

Abstract
Copy number variants (CNVs) account for the majority of human genomic diversity in terms of base coverage. Here, we have developed and applied a new method to combine high-resolution array comparative genomic hybridization (CGH) data with whole-genome DNA sequencing data to obtain a comprehensive catalog of common CNVs in Asian individuals. The genomes of 30 individuals from three Asian populations (Korean, Chinese and Japanese) were interrogated with an ultra-high-resolution array CGH platform containing 24 million probes. Whole-genome sequencing data from a reference genome (NA10851, with 28.3× coverage) and two Asian genomes (AK1, with 27.8× coverage and AK2, with 32.0× coverage) were used to transform the relative copy number information obtained from array CGH experiments into absolute copy number values. We discovered 5,177 CNVs, of which 3,547 were putative Asian-specific CNVs. These common CNVs in Asian populations will be a useful resource for subsequent genetic studies in these populations, and the new method of calling absolute CNVs will be essential for applying CNV data to personalized medicine.
Large-scale initiatives in sequencing individual genomes have targeted the identification of a broad range of genetic variants, from SNPs to structural genomic variants (including CNVs)1, to identify the genetic factors that contribute to an individual’s phenotype. However, current DNA sequencing strategies produce short reads (in a paired-end or non–paired-end manner), which places substantial limitations on accurately identifying structural genomic variants. Here, we have developed an integrated strategy to combine high-resolution array CGH (aCGH) information with whole-genome sequencing data to comprehensively identify and characterize common CNVs within three Asian populations.
We applied this strategy to the genomic DNA of 30 Asian females (ten Koreans (KOR), ten HapMap Chinese (CHB) and ten HapMap Japanese (JPT) individuals) to develop an accurate and comprehensive common CNV map for Asian populations that would complement a recently published CNV map for West African and European populations2. The genomic DNA of all 30 individuals was applied to a custom-designed aCGH platform comprising 24 million oligonucleotide probes (Supplementary Fig. 1). The platform was empirically determined to have an effective resolution to detect CNVs as small as 438 bp. The reference DNA source for our aCGH experiments in this study was the control individual (NA10851) used in previous CNV discovery studies3–5 (Fig. 1). We estimated that studying 30 individuals would provide 95.4% power to detect CNVs with a minor allele frequency of 5% among Asian populations.
Figure 1.
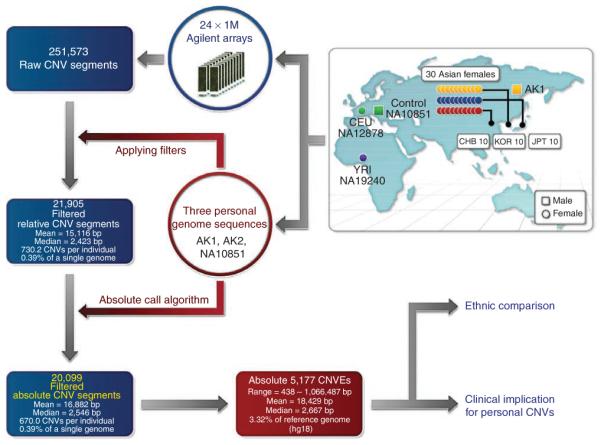
Overview of the CNV discovery project for Asian populations. The genomic DNA from ten Altaic Korean individuals, ten CHB HapMap individuals of Chinese ancestry, ten JPT HapMap individuals with Japanese ancestry and three platform-control comparison resource individuals (AK1, NA12878 and NA19240) were used for aCGH experiments. Genome sequence data from three subjects (AK1, AK2 and NA10851) were used to filter out false positive CNV calls and to obtain absolute CNV calls.
Among the 30 Asian individuals studied, we discovered 251,573 putative CNV segments. For algorithmic training purposes, we then conducted aCGH experiments on DNA from a Korean male, AK1, for whom whole-genome sequencing data was already available at 27.8× coverage5, and DNA from a Korean female, AK2, whose genome we sequenced at 32.0× coverage (data not shown, see URLs). Because all aCGH experiments used NA10851 as a reference DNA source, we also sequenced this individual at 28.3× coverage (see URLs). Using read-depth information from AK1 and NA10851, we developed criteria to optimize the concordance between aCGH and the DNA sequencing information. We subsequently used this read-depth data to further test and validate our filtering criteria (Supplementary Tables 1 and 2 and Supplementary Fig. 2). The filtered data included a total of 21,905 CNV segments from 30 Asian individuals. We found that smaller CNVs (<5 kb) with low log2 ratio were for the most part removed based on our filtering criteria.
Comparison of the aCGH results from the DNA of AK1 and AK2 with the read-depth information from whole-genome sequencing data on the same individuals suggested that approximately half of the CNV segments that were identified in AK1 and AK2 (as having relative genomic gains and losses by array CGH) actually had a copy number state of 2 in these individuals and a copy number state of <2 or >2 in the reference individual, NA10851 (Fig. 2a and Supplementary Figs. 3 and 4). The read-depth sequence information from NA10851 revealed that 10,980 out of the filtered 21,905 CNV segments might have been erroneously called because the control sample from NA10851 had copy number values not equal to 2 in these genomic regions (Fig. 2b and Supplementary Note). Out of the 10,980 ‘obscure’ CNV segments, in which NA10851 had copy number other than 2, 4,970 were determined to have a diploid copy number of two in the test individual (NA10851) after we converted them to absolute copy number states and removed them (Supplementary Fig. 4c–e). The remaining 6,010 CNV segments were found to have different copy number states from that predicted by aCGH (Fig. 2a). We also identified an additional 3,164 covert CNV segments that were initially missed by the aCGH experiments owing to their having identical copy numbers both in the test sample and NA10851(Supplementary Fig. 4f–i). By this method, we obtained absolute copy number states for a total of 20,099 CNV segments (Supplementary Table 3), in which 9,174 (3,164 from covert calls and 6,010 from obscure calls) were corrected by the read-depth sequence information for NA10851 (Fig. 2b).
Figure 2.

