

Abstract
Biochemical, epidemiological, and genetic findings demonstrate a link between cholesterol levels, processing of the amyloid precursor protein (APP), and Alzheimer's disease. In the present report, we identify the α-secretase ADAM 10 (a disintegrin and metalloprotease) as a major target of the cholesterol effects on APP metabolism. Treatment of various peripheral and neural cell lines with either the cholesterol-extracting agent methyl-β-cyclodextrin or the hydroxymethyl glutaryl-CoA reductase inhibitor lovastatin resulted in a drastic increase of secreted α-secretase cleaved soluble APP. This strong stimulatory effect was in the range obtained with phorbol esters and was further increased in cells overexpressing ADAM 10. In cells overexpressing APP, the increase of α-secretase activity resulted in a decreased secretion of Aβ peptides. Several mechanisms were elucidated as being the basis of enhanced α-secretase activity: increased membrane fluidity and impaired internalization of APP were responsible for the effect observed with methyl-β-cyclodextrin; treatment with lovastatin resulted in higher expression of the α-secretase ADAM 10. Our results demonstrate that cholesterol reduction promotes the nonamyloidogenic α-secretase pathway and the formation of neuroprotective α-secretase cleaved soluble APP by several mechanisms and suggest approaches to prevention of or therapy for Alzheimer's disease.
Amyloid-β peptides (Aβ), the principal proteinaceous components of amyloid plaques in brains of Alzheimer's disease (AD) patients, are derived from proteolytic cleavage of the amyloid precursor protein (APP), a type I integral membrane protein that is ubiquitously expressed. Both during and after its transport through the secretory pathway to the surface of cultured cells, a fraction of APP molecules undergoes specific endoproteolytic cleavage, most frequently by a scission between amino acids 16 and 17 of the Aβ region (1). This principal secretory cleavage is effected by (a) protease(s) designated as α-secretase(s). Soluble N-terminal APP fragments of 105–125 kDa are released into vesicle lumens and from the cell surface; similar species are readily detected in human plasma and cerebrospinal fluid (2). Recently, evidence has been provided that members of the ADAM family (a disintegrin and metalloprotease) act as α-secretases (3–5). For ADAM 10, basal and protein kinase C-stimulated α-secretase activity and many properties expected for the proteolytic processing of APP have been found (4).
The stimulation of α-secretase activity and an increase of α-secretase cleaved soluble APP (APPsα) might be beneficial for the treatment of AD for several reasons. In principle, proteolytic cleavage of APP within the Aβ sequence precludes the formation of the amyloid peptides derived from alternative proteolysis of APP with the β-secretase cleaving at the N terminus and the γ-secretase(s) at the C terminus of Aβ peptides (for a recent review of APP processing, see ref. 6). On the other hand, APPsα has trophic effects on cerebral neurons in culture (7), stimulates neurite outgrowth (8), and regulates synaptogenesis (9). This observation corresponds to the structural similarity of the APP-N-terminal domain with cysteine-rich growth factors (10). Furthermore, human secreted APP stabilizes neuronal calcium homeostasis and protects hippocampal and cortical neurons against the toxic effects of glutamate and Aβ peptides (11). It has been shown that APPsα can counteract the proapoptotic action of mutant presenilin-1 by activating the transcription factor NF-κB (12). In behavioral paradigms, Mezaine et al. (13) reported that APPsα has memory-enhancing effects in normal and amnestic mice.
Biochemical, epidemiological, and genetic findings link cholesterol levels and AD (14). The ɛ4 allele of apolipoprotein E, which increases the risk for early onset of the disease, is associated with higher plasma cholesterol levels (15). Several in vitro studies have demonstrated that high cholesterol concentrations in the medium of cultured cells inhibit the secretion of soluble APP (16–18). Aβ production in APP transfected hippocampal cells was decreased after cellular cholesterol depletion (19). In a transgenic mouse model, high dietary cholesterol accelerates AD-related pathologies, including Aβ deposition (20).
Different mechanisms and target secretases have been discussed to explain the relationship between cholesterol and APP proteolysis. The present study addresses the following questions: (i) Does removal of cellular cholesterol lead to a substantial increase of secreted APPsα? In particular, is the α-secretase activity of ADAM 10 regulated by cholesterol? Recent cytochemical studies support the role of ADAM 10 as an authentic α-secretase in mouse and human brain (21). (ii) What are the molecular mechanisms of α-secretase activity regulation by cholesterol? (iii) Is the increase of α-secretase activity after reduction of cellular cholesterol accompanied by a decrease of β-secretase activity and a reduction of Aβ production?
To answer these questions, we reduced cellular cholesterol by treatment with either methyl-β-cyclodextrin (MβCD) or lovastatin in several peripheral and neural cell lines, including human embryonic kidney (HEK) 293 cells overexpressing the α-secretase ADAM 10. The effect on APP processing, especially on α-secretase activity, was measured, and the influence of cholesterol depletion on membrane fluidity and APP internalization was determined. Furthermore, we examined the effect of cholesterol depletion on the localization of ADAM 10 in lipid rafts and on the level of its expression.
Materials and Methods
Materials.
Antibodies against APP were as follows: mouse IgG 6E10 (Senetek, St. Louis) detecting APPsα; rabbit antibody 192 wild type (S. Sinha, Elan Pharmaceuticals, San Francisco) against APP residues 591–596 detecting only β-secretase cleaved soluble APP (APPsβ); rabbit polyclonal antibody 5314 against the N-terminal part of APP (amino acids 444–592) (C. Haass, University of Munich, Germany); antihemagglutinin rabbit antibody Y-11 (Santa Cruz Biotechnology). Filipin, steroids, MβCD, and lovastatin were from Sigma. Lovastatin and steroid–MβCD complexes were prepared as described (22, 23). Incorporation of the steroids into the cholesterol-depleted cell membranes was determined by TLC analysis as described (24).
