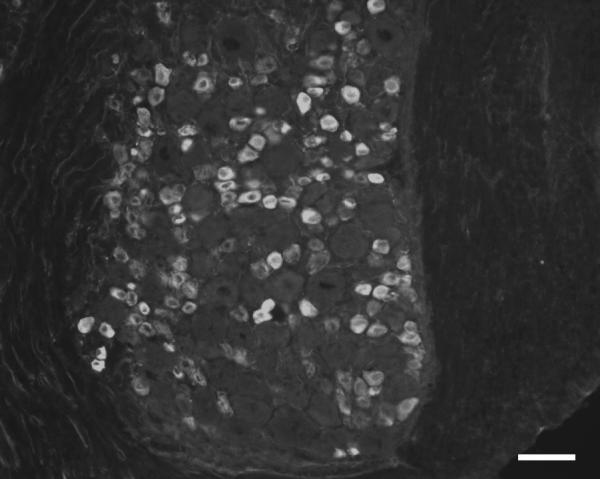
GLO1 is considered the key enzyme in anti-glycation defense because it is the rate-limiting step in the glyoxalase pathway and prevents the accumulation of reactive dicarbonyls [100, 101]. GLO1 is highly conserved with the enzyme being described in humans, mice, yeast, plants, insects, protozoa, fungi, and many bacterial strains [99]. Due to its critical function, GLO1 has been reported to be ubiquitously expressed in the cytosol of all cells [44, 99, 102, 103]. However, we have shown Glo1 is primarily expressed at high abundance in small, unmyelinated peptidergic neurons, a subset of DRG neurons that are responsible for pain transmission in the peripheral nervous system (Figure 3) [104]. The loss of peptidergic epidermal innervation has been shown to be correlated with the development of thermal and mechanical insensitivity [105].
Figure 3.

The DRG from C57BL/6 mice were stained with an antibody against Glo1. Glo1 is highly abundant in small, unmyelinated peptidergic neurons. This subpopulation of neurons is particularly important in transmitting noxious pain information. Genetic differences in the expression of GLO1 in human diabetic patients may protect this neuronal population from the damaging effects of AGEs. Scale bar = 50 μm.
Since GLO1 is the major detoxification system of reactive dicarbonyls, it is plausible that differences in production and activity of the enzyme influences AGE production and the development and/or modulation of diabetic neuropathy. Glo1 exists as a copy number variant (CNV) in many inbred strains mice where alterations in the genome that include either the gain or loss of sections of DNA result in expression differences [106, 107]. The region encompassing GLO1 has also been reported to be a CNV in humans [108]. Studies have also recognized various SNPs and null alleles of GLO1 in humans [109-111]. A study of patients with autism recognized the GLO1 rs2736654 SNP results in Ala111Glu in the mature GLO1 protein and reduced activity of the enzyme [112]. Another SNP that is located in the promoter region of GLO1 reduced promoter activity and was associated with the presence of nephropathy and retinopathy in type 2 diabetic patients [113]. These studies suggest genetic differences of GLO1 could contribute to either the susceptibility to or protection from the development of diabetic neuropathy.
While the role GLO1 in diabetic neuropathy has received limited attention, many studies related to other secondary complications have developed a clear understanding of the protective role of GLO1 in these tissues. In vitro overexpression of GLO1 in endothelial cells under hyperglycemic conditions reduced reactive dicarbonyls [114] and corrected defects in angiogenesis [115] and relaxation [116]. Overexpression in the lens and retinal capillary pericytes protected against hyperglycemia-induced protein modification [117] and apoptosis [118], respectively. Similarly, markers of oxidative damage were reduced in kidneys from diabetic transgenic rats overexpressing GLO1 [90].
Our studies have utilized two inbred strains of mice that are substrains of BALB/c mice. Since these mice were separated from the same parental strain, these two substrains likely have very similar genetic backgrounds. Indeed, the substrains are isogenic at all typed SNPs [106, 119]; however, BALB/cByJ mice have a nearly 10-fold higher abundance of GLO1 in the DRG due to a known CNV. Diabetic BALB/cJ mice with reduced GLO1 levels showed increased mechanical thresholds indicative of the development of insensate neuropathy, loss of epidermal fibers, and reduced amounts of components of mitochondrial oxidative phosphorylation proteins including Complex I and V. However, diabetic BALB/cByJ that have higher levels of GLO1 due to increased copy numbers were protected from these indices of diabetic neuropathy.
While a large proportion of patients develop altered peripheral sensation, others, nearly 30-40% of patients with diabetes, do not develop overt neuropathy signs and symptoms even after years of diabetes mellitus. A recent study highlights and complements our studies suggesting certain patients with diabetes mellitus may be protected from secondary complications due to differences in their individual genetic susceptibility. A study of Joslin Gold Medalists, patients who have survived type 1 diabetes mellitus for over 50 years, determined that current glycemic control was unrelated to the development of diabetic complications [120]. However, those patients with higher concentrations of AGEs, including methylglyoxal-derived N-(carboxyethyl)lysine, were 2.5 times more likely to suffer from neuropathy [120]. Those authors and others have suggested that certain patients may have an abundance of protective mechanisms that allow them to remain complication-free [121].