Modulation of Cellular Cholesterol.
Neuroblastoma cells SH-SY5Y were grown in DME/Ham's F-12 medium with 15% FCS, and all other cells were grown in DMEM supplemented with 10% FCS. All cells were grown to near confluency on 10-cm dishes coated with poly(L)lysine. Cells were depleted of cholesterol or enriched with cholesterol by incubation for 30 min at 37°C with 10 mM MβCD or 0.3 mM cholesterol–MβCD complex, respectively (23). After washing (3×) with serum-free DMEM, cells were incubated for 4 h in serum-free DMEM containing fatty acid-free BSA (10 μg/ml). Medium was collected and analyzed for the amount of APPsα, as described below. For exchange of cholesterol by other steroids, cholesterol-depleted cells were reloaded with the steroids by incubation with the steroid–MβCD complexes (0.3 mM) for 30 min. Cells for the lovastatin experiments were cultured for 20 h with DMEM supplemented with 10% lipid-deficient serum and 1 μM lovastatin. Then the medium was replaced with serum-free DMEM containing fatty acid-free BSA (10 μg/ml) and 1 μM lovastatin. In another series of experiments, cells were incubated for 4 h with filipin (1 μg/ml) in serum-free DMEM containing fatty acid-free BSA (10 μg/ml). Medium was collected and analyzed for the amount of APPsα.
Immunoblot Analysis of Secreted APPsα and -β.
Cell supernatants were collected, and proteins were precipitated with 10% (vol/vol) trichloroacetic acid (TCA). Immunoblot analysis of secreted APPsα was performed as described (4). For APPsα detection, membranes were probed with antibody 6E10 (1:2,000) followed by a 35S-labeled anti-mouse IgG antibody. The radioactive bands corresponding to APPsα were quantified with the Bio-Imaging Analyzer BAS-1800 (Fuji). The protein content of each cell culture dish was determined by the method of Bradford (25), and the values of the radioactive bands were normalized to the protein amount. For APPsβ determination, membranes were probed with antibody 192 wild type, followed by alkaline phosphatase-coupled anti-rabbit IgG secondary antibodies (Tropix, Bedford, MA) and Western-Star chemiluminescent detection system (Tropix).
Analysis of Aβ40.
Human astroglioma cells were treated with 10 mM MβCD, as described. Culture medium (400 μl) was collected and assayed for Aβ40 by using a previously described ELISA assay (26).
Cell Surface Biotinylation and Detection of Biotinylated APP.
Cells were incubated in PBS containing 1 mg/ml of sulfo-NHS-LC-Biotin (Pierce) for 30 min at 4°C. After biotinylation, cells were washed with serum-free DMEM, then incubated for 30 min with 10 mM MβCD for cholesterol depletion or with serum-free DMEM. Cells were incubated for 4 h in serum-free DMEM containing fatty acid-free BSA. Media samples were precipitated with TCA to detect all APPs or were incubated with streptavidin–agarose beads for 2 h at 4°C to detect biotinylated APPs. Biotinylated APPsα and APPsα were detected with 6E10 antibody, as described above.
Confocal Microscopy.
HEK cells overexpressing ADAM 10 (HEK ADAM 10) cells (4) grown on poly(L)lysine-coated glass coverslips were treated with 10 mM MβCD, as described. After incubation with anti-N-terminal APP 5314 antibody, cells were fixed with 4% paraformaldehyde and stained for visualization with Cy3-coupled anti-rabbit IgG antibody (Jackson ImmunoResearch). Confocal microscopy was performed on a Leica microscope TCS-SP with a 40 × 1.0 oil immersion objective.
Flotation Gradients.
HEK ADAM 10 cells grown to confluency in three 10-cm dishes were depleted of cholesterol by incubation with MβCD for 30 min or treated with serum-free medium, washed with ice-cold PBS, scraped in PBS, and spun down at 750 × g at 4°C. Fractionations were carried out as described by Harder et al. (27) with minor modifications. Twelve fractions from the top of the gradient were collected and precipitated with TCA. Hemagglutinin-tagged ADAM 10 was detected by immunoblot with antihemagglutinin antibody Y-11, followed by alkaline phosphatase-coupled secondary antibodies and the Western-Star chemiluminescent detection system (Tropix).
Analytical Methods.
For lipid determination, the samples were extracted with chloroform–methanol according to the method of Bligh and Dyer (28). Cholesterol was assayed spectrophotometrically by using the Boehringer Roche Diagnostic kit. Steady-state anisotropy measurements were performed as described (24).
Results
Effect of Cholesterol Depletion on the α-Secretase Activity ADAM 10 in HEK Cells.
Studies with a dominant negative form of the disintegrin metalloprotease ADAM 10 have provided evidence that the α-secretase activity in HEK 293 cells is mainly because of the activity of ADAM 10 (4). The influence of cholesterol on the activity of the endogenous α-secretase in HEK cells and after overexpression of ADAM 10 was examined after depletion of cellular cholesterol with MβCD. Treatment of HEK cells with 10 mM MβCD for 30 min resulted in removal of 63 ± 8% of cholesterol (n = 6). After cholesterol depletion, cells were incubated for 4 h, and the release of APPsα into the medium was monitored with the antibody 6E10. APPsα was released by HEK cells into the medium approximately three times more after cholesterol depletion (Fig. 1, lane 2). HEK ADAM 10 showed an already 3-fold enhanced α-secretase activity (Fig. 1, lane 3) as compared with untransfected HEK cells. Treatment with MβCD yielded a further 3-fold increase in secreted APPsα (Fig. 1, lane 4).
Figure 1.

Influence of cholesterol depletion on the secretion of APPsα from HEK and HEK ADAM 10 cells. (A) Cells were incubated in the presence of 10 mM MβCD for 30 min (lanes 2 and 4). After 4 h, the medium was collected, and proteins were precipitated and subjected to immunoblot analysis with the antibody 6E10 followed by a 35S-labeled anti-mouse IgG antibody. (B) Quantitative analysis of secreted APPsα. Bars indicate mean ± SD (n = 4). **, P < 0.001 (Student's t test).
In another series of experiments, maximal stimulation of α-secretase activity after cholesterol depletion was up to even 6-fold in HEK cells (see below, Fig. 2) and again up to 3-fold in HEK ADAM 10 cells. In comparison, stimulation of HEK and HEK ADAM 10 cells with the phorbol ester phorbol-12-myristate 13-acetate (PMA) resulted in a roughly 4-fold increase of α-secretase activity in HEK cells and a 2-fold increase in HEK ADAM 10 cells (not shown). A stronger relative stimulation of the α-secretase activity by PMA in HEK cells than in HEK ADAM 10 cells has been observed before (4). Together, these results demonstrate the strong stimulatory effect of cholesterol depletion on the α-secretase activity of ADAM 10, that is in the range obtained with phorbol esters and may be limited by the enzyme to substrate ratio.
Figure 2.

Relation between cholesterol, membrane fluidity, and α-secretase activity. (A) Secretion of APPsα as a function of cellular cholesterol amount and of fluorescence anisotropy in membranes of HEK cells. HEK cells were incubated with 10 mM MβCD for various times (0, 2, 5, 10, 20, 30 min) to remove defined cholesterol amounts. After 4 h, the medium was collected, and secreted APPsα was identified as described in Fig. 1. The cell pellet was split for determination of cellular cholesterol and protein content and for measurements of 1,6-diphenyl-1,3,5-hexatriene steady-state fluorescence anisotropy in cell membranes. The data are expressed as percentage of control and are the averages ± SD of at least three experiments. (B) Influence of steroids on secretion of APPsα from HEK ADAM 10 cells, which were incubated in the presence of 10 mM MβCD for 30 min at 37°C. Subsequently, cells were incubated in the presence of 0.3 mM steroid–MβCD complexes for 30 min at 37°C.
Influence of Plasma Membrane Fluidity on α-Secretase Activity.
To determine whether the stimulatory effect of cholesterol removal on the α-secretase activity was because of an influence of cholesterol on the plasma membrane fluidity, the α-secretase activity and the membrane fluidity were analyzed in dependence of the cellular cholesterol content. Cells were pretreated with 10 mM MβCD for various times (2–30 min) to remove stepwise defined amounts of cholesterol. In cell membranes, the “steady-state” fluorescence anisotropy of 1,6-diphenyl-1,3,5-hexatriene was measured, which is inversely related to membrane fluidity. The activity of the α-secretase showed a very similar dependence on the cellular cholesterol content and on fluorescence anisotropy (Fig. 2A). The activity of the α-secretase first slightly increased in parallel with the removal of cholesterol. However, when the cholesterol amount was lower than 60% of the initial amount, the α-secretase activity sharply increased. As expected, membranes prepared from cholesterol-depleted cells had significantly reduced fluorescence anisotropy values, i.e., increased membrane fluidity. A strong increase in α-secretase activity was observed when the fluorescence anisotropy of the cell membranes had been reduced to less than r = 0.167 (Fig. 2A). A similar dependence of α-secretase activity was observed for HEK cells overexpressing ADAM 10.
Substitution of cholesterol in isolated plasma membranes of HEK cells by the steroid 4-cholesten-3-one has only a minor effect on membrane fluidity, whereas substitution of cholesterol by lanosterol increases membrane fluidity (24). We therefore analyzed the influence of these steroids on the α-secretase activity. Reloading of cholesterol-depleted HEK ADAM 10 cells with 4-cholesten-3-one reduced the amount of APPsα secreted by HEK ADAM 10 cells to nearly the same level as compared with cells reloaded with cholesterol (Fig. 2B, bars 3 and 4). In contrast, when cholesterol-depleted HEK ADAM 10 cells were reloaded with lanosterol, no reduction of α-secretase activity occurred (Fig. 2B, bars 2 and 5). The same effects were observed for HEK cells. Thus, these data demonstrate that the effect of cholesterol on the α-secretase activity is reversible, dependent on membrane fluidity, and that replacement of cholesterol by its precursor lanosterol strongly enhances α-secretase activity.
Inhibition of APP Internalization After Cholesterol Depletion.
Recently it has been shown that cholesterol is essential for clathrin-dependent endocytosis of transferrin receptors: cholesterol depletion by using cyclodextrin inhibited clathrin-coated pit budding and endocytosis of transferrin receptors (29, 30). As mutations of APP that inhibit its endocytosis result in enhanced secretion of soluble APP (31), we studied the influence of cholesterol depletion on APP endocytosis in HEK ADAM 10 cells by confocal microscopy and by cell surface biotinylation experiments.