Therapeutic Strategies to Reduce AGEs
Therapeutic modalities can intervene at multiple levels to reduce either formation or the toxic effects of AGEs. Treatments that breakdown and/or prevent AGE crosslinking or interfere with the AGE-RAGE pathway may have benefit as clinical interventions [36]. The initial approach, however, is to reduce the formation of AGEs. Aminoguanidine, the best characterized compound, is a non-specific inhibitor of AGEs. While early clinical trials showed promising therapeutic potential [122], the ACTION II trial was terminated early due to lack of efficacy and safety concerns [123]. Despite these findings, other compounds and drugs have shown promise, both experimentally and clinically, in protecting against and/or reducing diabetic complications. Pyridoxamine showed benefit in clinical studies of diabetic nephropathy including reductions in urinary N(epsilon)-(carboxymethyl)lysine and N(epsilon)-(carboxyethyl)lysine [124]. Metformin, a common treatment for type 2 diabetes, was able to reduce levels of serum reactive dicarbonyls and AGEs in type 2 diabetic patients [125, 126]. Other compounds including aspirin, pioglitazone, benfotiamine, angiotensin converting enzyme inihbitors, angiostensin II-receptor blockers, and thiamine have also been shown to have anti-AGE effects [127]. Soluble RAGE (sRAGE) has shown promising results as a decoy receptor for AGEs by preventing the development of sensory deficits in diabetic mice with chronic administration of sRAGE [128]. A recent study using Akita mice, a spontaneous type 1 diabetic model, found fisetin, a naturally-occurring flavone, increased Glo1 expression and activity and increased the synthesis of glutathione [129]. Treatment reduced methylglyoxal modification of proteins and protected against kidney damage in these mice [129]. Investigation into other compounds like fisetin that increase GLO1 levels or activity could have profound clinical impact on reducing the incidence of diabetic complications, including diabetic neuropathy.
Conclusion
Though the pathogenesis of diabetic neuropathy involves perturbations in a number of metabolic pathways, AGEs likely play a large role in the development and/or progression of diabetic neuropathy. New research has identified neuronal populations that express enzymes to combat AGE formation, deposition, and accumulation. Genetic differences may play a key role in the vulnerability to diabetic neuropathy associated with AGE damage. Future approaches should include determination of genetic differences in humans, particularly in patients with diabetic neuropathy, which could underlie the variability in expression or activity of anti-AGE systems, like GLO1. These systems are a promising therapeutic target that could be used as an intervention in diabetic neuropathy.
Acknowledgements
This work was supported by the Juvenile Diabetes Research Foundation and NIH RO1NS43314 to D.E. Wright, and by the Kansas IDDRC, P30 NICHD HD 002528.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors have read the policy on disclosures of potential conflicts of interest. All authors have nothing to disclose including any financial or personal relationship with organizations that could potentially be perceived as influencing the described research.
References
- 1.Centers for Disease Control and Prevention . National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States. 2011. In: Atlanta GA, editor. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2011. [Google Scholar]
- 2.van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91:79–118. doi: 10.1152/physrev.00003.2010. [DOI] [PubMed] [Google Scholar]
- 3.Said G. Diabetic neuropathy--a review. Nat Clin Pract Neurol. 2007;3:331–40. doi: 10.1038/ncpneuro0504. [DOI] [PubMed] [Google Scholar]
- 4.Boulton AJ, Vileikyte L, Ragnarson-Tennvall G, Apelqvist J. The global burden of diabetic foot disease. Lancet. 2005;366:1719–24. doi: 10.1016/S0140-6736(05)67698-2. [DOI] [PubMed] [Google Scholar]
- 5.Vinik AI, Park TS, Stansberry KB, Pittenger GL. Diabetic neuropathies. Diabetologia. 2000;43:957–73. doi: 10.1007/s001250051477. [DOI] [PubMed] [Google Scholar]
- 6.Edwards JL, Vincent AM, Cheng HT, Feldman EL. Diabetic neuropathy: mechanisms to management. Pharmacol Ther. 2008;120:1–34. doi: 10.1016/j.pharmthera.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Little AA, Edwards JL, Feldman EL. Diabetic neuropathies. Pract Neurol. 2007;7:82–92. [PubMed] [Google Scholar]
- 8.Zochodne DW, Ramji N, Toth C. Neuronal targeting in diabetes mellitus: a story of sensory neurons and motor neurons. Neuroscientist. 2008;14:311–8. doi: 10.1177/1073858408316175. [DOI] [PubMed] [Google Scholar]