After treatment of HEK ADAM 10 cells with MβCD, confocal microscopy of the nonpermeabilized cells with antibody 5314 directed against an extracellular region of APP revealed an increased amount of APP on the cell surface (Fig. 3A Right) as compared with untreated cells (Fig. 3A Left). To determine the amount of APPsα produced by α-secretase cleavage on the cell surface, HEK ADAM 10 cells were first biotinylated and then incubated with MβCD to remove cholesterol. Biotinylated proteins secreted into the medium were bound to streptavidin–agarose beads, whereas the total secreted proteins were precipitated with TCA. As can be seen in Fig. 3B, after cholesterol depletion a ≈2-fold increase of both secreted biotinylated APPsα and total APPsα was detected in the cell medium. The results demonstrate that removing cholesterol from the plasma membrane inhibits endocytosis of APP and increases the amount of APP at the cell surface, where it could undergo enhanced α-secretase cleavage.
Figure 3.

Effect of cholesterol depletion on APP internalization and APPsα secretion. (A) Localization of APP on the cell surface of cholesterol-depleted HEK ADAM 10 cells, which were incubated in serum-free medium (control, Left) or in serum-free medium with 10 mM MβCD for 30 min at 37°C (Right). Cells were stained for APP with the anti-N-terminal APP antibody 5314 and visualized with Cy3-coupled anti-rabbit IgG. (B) Influence of cholesterol depletion on the secretion of biotinylated APPsα. HEK ADAM 10 cells were first biotinylated, then treated with MβCD. Secreted biotinylated APPsα was bound to streptavidin–agarose beads and, in parallel, total secreted APPsα was collected by TCA precipitation. Quantitative analysis of secreted biotinylated APPsα and APPsα was performed as described above. Statistical significance between control cells and MβCD-treated cells was determined by Student's unpaired t test (*, P < 0.05).
Localization of ADAM 10 in Detergent-Soluble and Insoluble Membrane Compartments.
It was proposed that α-secretase cleavage of APP occurs preferentially in caveolae, cholesterol-rich plasma membrane microdomains (32). Therefore, we analyzed whether the α-secretase ADAM 10 is insoluble in Triton X-100 at 4°C, a property shared with transmembrane proteins associating with sphingolipid-cholesterol rafts (33). After separation in an OptiPrep step gradient, both forms of ADAM 10, the full-length precursor (90 kDa) and the proteolytically active form lacking the prodomain (64 kDa), were present mainly in fractions of higher density with the majority of ADAM 10 as proenzyme. Only a low amount of proenzyme was detected in detergent-insoluble low-density complexes (Fig. 4A, fractions 2 and 3). After cholesterol depletion with cyclodextrin whereby association of proteins with rafts is prevented, both ADAM 10 forms were completely localized in high-density fractions (Fig. 4A).
Figure 4.

Effect of cholesterol depletion on the distribution of the α-secretase ADAM 10 in detergent-insoluble lipid rafts and in caveolae. (A) Localization of ADAM 10 in lipid rafts. HEK ADAM 10 cells with or without treatment for 30 min with 10 mM MβCD were extracted with 2% Triton X-100. After flotation in an OptiPrep step-gradient, the fractions were TCA precipitated and analyzed by Western blot with antihemagglutinin antibody for ADAM 10 detection. (B) HEK ADAM 10 cells were incubated for 4 h with filipin (1 μg/ml). Then the cell medium was collected and the secreted APPsα was quantified as described above. Statistical significance between control cells and filipin-treated cells was determined by Student's unpaired t test (*, P < 0.05).
We also examined the effects of the sterol-binding agent filipin on APPsα secretion. Filipin binds to cholesterol, a major component of glycolipid microdomains and caveolae, and disrupts caveolar structure and function (34, 35). After filipin treatment, both HEK and HEK ADAM 10 cells exhibited an increase in α-secretase activity: about 70% more APPsα was detected as compared with untreated cells (Fig. 4B). Conclusively, cholesterol depletion as well as filipin treatment of HEK cells leads to a disruption of rafts and caveolae on the one hand and to an increase in α-secretase activity on the other.
Effect of Cholesterol Depletion on the α-Secretase Activity and β-Amyloid Peptide Production in Neural Cell Lines.
Because there is evidence that APP processing might be cell type-dependent, we determined whether cholesterol has an effect on the α-secretase activity in two neural cell lines. Treatment of human neuroglioma H4 cells with 10 mM MβCD resulted in a removal of 67 ± 5% (n = 6) of cholesterol and in a 2.7 ± 0.8 (n = 6) relative increase of APPsα. Human astroglioma U373 cells overexpressing APP showed already a high basal level of APPsα in the medium. Therefore the effect of cholesterol removal (66 ± 1%, n = 4) was less intense (1.5-fold increase of APPsα; Fig. 5). To examine the relationship between cholesterol levels and β-amyloid production, the amount of β-amyloid peptide (1–40) was determined. Human astroglioma U373 cells overexpressing APP were chosen (because many assays are not sensitive enough to detect Aβ peptides in the medium of cells containing low amounts of endogenously expressed APP). Treatment with 10 mM MβCD for 30 min reduced the secretion of Aβ/40 by 40–45% (Fig. 5). To determine whether the increase in α-secretase activity is accompanied by a decrease in β-secretase activity, we analyzed cell extracts with the antibody 192 wild type, which recognizes APPsβ. Treatment with MβCD resulted in a significant reduction of secreted APPsβ by 50–60% (Fig. 5).
Figure 5.