- 9.DCCT The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–86. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 10.Yagihashi S, Yamagishi S, Wada R. Pathology and pathogenetic mechanisms of diabetic neuropathy: correlation with clinical signs and symptoms. Diabetes Res Clin Pract. 2007;77(Suppl 1):S184–9. doi: 10.1016/j.diabres.2007.01.054. [DOI] [PubMed] [Google Scholar]
- 11.Said G, Slama G, Selva J. Progressive centripetal degeneration of axons in small fibre diabetic polyneuropathy. Brain. 1983;106(Pt 4):791–807. doi: 10.1093/brain/106.4.791. [DOI] [PubMed] [Google Scholar]
- 12.Malik RA, Tesfaye S, Newrick PG, Walker D, Rajbhandari SM, Siddique I, et al. Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia. 2005;48:578–85. doi: 10.1007/s00125-004-1663-5. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy JM, Zochodne DW. Impaired peripheral nerve regeneration in diabetes mellitus. J Peripher Nerv Syst. 2005;10:144–57. doi: 10.1111/j.1085-9489.2005.0010205.x. [DOI] [PubMed] [Google Scholar]
- 14.Kennedy JM, Zochodne DW. The regenerative deficit of peripheral nerves in experimental diabetes: its extent, timing and possible mechanisms. Brain. 2000;123(Pt 10):2118–29. doi: 10.1093/brain/123.10.2118. [DOI] [PubMed] [Google Scholar]
- 15.Vincent AM, Russell JW, Low P, Feldman EL. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr Rev. 2004;25:612–28. doi: 10.1210/er.2003-0019. [DOI] [PubMed] [Google Scholar]
- 16.Pambianco G, Costacou T, Strotmeyer E, Orchard TJ. The assessment of clinical distal symmetric polyneuropathy in type 1 diabetes: A comparison of methodologies from the Pittsburgh Epidemiology of Diabetes Complications Cohort. Diabetes Res Clin Pract. 2011 doi: 10.1016/j.diabres.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.England JD, Gronseth GS, Franklin G, Miller RG, Asbury AK, Carter GT, et al. Distal symmetric polyneuropathy: a definition for clinical research: report of the American Academy of Neurology, the American Association of Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2005;64:199–207. doi: 10.1212/01.WNL.0000149522.32823.EA. [DOI] [PubMed] [Google Scholar]
- 18.Arezzo JC, Zotova E. Electrophysiologic measures of diabetic neuropathy: mechanism and meaning. Int Rev Neurobiol. 2002;50:229–55. doi: 10.1016/s0074-7742(02)50079-9. [DOI] [PubMed] [Google Scholar]
- 19.Kles KA, Bril V. Diagnostic tools for diabetic sensorimotor polyneuropathy. Curr Diabetes Rev. 2006;2:353–61. doi: 10.2174/157339906777950598. [DOI] [PubMed] [Google Scholar]
- 20.Gibbons C, Freeman R. The evaluation of small fiber function-autonomic and quantitative sensory testing. Neurol Clin. 2004;22:683–702. vii. doi: 10.1016/j.ncl.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Sinnreich M, Taylor BV, Dyck PJ. Diabetic neuropathies. Classification, clinical features, and pathophysiological basis. Neurologist. 2005;11:63–79. doi: 10.1097/01.nrl.0000156314.24508.ed. [DOI] [PubMed] [Google Scholar]
- 22.Zochodne DW. Diabetic neuropathies: features and mechanisms. Brain Pathol. 1999;9:369–91. doi: 10.1111/j.1750-3639.1999.tb00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Obrosova IG. Diabetic painful and insensate neuropathy: pathogenesis and potential treatments. Neurotherapeutics. 2009;6:638–47. doi: 10.1016/j.nurt.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorensen L, Molyneaux L, Yue DK. Insensate versus painful diabetic neuropathy: the effects of height, gender, ethnicity and glycaemic control. Diabetes Res Clin Pract. 2002;57:45–51. doi: 10.1016/s0168-8227(02)00010-4. [DOI] [PubMed] [Google Scholar]
- 25.Sugimoto K, Murakawa Y, Sima AA. Diabetic neuropathy--a continuing enigma. Diabetes Metab Res Rev. 2000;16:408–33. doi: 10.1002/1520-7560(200011/12)16:6<408::aid-dmrr158>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 26.Tesfaye S, Kempler P. Painful diabetic neuropathy. Diabetologia. 2005;48:805–7. doi: 10.1007/s00125-005-1721-7. [DOI] [PubMed] [Google Scholar]
- 27.Malik RA, Veves A, Walker D, Siddique I, Lye RH, Schady W, et al. Sural nerve fibre pathology in diabetic patients with mild neuropathy: relationship to pain, quantitative sensory testing and peripheral nerve electrophysiology. Acta Neuropathol. 2001;101:367–74. doi: 10.1007/s004010000287. [DOI] [PubMed] [Google Scholar]
- 28.Pop-Busui R, Herman WH, Feldman EL, Low PA, Martin CL, Cleary PA, et al. DCCT and EDIC studies in type 1 diabetes: lessons for diabetic neuropathy regarding metabolic memory and natural history. Curr Diab Rep. 2010;10:276–82. doi: 10.1007/s11892-010-0120-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albers JW, Herman WH, Pop-Busui R, Feldman EL, Martin CL, Cleary PA, et al. Effect of prior intensive insulin treatment during the Diabetes Control and Complications Trial (DCCT) on peripheral neuropathy in type 1 diabetes during the Epidemiology of Diabetes Interventions and Complications (EDIC) Study. Diabetes Care. 2010;33:1090–6. doi: 10.2337/dc09-1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tesfaye S, Stevens LK, Stephenson JM, Fuller JH, Plater M, Ionescu-Tirgoviste C, et al. Prevalence of diabetic peripheral neuropathy and its relation to glycaemic control and potential risk factors: the EURODIAB IDDM Complications Study. Diabetologia. 1996;39:1377–84. doi: 10.1007/s001250050586. [DOI] [PubMed] [Google Scholar]
- 31.The Diabetes Control and Complications Trial Research Group The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann Intern Med. 1995;122:561–8. doi: 10.7326/0003-4819-122-8-199504150-00001. [DOI] [PubMed] [Google Scholar]
- 32.Martin CL, Albers J, Herman WH, Cleary P, Waberski B, Greene DA, et al. Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes Care. 2006;29:340–4. doi: 10.2337/diacare.29.02.06.dc05-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peppa M, Stavroulakis P, Raptis SA. Advanced glycoxidation products and impaired diabetic wound healing. Wound Repair Regen. 2009;17:461–72. doi: 10.1111/j.1524-475X.2009.00518.x. [DOI] [PubMed] [Google Scholar]
- 34.Ulrich P, Cerami A. Protein glycation, diabetes, and aging. Recent Prog Horm Res. 2001;56:1–21. doi: 10.1210/rp.56.1.1. [DOI] [PubMed] [Google Scholar]
- 35.Nass N, Bartling B, Navarrete Santos A, Scheubel RJ, Borgermann J, Silber RE, et al. Advanced glycation end products, diabetes and ageing. Z Gerontol Geriatr. 2007;40:349–56. doi: 10.1007/s00391-007-0484-9. [DOI] [PubMed] [Google Scholar]
- 36.Huijberts MS, Schaper NC, Schalkwijk CG. Advanced glycation end products and diabetic foot disease. Diabetes Metab Res Rev. 2008;24:S19–S24. doi: 10.1002/dmrr.861. [DOI] [PubMed] [Google Scholar]
- 37.Koenig RJ, Peterson CM, Jones RL, Saudek C, Lehrman M, Cerami A. Correlation of glucose regulation and hemoglobin AIc in diabetes mellitus. N Engl J Med. 1976;295:417–20. doi: 10.1056/NEJM197608192950804. [DOI] [PubMed] [Google Scholar]
- 38.Kankova K. Diabetic threesome (hyperglycaemia, renal function and nutrition) and advanced glycation end products: evidence for the multiple-hit agent? Proc Nutr Soc. 2008;67:60–74. doi: 10.1017/S0029665108006034. [DOI] [PubMed] [Google Scholar]
- 39.Munch G, Westcott B, Menini T, Gugliucci A. Advanced glycation endproducts and their pathogenic roles in neurological disorders. Amino Acids. 2010 doi: 10.1007/s00726-010-0777-y. [DOI] [PubMed] [Google Scholar]
- 40.Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. 2005;15:16R–28R. doi: 10.1093/glycob/cwi053. [DOI] [PubMed] [Google Scholar]
- 41.Turk Z. Glycotoxines, carbonyl stress and relevance to diabetes and its complications. Physiol Res. 2010;59:147–56. doi: 10.33549/physiolres.931585. [DOI] [PubMed] [Google Scholar]
- 42.Phillips SA, Thornalley PJ. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur J Biochem. 1993;212:101–5. doi: 10.1111/j.1432-1033.1993.tb17638.x. [DOI] [PubMed] [Google Scholar]
- 43.Thornalley PJ. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification--a role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol. 1996;27:565–73. doi: 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- 44.Xue M, Rabbani N, Thornalley PJ. Glyoxalase in ageing. Semin Cell Dev Biol. 2011;22:293–301. doi: 10.1016/j.semcdb.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 45.Thornalley PJ. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems--role in ageing and disease. Drug Metabol Drug Interact. 2008;23:125–50. doi: 10.1515/dmdi.2008.23.1-2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oya T, Hattori N, Mizuno Y, Miyata S, Maeda S, Osawa T, et al. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxalarginine adducts. J Biol Chem. 1999;274:18492–502. doi: 10.1074/jbc.274.26.18492. [DOI] [PubMed] [Google Scholar]
- 47.Beisswenger PJ, Howell SK, Nelson RG, Mauer M, Szwergold BS. Alpha-oxoaldehyde metabolism and diabetic complications. Biochem Soc Trans. 2003;31:1358–63. doi: 10.1042/bst0311358. [DOI] [PubMed] [Google Scholar]
- 48.Fleming TH, Humpert PM, Nawroth PP, Bierhaus A. Reactive Metabolites and AGE/RAGE-Mediated Cellular Dysfunction Affect the Aging Process - A Mini-Review. Gerontology. 2010 doi: 10.1159/000322087. [DOI] [PubMed] [Google Scholar]
- 49.Mirza MA, Kandhro AJ, Memon SQ, Khuhawar MY, Arain R. Determination of glyoxal and methylglyoxal in the serum of diabetic patients by MEKC using stilbenediamine as derivatizing reagent. Electrophoresis. 2007;28:3940–7. doi: 10.1002/elps.200700129. [DOI] [PubMed] [Google Scholar]