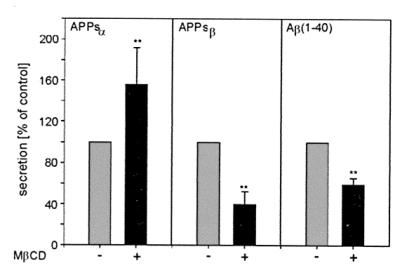
Influence of cholesterol depletion on the secretion of APPsα, APPsβ, and Aβ from human astroglioma (U373) cells. U373 cells were incubated for 30 min with 10 mM MβCD for cholesterol depletion. After 4 h incubation, medium was collected, and quantitative analysis of secreted APPsα and APPsβ was performed as described in Materials and Methods. For Aβ detection, a highly specific ELISA assay was performed. The results are expressed as percentage of secreted APP fragments in control HEK cells and are the averages ± SD of at least four experiments. Statistical significance between control cells and MβCD-treated cells was determined by Student's unpaired t test (**, P < 0.001).
Influence of Lovastatin on α-Secretase Activity and ADAM 10 Expression.
For deprivation of the cellular cholesterol by inhibiting cholesterol synthesis, cells were cultured for 20 h in a lipid-deficient medium in the presence of 1 μM lovastatin, a potent inhibitor of 3-hydroxy-3-methylglutaryl-CoA reductase. This treatment reduced cholesterol content by ≈50% as compared with cells cultured in regular medium. As shown in Table 1, lovastatin treatment resulted in an increase of α-secretase activity in all tested peripheral and neural cell types. In the human astroglioma cells U373 overexpressing APP, we observed a significant (≈20%) reduction of Aβ secretion after lovastatin treatment. Increased α-secretase activity was also observed when 200 μM mevalonate was included in the medium to provide for nonsterol pathways, such as protein isoprenylation.
Table 1.
Effect of lovastatin on APPsα secretion in different cell lines
| Cell line | APPsα (% of control) |
|---|---|
| HEK 293 | 272 ± 46* |
| HEK ADAM 10 | 190 ± 20** |
| U373 | 151 ± 7** |
| H4 | 139 ± 4** |
| SH-SY5Y (APP751/770) | 171 ± 4** |
| SH-SY5Y (APP695) | 221 ± 9** |
Cells were cultured in DMEM supplemented with 10% lipid-deficient serum and 1 μM lovastatin for 20 h. After 4 h, the medium was collected, and APPsα was determined as described. Control cells were cultured without lovastatin.
Data are expressed as percentage of control and represent the mean ± SD for three independent determinations. *, P < 0.005; **, P < 0.001 (Student's t test).
In another series of experiments, we investigated whether exogenous cholesterol could prevent the increase of α-secretase activity by treatment with lovastatin. First we analyzed the effect of cholesterol enrichment on α-secretase activity. For that purpose, HEK ADAM 10 cells cultured in lipid-deficient medium were incubated with 0.3 mM MβCD–cholesterol complex for 20 h, which increased cellular cholesterol (≈170%) and caused 30% inhibition of the α-secretase activity (Fig. 6A, bar 2). Thus, increasing the cellular cholesterol content reduces the activity of ADAM 10. Simultaneous incubation of HEK ADAM 10 cells with 0.3 mM cholesterol-cyclodextrin complex and 1 μM lovastatin for 20 h resulted in an increased cholesterol content (≈200%) and prevented the increase in α-secretase activity by lovastatin (Fig. 6A, bars 3 and 4). Treatment of HEK and HEK ADAM 10 cells with lovastatin (20 h) resulted in increased expression of ADAM 10. In HEK ADAM 10 cells, we observed an increased amount (30–70%) of both the proform and the mature form of enzyme (Fig. 6B). Treatment with neither lovastatin nor MβCD affected the level of APP holoprotein.
Figure 6.

Influence of lovastatin on activity and expression of the α-secretase ADAM 10 in HEK ADAM 10 cells. (A) Reversal of the lovastatin effect on APPsα secretion in HEK ADAM 10 cells by cholesterol. All cells were cultured in lipid-deficient medium either in the absence (lane 1) or in the presence of 0.3 mM cholesterol–MβCD complex (lane 2), in the presence of 1 μM lovastatin (lane 3), or in the presence of 1 μM lovastatin and 0.3 mM cholesterol–MβCD complex (lane 4). The medium was analyzed for secreted APPsα, as described in Fig. 1. Statistical significance between control cells and treated cells was determined by Student's unpaired t test (*, P < 0.01; **, P < 0.005)). (B) Membranes (200 μg of each) prepared from cells cultured in lipid-deficient serum in the absence (lane 1) or in the presence of 1 μM lovastatin were separated by electrophoresis and transferred to PVDF membranes. For detection of ADAM 10, membranes were probed with an antihemagglutinin antibody (Y-11).
Discussion
Despite several reports on the influence of cholesterol on the processing of APP, the detailed molecular mechanisms have not yet been elucidated. In the present report, we identify the α-secretase ADAM 10 as a major target of the cholesterol effects and provide evidence for several mechanisms as the basis for the strong increase of α-secretase activity induced by reduction of cellular cholesterol.
Treatment of various peripheral and neural cell lines with either MβCD or lovastatin resulted in a drastic increase of secreted APPsα. In HEK cells, where ADAM 10 has been identified as the major enzyme responsible for α-secretase activity (4), a 3- to 6-fold increase in generation of APPsα was observed after cholesterol depletion. This strong stimulatory effect of cholesterol depletion is in the range obtained with phorbol esters (4). Overexpression of ADAM 10 led to a several-fold enhanced α-secretase activity, which was further increased after cholesterol depletion. A lower increase of α-secretase activity was observed in cells overexpressing APP. The high concentration of substrate leads to high basal α-secretase activity, probably because of enzyme saturation, and thus limits an additional increase of α-secretase activity.