- 50.Han Y, Randell E, Vasdev S, Gill V, Curran M, Newhook LA, et al. Plasma advanced glycation endproduct, methylglyoxal-derived hydroimidazolone is elevated in young, complication-free patients with Type 1 diabetes. Clin Biochem. 2009;42:562–9. doi: 10.1016/j.clinbiochem.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 51.Beisswenger PJ, Drummond KS, Nelson RG, Howell SK, Szwergold BS, Mauer M. Susceptibility to diabetic nephropathy is related to dicarbonyl and oxidative stress. Diabetes. 2005;54:3274–81. doi: 10.2337/diabetes.54.11.3274. [DOI] [PubMed] [Google Scholar]
- 52.Ahmed N, Babaei-Jadidi R, Howell SK, Beisswenger PJ, Thornalley PJ. Degradation products of proteins damaged by glycation, oxidation and nitration in clinical type 1 diabetes. Diabetologia. 2005;48:1590–603. doi: 10.1007/s00125-005-1810-7. [DOI] [PubMed] [Google Scholar]
- 53.Karachalias N, Babaei-Jadidi R, Ahmed N, Thornalley PJ. Accumulation of fructosyl-lysine and advanced glycation end products in the kidney, retina and peripheral nerve of streptozotocin-induced diabetic rats. Biochem Soc Trans. 2003;31:1423–5. doi: 10.1042/bst0311423. [DOI] [PubMed] [Google Scholar]
- 54.Ahmed N. Advanced glycation endproducts--role in pathology of diabetic complications. Diabetes Res Clin Pract. 2005;67:3–21. doi: 10.1016/j.diabres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 55.Stitt AW. AGEs and diabetic retinopathy. Invest Ophthalmol Vis Sci. 2010;51:4867–74. doi: 10.1167/iovs.10-5881. [DOI] [PubMed] [Google Scholar]
- 56.Bohlender JM, Franke S, Stein G, Wolf G. Advanced glycation end products and the kidney. Am J Physiol Renal Physiol. 2005;289:F645–59. doi: 10.1152/ajprenal.00398.2004. [DOI] [PubMed] [Google Scholar]
- 57.Thornalley PJ. Dicarbonyl intermediates in the maillard reaction. Ann N Y Acad Sci. 2005;1043:111–7. doi: 10.1196/annals.1333.014. [DOI] [PubMed] [Google Scholar]
- 58.Haslbeck KM, Schleicher ED, Friess U, Kirchner A, Neundorfer B, Heuss D. N(epsilon)-Carboxymethyllysine in diabetic and non-diabetic polyneuropathies. Acta Neuropathol. 2002;104:45–52. doi: 10.1007/s00401-002-0518-8. [DOI] [PubMed] [Google Scholar]
- 59.Lukic IK, Humpert PM, Nawroth PP, Bierhaus A. The RAGE pathway: activation and perpetuation in the pathogenesis of diabetic neuropathy. Ann N Y Acad Sci. 2008;1126:76–80. doi: 10.1196/annals.1433.059. [DOI] [PubMed] [Google Scholar]
- 60.Sugimoto K, Nishizawa Y, Horiuchi S, Yagihashi S. Localization in human diabetic peripheral nerve of N(epsilon)-carboxymethyllysine-protein adducts, an advanced glycation endproduct. Diabetologia. 1997;40:1380–7. doi: 10.1007/s001250050839. [DOI] [PubMed] [Google Scholar]
- 61.Ryle C, Donaghy M. Non-enzymatic glycation of peripheral nerve proteins in human diabetics. J Neurol Sci. 1995;129:62–8. doi: 10.1016/0022-510x(94)00251-i. [DOI] [PubMed] [Google Scholar]
- 62.Misur I, Zarkovic K, Barada A, Batelja L, Milicevic Z, Turk Z. Advanced glycation endproducts in peripheral nerve in type 2 diabetes with neuropathy. Acta Diabetol. 2004;41:158–66. doi: 10.1007/s00592-004-0160-0. [DOI] [PubMed] [Google Scholar]
- 63.Yu Y, Thorpe SR, Jenkins AJ, Shaw JN, Sochaski MA, McGee D, et al. Advanced glycation end-products and methionine sulphoxide in skin collagen of patients with type 1 diabetes. Diabetologia. 2006;49:2488–98. doi: 10.1007/s00125-006-0355-8. [DOI] [PubMed] [Google Scholar]
- 64.Meerwaldt R, Links TP, Graaff R, Hoogenberg K, Lefrandt JD, Baynes JW, et al. Increased accumulation of skin advanced glycation end-products precedes and correlates with clinical manifestation of diabetic neuropathy. Diabetologia. 2005;48:1637–44. doi: 10.1007/s00125-005-1828-x. [DOI] [PubMed] [Google Scholar]
- 65.Hwang JS, Shin CH, Yang SW. Clinical implications of N epsilon-(carboxymethyl)lysine, advanced glycation end product, in children and adolescents with type 1 diabetes. Diabetes Obes Metab. 2005;7:263–7. doi: 10.1111/j.1463-1326.2004.00398.x. [DOI] [PubMed] [Google Scholar]
- 66.Araszkiewicz A, Naskret D, Niedzwiecki P, Samborski P, Wierusz-Wysocka B, Zozulinska-Ziolkiewicz D. Increased Accumulation of Skin Advanced Glycation End Products Is Associated with Microvascular Complications in Type 1 Diabetes. Diabetes Technol Ther. 2011 doi: 10.1089/dia.2011.0043. [DOI] [PubMed] [Google Scholar]
- 67.Toth C, Rong LL, Yang C, Martinez J, Song F, Ramji N, et al. Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes. 2008;57:1002–17. doi: 10.2337/db07-0339. [DOI] [PubMed] [Google Scholar]
- 68.Vincent AM, Perrone L, Sullivan KA, Backus C, Sastry AM, Lastoskie C, et al. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology. 2007;148:548–58. doi: 10.1210/en.2006-0073. [DOI] [PubMed] [Google Scholar]
- 69.Bierhaus A, Haslbeck KM, Humpert PM, Liliensiek B, Dehmer T, Morcos M, et al. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J Clin Invest. 2004;114:1741–51. doi: 10.1172/JCI18058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toth C, Martinez J, Zochodne DW. RAGE, diabetes, and the nervous system. Curr Mol Med. 2007;7:766–76. doi: 10.2174/156652407783220705. [DOI] [PubMed] [Google Scholar]
- 71.Ahmed N, Thornalley PJ. Advanced glycation endproducts: what is their relevance to diabetic complications? Diabetes Obes Metab. 2007;9:233–45. doi: 10.1111/j.1463-1326.2006.00595.x. [DOI] [PubMed] [Google Scholar]
- 72.Ramasamy R, Yan SF, Herold K, Clynes R, Schmidt AM. Receptor for advanced glycation end products: fundamental roles in the inflammatory response: winding the way to the pathogenesis of endothelial dysfunction and atherosclerosis. Ann N Y Acad Sci. 2008;1126:7–13. doi: 10.1196/annals.1433.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morcos M, Du X, Pfisterer F, Hutter H, Sayed AA, Thornalley P, et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. 2008;7:260–9. doi: 10.1111/j.1474-9726.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 74.Chougale AD, Bhat SP, Bhujbal SV, Zambare MR, Puntambekar S, Somani RS, et al. Proteomic Analysis of Glycated Proteins from Streptozotocin-Induced Diabetic Rat Kidney. Mol Biotechnol. 2011 doi: 10.1007/s12033-011-9409-3. [DOI] [PubMed] [Google Scholar]
- 75.Mendez JD, Xie J, Aguilar-Hernandez M, Mendez-Valenzuela V. Molecular susceptibility to glycation and its implication in diabetes mellitus and related diseases. Mol Cell Biochem. 2010;344:185–93. doi: 10.1007/s11010-010-0541-3. [DOI] [PubMed] [Google Scholar]
- 76.Rabbani N, Thornalley PJ. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids. 2010 doi: 10.1007/s00726-010-0783-0. [DOI] [PubMed] [Google Scholar]
- 77.Lee HJ, Howell SK, Sanford RJ, Beisswenger PJ. Methylglyoxal can modify GAPDH activity and structure. Ann N Y Acad Sci. 2005;1043:135–45. doi: 10.1196/annals.1333.017. [DOI] [PubMed] [Google Scholar]
- 78.Beisswenger PJ, Howell SK, Smith K, Szwergold BS. Glyceraldehyde-3-phosphate dehydrogenase activity as an independent modifier of methylglyoxal levels in diabetes. Biochim Biophys Acta. 2003;1637:98–106. doi: 10.1016/s09254439(02)00219-3. [DOI] [PubMed] [Google Scholar]
- 79.Riboulet-Chavey A, Pierron A, Durand I, Murdaca J, Giudicelli J, Van Obberghen E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes. 2006;55:1289–99. doi: 10.2337/db05-0857. [DOI] [PubMed] [Google Scholar]
- 80.Schalkwijk CG, Brouwers O, Stehouwer CD. Modulation of insulin action by advanced glycation endproducts: a new player in the field. Horm Metab Res. 2008;40:614–9. doi: 10.1055/s-0028-1082085. [DOI] [PubMed] [Google Scholar]
- 81.Bento CF, Marques F, Fernandes R, Pereira P. Methylglyoxal alters the function and stability of critical components of the protein quality control. PLoS One. 2010;5:e13007. doi: 10.1371/journal.pone.0013007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Queisser MA, Yao D, Geisler S, Hammes HP, Lochnit G, Schleicher ED, et al. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes. 2010;59:670–8. doi: 10.2337/db08-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schalkwijk CG, van Bezu J, van der Schors RC, Uchida K, Stehouwer CD, van Hinsbergh VW. Heat-shock protein 27 is a major methylglyoxal-modified protein in endothelial cells. FEBS Lett. 2006;580:1565–70. doi: 10.1016/j.febslet.2006.01.086. [DOI] [PubMed] [Google Scholar]
- 84.Scheffler IE. Mitochondria make a come back. Adv Drug Deliv Rev. 2001;49:3–26. doi: 10.1016/s0169-409x(01)00123-5. [DOI] [PubMed] [Google Scholar]
- 85.Han XJ, Tomizawa K, Fujimura A, Ohmori I, Nishiki T, Matsushita M, et al. Regulation of mitochondrial dynamics and neurodegenerative diseases. Acta Med Okayama. 2011;65:1–10. doi: 10.18926/AMO/43824. [DOI] [PubMed] [Google Scholar]
- 86.Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–66. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lu B. Mitochondrial dynamics and neurodegeneration. Curr Neurol Neurosci Rep. 2009;9:212–9. doi: 10.1007/s11910-009-0032-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fernyhough P, Roy Chowdhury SK, Schmidt RE. Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev Endocrinol Metab. 2010;5:39–49. doi: 10.1586/eem.09.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rosca MG, Mustata TG, Kinter MT, Ozdemir AM, Kern TS, Szweda LI, et al. Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol Renal Physiol. 2005;289:F420–30. doi: 10.1152/ajprenal.00415.2004. [DOI] [PubMed] [Google Scholar]