From this study and two other reports (16, 19), it can be concluded that an increase of APP holoprotein does not contribute to the increase of α-secretase activity by cholesterol depletion. According to our results, the regulatory role of cholesterol for α-secretase activity can be attributed to the following mechanisms: (i) The MβCD-mediated removal of cholesterol from the plasma membranes significantly increased the membrane fluidity, as shown by steady-state anisotropy measurements. Cholesterol depletion below a critical concentration (about 60% of the initial quantity) caused a significantly enhanced α-secretase activity along with increased membrane fluidity. This could increase the lateral movement of APP and the α-secretase within the membrane. The influence of membrane fluidity on α-secretase activity was confirmed in experiments, where cholesterol was substituted by other steroids. Substitution by lanosterol led to an increase in membrane fluidity and to a several-fold increase in α-secretase activity. Thus, α-secretase activity might be enhanced by inhibitors or genetic defects (36) of postlanosterol cholesterol biosynthesis.
(ii) APP as a transmembrane protein may reside at the cell surface (37) and is reinternalized via clathrin-coated pits (38, 39) to the endosomal–lysosomal pathway (40). Several reports support a principally cell surface localization for α-secretase activity (40, 41). By two different approaches, we demonstrate an inhibition of APP endocytosis and an increased amount of APP on the cell surface after cholesterol depletion with MβCD. This treatment resulted in an increased α-secretase cleavage of APP on the cell surface.
(iii) Treatment of peripheral and neural cells with lovastatin, an inhibitor of hydroxymethyl glutaryl-CoA reductase, the rate-limiting enzyme in cholesterol synthesis, also resulted in a strong increase of the α-secretase activity. Secretion of APPsα was prevented by complementation of cells with cholesterol, which suggests that the effect of lovastatin on α-secretase activity was because of its reduction of cholesterol synthesis. Because the membrane-impermeable cyclodextrin removes cell-surface cholesterol, alternative or additional mechanisms might be responsible for the effect of lovastatin on the α-secretase activity. So we did not find an influence of lovastatin on membrane fluidity. After 20-h treatment with lovastatin, we observed an increase in the expression of the mature form and the proform of ADAM 10. No significant change of ADAM 10 expression was found after treatment with MβCD, which, however, was limited to 30 min because of cell damages after prolonged exposure. It will be important to study by which molecular mechanism the statin-reduced cellular concentration of cholesterol increases the expression of ADAM 10.
Lipid rafts have been proposed as the site for the proteolytic processing of APP. Currently there are conflicting reports as to whether APP is present in lipid rafts (19, 42–44). Studies with peripheral cell lines led to the conclusion that α-secretase cleavage occurs at the cell surface and that cholesterol-rich caveolae microdomains may play a role in the α-secretase-mediated proteolysis of APP in vivo (32). Detergent insolubility in Triton-X-100 at 4°C was used in our study as the basis for isolating lipid rafts. Only a minor fraction of the ADAM 10 immature proform associated with rafts in HEK cells. This was prevented by cholesterol depletion, i.e., under conditions where an increased α-secretase activity was observed. Furthermore, filipin treatment that causes destruction of caveolae structures also led to a substantial increase of α-secretase activity. From these results, we conclude that only a minor part of APP could be cleaved by the α-secretase within rafts or caveolae microdomains. However, because a fraction of the proform of ADAM 10 is localized in rafts, cholesterol-rich domains might play a role for maturation and transport of ADAM 10 in the secretory pathway. For APP, it has been shown that exposure of neural cells to cholesterol interfered with its glycosylation and decreased its secretion (18).
In cells overexpressing APP, we could demonstrate that the increase of α-secretase activity after cholesterol reduction resulted in a decreased secretion of Aβ peptides. For cyclodextrin treatment of human astroglioma cells, we observed a similar reduction of secreted APPsβ. The decreased β-secretase activity after cholesterol depletion could be a result of the reduced amount of the substrate available for β-secretase cleavage. The β-secretase β-site APP-cleaving enzyme (BACE) was found to circulate between the cell membrane and endosomes (45). In situ hybridization provided evidence for the coexpression of β-APP with BACE and ADAM 10 in human cortical neurons (21). Thus cholesterol depletion leads to an increased nonamyloidogenic α-secretase cleavage accompanied by a decreased concentration of Aβ peptides. An inhibition of Aβ production after cholesterol depletion of hippocampal neurons overexpressing APP has also been demonstrated (19). The authors proposed that inhibition of the β-secretase activity in lipid rafts might be responsible for decreased Aβ production. An increase in APPsα secretion was not detectable probably because they analyzed the processing of overexpressed APP in cells with only a limited survival time after viral infections.
Several potential mechanisms for the effect of ApoE4 in increasing Aβ deposition have been published, e.g., decreased inhibition of Aβ fibrilliogenesis by ApoE4 vs. ApoE3 (for a review, see ref. 46). A recent study has shown that apolipoprotein E promotes the efflux of cholesterol and phosphatidyl choline from both astrocytes and neurons. The order of potency of the isoforms as lipid acceptors was ApoE2 > ApoE3 > ApoE4 in neurons (47). From our results, one may conclude that the lower cholesterol efflux mediated by ApoE4 contributes to a reduction of α-secretase activity and to pathogenesis of AD.