- 90.Brouwers O, Niessen PM, Ferreira I, Miyata T, Scheffer PG, Teerlink T, et al. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation endproducts and oxidative stress in diabetic rats. J Biol Chem. 2010 doi: 10.1074/jbc.M110.144097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rosca MG, Monnier VM, Szweda LI, Weiss MF. Alterations in renal mitochondrial respiration in response to the reactive oxoaldehyde methylglyoxal. Am J Physiol Renal Physiol. 2002;283:F52–9. doi: 10.1152/ajprenal.00302.2001. [DOI] [PubMed] [Google Scholar]
- 92.Rabbani N, Thornalley PJ. Dicarbonyls linked to damage in the powerhouse: glycation of mitochondrial proteins and oxidative stress. Biochem Soc Trans. 2008;36:1045–50. doi: 10.1042/BST0361045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schlotterer A, Kukudov G, Bozorgmehr F, Hutter H, Du X, Oikonomou D, et al. C. elegans as model for the study of high glucose- mediated life span reduction. Diabetes. 2009;58:2450–6. doi: 10.2337/db09-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zherebitskaya E, Akude E, Smith DR, Fernyhough P. Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes. 2009;58:1356–64. doi: 10.2337/db09-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chowdhury SK, Zherebitskaya E, Smith DR, Akude E, Chattopadhyay S, Jolivalt CG, et al. Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes. 2010;59:1082–91. doi: 10.2337/db09-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Akude E, Zherebitskaya E, Chowdhury SK, Smith DR, Dobrowsky RT, Fernyhough P. Diminished superoxide generation is associated with respiratory chain dysfunction and changes in the mitochondrial proteome of sensory neurons from diabetic rats. Diabetes. 2011;60:288–97. doi: 10.2337/db10-0818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mannervik B. Molecular enzymology of the glyoxalase system. Drug Metabol Drug Interact. 2008;23:13–27. doi: 10.1515/dmdi.2008.23.1-2.13. [DOI] [PubMed] [Google Scholar]
- 98.Racker E. The mechanism of action of glyoxalase. J Biol Chem. 1951;190:685–96. [PubMed] [Google Scholar]
- 99.Thornalley PJ. Glyoxalase I--structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans. 2003;31:1343–8. doi: 10.1042/bst0311343. [DOI] [PubMed] [Google Scholar]
- 100.Kuhla B, Boeck K, Luth HJ, Schmidt A, Weigle B, Schmitz M, et al. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol Aging. 2006;27:815–22. doi: 10.1016/j.neurobiolaging.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 101.Thornalley PJ. The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem J. 1990;269:1–11. doi: 10.1042/bj2690001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rabbani N, Thornalley PJ. Glyoxalase in diabetes, obesity and related disorders. Semin Cell Dev Biol. 2011 doi: 10.1016/j.semcdb.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 103.Thornalley PJ. Glutathione-dependent detoxification of alpha-oxoaldehydes by the glyoxalase system: involvement in disease mechanisms and antiproliferative activity of glyoxalase I inhibitors. Chem Biol Interact. 1998;111-112:137–51. doi: 10.1016/s0009-2797(97)00157-9. [DOI] [PubMed] [Google Scholar]
- 104.Jack MM, Ryals JM, Wright DE. Characterisation of glyoxalase I in a streptozocin-induced mouse model of diabetes with painful and insensate neuropathy. Diabetologia. 2011;54:2174–82. doi: 10.1007/s00125-011-2196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Johnson MS, Ryals JM, Wright DE. Early loss of peptidergic intraepidermal nerve fibers in an STZ-induced mouse model of insensate diabetic neuropathy. Pain. 2008;140:35–47. doi: 10.1016/j.pain.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Williams Rt, Lim JE, Harr B, Wing C, Walters R, Distler MG, et al. A common and unstable copy number variant is associated with differences in Glo1 expression and anxiety-like behavior. PLoS One. 2009;4:e4649. doi: 10.1371/journal.pone.0004649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Graubert TA, Cahan P, Edwin D, Selzer RR, Richmond TA, Eis PS, et al. A high-resolution map of segmental DNA copy number variation in the mouse genome. PLoS Genet. 2007;3:e3. doi: 10.1371/journal.pgen.0030003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sparkes RS, Sparkes MC, Crist M, Anderson CE. Glyoxalase I “null” allele in a new family: identification by abnormal segregation pattern and quantitative assay. Hum Genet. 1983;64:146–7. doi: 10.1007/BF00327112. [DOI] [PubMed] [Google Scholar]
- 110.Gale CP, Futers TS, Summers LK. Common polymorphisms in the glyoxalase-1 gene and their association with pro-thrombotic factors. Diab Vasc Dis Res. 2004;1:34–9. doi: 10.3132/dvdr.2004.004. [DOI] [PubMed] [Google Scholar]
- 111.Gale CP, Grant PJ. The characterisation and functional analysis of the human glyoxalase-1 gene using methods of bioinformatics. Gene. 2004;340:251–60. doi: 10.1016/j.gene.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 112.Barua M, Jenkins EC, Chen W, Kuizon S, Pullarkat RK, Junaid MA. Glyoxalase I polymorphism rs2736654 causing the Ala111Glu substitution modulates enzyme activity-implications for autism. Autism Res. 2011 doi: 10.1002/aur.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wu JC, Li XH, Peng YD, Wang JB, Tang JF, Wang YF. Association of Two Glyoxalase 1 Gene Polymorphisms with Nephropathy and Retinopathy in Type 2 Diabetes. J Endocrinol Invest. 2011 doi: 10.3275/7856. [DOI] [PubMed] [Google Scholar]
- 114.Shinohara M, Thornalley PJ, Giardino I, Beisswenger P, Thorpe SR, Onorato J, et al. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest. 1998;101:1142–7. doi: 10.1172/JCI119885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ahmed U, Dobler D, Larkin SJ, Rabbani N, Thornalley PJ. Reversal of hyperglycemia-induced angiogenesis deficit of human endothelial cells by overexpression of glyoxalase 1 in vitro. Ann N Y Acad Sci. 2008;1126:262–4. doi: 10.1196/annals.1433.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Brouwers O, Niessen PM, Haenen G, Miyata T, Brownlee M, Stehouwer CD, et al. Hyperglycaemia-induced impairment of endothelium-dependent vasorelaxation in rat mesenteric arteries is mediated by intracellular methylglyoxal levels in a pathway dependent on oxidative stress. Diabetologia. 2010;53:989–1000. doi: 10.1007/s00125-010-1677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gangadhariah MH, Mailankot M, Reneker L, Nagaraj RH. Inhibition of methylglyoxal-mediated protein modification in glyoxalase I overexpressing mouse lenses. J Ophthalmol. 2010;2010:274317. doi: 10.1155/2010/274317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Miller AG, Smith DG, Bhat M, Nagaraj RH. Glyoxalase I is critical for human retinal capillary pericyte survival under hyperglycemic conditions. J Biol Chem. 2006;281:11864–71. doi: 10.1074/jbc.M513813200. [DOI] [PubMed] [Google Scholar]
- 119.Velez L, Sokoloff G, Miczek KA, Palmer AA, Dulawa SC. Differences in aggressive behavior and DNA copy number variants between BALB/cJ and BALB/cByJ substrains. Behav Genet. 2010;40:201–10. doi: 10.1007/s10519-009-9325-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sun JK, Keenan HA, Cavallerano JD, Asztalos BF, Schaefer EJ, Sell DR, et al. Protection from retinopathy and other complications in patients with type 1 diabetes of extreme duration: the joslin 50-year medalist study. Diabetes Care. 2011;34:968–74. doi: 10.2337/dc10-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vinik A. The question is, my dear watson, why did the dog not bark?: the joslin 50-year medalist study. Diabetes Care. 2011;34:1060–3. doi: 10.2337/dc11-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bolton WK, Cattran DC, Williams ME, Adler SG, Appel GB, Cartwright K, et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. 2004;24:32–40. doi: 10.1159/000075627. [DOI] [PubMed] [Google Scholar]
- 123.Freedman BI, Wuerth JP, Cartwright K, Bain RP, Dippe S, Hershon K, et al. Design and baseline characteristics for the aminoguanidine Clinical Trial in Overt Type 2 Diabetic Nephropathy (ACTION II). Control Clin Trials. 1999;20:493–510. doi: 10.1016/s0197-2456(99)00024-0. [DOI] [PubMed] [Google Scholar]
- 124.Williams ME, Bolton WK, Khalifah RG, Degenhardt TP, Schotzinger RJ, McGill JB. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol. 2007;27:605–14. doi: 10.1159/000108104. [DOI] [PubMed] [Google Scholar]
- 125.Kanazawa I, Yamamoto M, Yamaguchi T, Sugimoto T. Effects of Metformin and Pioglitazone on Serum Pentosidine Levels in Type 2 Diabetes Mellitus. Exp Clin Endocrinol Diabetes. 2011 doi: 10.1055/s-0030-1267953. [DOI] [PubMed] [Google Scholar]
- 126.Engelen L, Lund SS, Ferreira I, Tarnow L, Parving HH, Gram J, et al. Improved glycemic control induced by both metformin and repaglinide is associated with a reduction in blood levels of 3-deoxyglucosone in nonobese patients with type 2 diabetes. Eur J Endocrinol. 2011;164:371–9. doi: 10.1530/EJE-10-0851. [DOI] [PubMed] [Google Scholar]
- 127.Desai K, Wu L. Methylglyoxal and advanced glycation endproducts: new therapeutic horizons? Recent Pat Cardiovasc Drug Discov. 2007;2:89–99. doi: 10.2174/157489007780832498. [DOI] [PubMed] [Google Scholar]
- 128.Yan SF, Ramasamy R, Schmidt AM. Soluble RAGE: therapy and biomarker in unraveling the RAGE axis in chronic disease and aging. Biochem Pharmacol. 2010;79:1379–86. doi: 10.1016/j.bcp.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Maher P, Dargusch R, Ehren JL, Okada S, Sharma K, Schubert D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS One. 2011;6:e21226. doi: 10.1371/journal.pone.0021226. [DOI] [PMC free article] [PubMed] [Google Scholar]