In conclusion, we provided evidence for several mechanisms that are responsible for the large increase of nonamyloidogenic α-secretase cleavage of APP. Increased membrane fluidity and impaired internalization of APP are responsible for the increased α-secretase activity after acute cholesterol depletion by treatment with cyclodextrin. The hydroxymethyl glutaryl-CoA-reductase inhibitor lovastatin increases the expression of the α-secretase ADAM 10 by yet-not-elucidated mechanisms. Therapeutic intervention with statins that have been shown to cross the blood–brain barrier (48) might therefore be helpful to those AD patients at early stages of the disease. Recently an epidemiological study demonstrated that individuals over age 50 had a substantially lower risk of developing dementia after statin treatment (49). Studies with the transgenic mouse model for AD amyloidosis suggest that diet can be used to modulate the risk of developing AD (20). From the results of our study, additional interventions seem possible to increase the α-secretase activity and the amount of secreted APPsα known to have neuroprotective and growth promoting properties.
Acknowledgments
We thank Drs. C. Haass and S. Sinha for providing antibodies and Drs. K. Mendla and H. Romig (Boehringer Ingelheim) for providing human astroglioma cell lines and for the determination of Aβ. We also appreciate the technical help of K. Gajda and the computing advice and confocal microscopy of F. Bender (University of Mainz, Germany). This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 474 and Fa 122/5–1) and Fonds der Chemischen Industrie.
Abbreviations
- Aβ
amyloid β peptide
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- APPsα
α-secretase cleaved soluble APP
- APPsβ
β-secretase cleaved soluble APP
- HEK
human embryonic kidney
- MβCD
methyl-β-cyclodextrin
- TCA
trichloroacetic acid
- HEK ADAM 10
HEK cells overexpressing ADAM 10
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 5371.
References
- 1.Esch F S, Keim P S, Beattie E C, Blacher R W, Culwell A R, Oltersdorf T, McClure D, Ward P J. Science. 1990;248:1122–1124. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- 2.Palmert M R, Podlisny M B, Witker D S, Oltersdorf T, Younkin S H, Selkoe D L, Younkin S G. Proc Natl Acad Sci USA. 1998;86:6338–6342. doi: 10.1073/pnas.86.16.6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buxbaum J D, Liu K N, Luo Y, Slack JL, Stocking K L, Peschon J J, Johnson R S, Castner B J, Cerretti D P, Black R A. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 4.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Proc Natl Acad Sci USA. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koike H, Tomioka S, Sorimachi H, Saido T C, Maruyama K, Okuyama A, Fujisawa-Sehara A, Ohno S, Suzuki K, Ishiura S. Biochem J. 1999;343:371–375. [PMC free article] [PubMed] [Google Scholar]
- 6.De Strooper B, Annaert W. J Cell Sci. 2000;113:1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- 7.Araki W, Kitaguchi N, Tokushima Y, Ishii K, Aratake H, Shimohama S, Nakamura S, Kimura J. Biochem Biophys Res Commun. 1991;181:265–271. doi: 10.1016/s0006-291x(05)81412-3. [DOI] [PubMed] [Google Scholar]
- 8.Small D H, Nurcombe V, Reed G, Clarris H, Moir R, Beyreuther K, Masters C L. J Neurosci. 1994;14:2117–2127. doi: 10.1523/JNEUROSCI.14-04-02117.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morimoto T, Ohsawa I, Takamura C, Ishiguro M, Kohsaka S. J Neurosci Res. 1998;51:185–195. doi: 10.1002/(SICI)1097-4547(19980115)51:2<185::AID-JNR7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 10.Rossjohn J, Cappai R, Feil S C, Henry A, McKinstry W J, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters C L, Parker M W. Nat Struct Biol. 1999;6:327–331. doi: 10.1038/7562. [DOI] [PubMed] [Google Scholar]
- 11.Furukawa K S, Barger E, Blalock E, Mattson M. Nature (London) 1996;379:74–78. doi: 10.1038/379074a0. [DOI] [PubMed] [Google Scholar]
- 12.Guo Q, Robinson N, Mattson M P. J Biol Chem. 1998;273:12341–12351. doi: 10.1074/jbc.273.20.12341. [DOI] [PubMed] [Google Scholar]
- 13.Meziane H, Dodart J C, Mathis C, Little S, Clemens J, Paul S M, Ungerer A. Proc Natl Acad Sci USA. 1998;95:12683–12688. doi: 10.1073/pnas.95.21.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dartigues J F, Letenneur L. Curr Opin Neurol. 2000;13:385–389. doi: 10.1097/00019052-200008000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Sing C F, Davignon J. Am J Hum Genet. 1985;37:268–286. [PMC free article] [PubMed] [Google Scholar]
- 16.Bodovitz S, Klein W L. J Biol Chem. 1996;271:4436–4440. doi: 10.1074/jbc.271.8.4436. [DOI] [PubMed] [Google Scholar]
- 17.Racchi M, Baetta R, Salvietti N, Ianna P, Franceschini G, Paoletti R, Fumagalli R, Govoni S, Trabucchi M, Soma M. Biochem J. 1997;322:893–898. doi: 10.1042/bj3220893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galbete J L, Rodriguez-Martin T, Peressini E, Modena P, Bianchi R, Forloni G. Biochem J. 2000;348:307–313. [PMC free article] [PubMed] [Google Scholar]
- 19.Simons M, Keller P, De Strooper B, Beyreuther K, Dotti C G, Simons K. Proc Natl Acad Sci USA. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Refolo L M, Pappolla M A, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint G S, Sambamurti K, Duff K. Neurobiol Dis. 2000;7:321–331. doi: 10.1006/nbdi.2000.0304. [DOI] [PubMed] [Google Scholar]
- 21.Marcinkiewicz M, Seidah N G. J Neurochem. 2000;75:2133–2143. doi: 10.1046/j.1471-4159.2000.0752133.x. [DOI] [PubMed] [Google Scholar]
- 22.Cutts J L, Melnykovych G. Biochim Biophys Acta. 1988;961:65–72. doi: 10.1016/0005-2760(88)90131-2. [DOI] [PubMed] [Google Scholar]
- 23.Klein U, Gimpl G, Fahrenholz F. Biochemistry. 1995;34:13784–13793. doi: 10.1021/bi00042a009. [DOI] [PubMed] [Google Scholar]
- 24.Gimpl G, Burger K, Fahrenholz F. Biochemistry. 1997;36:10959–10974. doi: 10.1021/bi963138w. [DOI] [PubMed] [Google Scholar]
- 25.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 26.Steiner H, Capell A, Pesold B, Citron M, Kloetzel P, Selkoe D J, Romig H, Mendla K, Haass C. J Biol Chem. 1998;273:32322–32331. doi: 10.1074/jbc.273.48.32322. [DOI] [PubMed] [Google Scholar]
- 27.Harder T, Scheiffele P, Verkade P, Simons K. J Cell Biol. 1998;141:929–942. doi: 10.1083/jcb.141.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bligh E D, Dyer W J. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 29.Subtil A, Gaidarow I, Kobylarz K, Lampson M A, Keen J H, McGraw T E. Proc Natl Acad Sci USA. 1999;96:6775–6780. doi: 10.1073/pnas.96.12.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodal S K, Skretting G, Garred O, Vilhardt F, van Deurs B, Sandvig K. Mol Biol Cell. 1999;10:961–974. doi: 10.1091/mbc.10.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez R G, Soriano S, Hayes J D, Ostaszewski B, Xia W, Selkoe D J, Chen X, Stokin G B, Koo H. J Biol Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- 32.Ikezu T, Trapp B D, Song K S, Schlegel A, Lisanti M P, Okamoto T. J Biol Chem. 1998;273:10485–10495. doi: 10.1074/jbc.273.17.10485. [DOI] [PubMed] [Google Scholar]
- 33.Simons K, Ikonen E. Nature (London) 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 34.Rothberg K G, Heuser J E, Donzell W C, Ying Y S, Glenney J R, Anderson R G W. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- 35.Smart E J, Ying YS, Conrad P A, Anderson R G W. J Cell Biol. 1994;127:1185–1197. doi: 10.1083/jcb.127.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moebius F F, Fitzky B U, Glossmann H. Trends Endocrinol Metab. 2000;11:106–114. doi: 10.1016/s1043-2760(00)00235-6. [DOI] [PubMed] [Google Scholar]
- 37.Shivers B D, Hilbich C, Multhaup G, Salbaum M, Beyreuther K, Seeburg P H. EMBO J. 1988;7:1365–1370. doi: 10.1002/j.1460-2075.1988.tb02952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nordstedt C, Caporaso G L, Thyberg J, Gandy S E, Greengard P. J Biol Chem. 1993;268:608–612. [PubMed] [Google Scholar]
- 39.Yamazaki T, Koo E H, Selkoe D J. J Cell Sci. 1996;109:999–1008. doi: 10.1242/jcs.109.5.999. [DOI] [PubMed] [Google Scholar]
- 40.Haass C, Koo E H, Mellon A, Hung A Y, Selkoe D J. Nature (London) 1992;357:500–503. doi: 10.1038/357500a0. [DOI] [PubMed] [Google Scholar]
- 41.Parvathy S, Hussain I, Karran E H, Turner A J, Hooper N M. Biochemistry. 1999;38:9728–9734. doi: 10.1021/bi9906827. [DOI] [PubMed] [Google Scholar]
- 42.Lee S J, Liyanage U, Bickel P E, Xia W, Lansbury P T, Kosik S S. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- 43.Bouillot C, Prochiantz A, Rougon G, Allinquant B. J BiolChem. 1996;271:7640–7644. doi: 10.1074/jbc.271.13.7640. [DOI] [PubMed] [Google Scholar]
- 44.Parkin E T, Hussain I, Hooper N M. J Neurochem. 1997;69:2179–2188. doi: 10.1046/j.1471-4159.1997.69052179.x. [DOI] [PubMed] [Google Scholar]
- 45.Huse J T, Pijak D S, Leslie G J, Lee V M Y, Doms R W. J Biol Chem. 2000;275:33729–33737. doi: 10.1074/jbc.M004175200. [DOI] [PubMed] [Google Scholar]
- 46.Weisgraber K H, Mahley R W. FASEB J. 1996;10:1485–1494. doi: 10.1096/fasebj.10.13.8940294. [DOI] [PubMed] [Google Scholar]
- 47.Michikawa M, Fan Q W, Isobe I, Yanagisawa K. J Neurochem. 2000;74:1008–1016. doi: 10.1046/j.1471-4159.2000.0741008.x. [DOI] [PubMed] [Google Scholar]
- 48.Seheki A, Terasaki T, Tamai I, Tsuji A. Pharm Res. 1994;11:305–311. doi: 10.1023/a:1018975928974. [DOI] [PubMed] [Google Scholar]
- 49.Jick H, Zornberg G L, Jick S S, Seshadri S, Drachman D A. Lancet. 2000;356:1627–1631. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]