Original approach for calling absolute copy number status. (a) Right: The top, panel shows aCGH data for a genomic region on chromosome 3 in AK1 as compared to the reference sample, NA10851. The second panel down shows read-depth information for the same genomic region, derived from whole-genome sequencing data of NA10851. The third panel down is a ‘corrected’ absolute copy number result for this genomic region in AK1 using the absolute copy number algorithm analysis method described in this study. The bottom panel displays the same genomic region for AK1 using read-depth information derived from whole-genome sequencing data. (b) Comparison between relative copy number states and absolute copy number values for CNV segments, before and after corrections for NA10851 copy number states. Out of the total 21,905 CNVs identified in the 30 Asian individuals by aCGH (that is, by relative copy number states), the relative copy number values of 10,925 were not affected by CNVs in the NA10851 reference. Among 10,980 ‘obscure’ CNVs, 4,970 were determined to be non-CNVs by absolute calls and were removed from the final list of CNVs. An additional 3,164 CNVs, which were considered ‘covert’ CNVs and were initially missed by the aCGH experiments, were also identified by the absolute copy number state calling algorithm.
This correction also changed the ratio of total count of copy number losses and gains, with 72.6% of all variants actually having a copy number of <2 per diploid cell (copy number loss), as compared to 44.4% before correction (Fig. 3a,b and Supplementary Fig. 5). This corrected ratio is more compatible with a recent report by Conrad et al.2, in which researchers obtained absolute copy number states by clustering aCGH data from 450 samples. The average lengths of CNV segments with copy number losses and gains were 11.8 kb and 30.3 kb, respectively (Fig. 3b). In genic regions, copy number gains were more frequently recorded than copy number losses, which may be due to the fact that in these regions copy number gains are less likely to be deleterious and therefore less likely to incur a penalty in evolutionary selection6.
Figure 3.

Frequency of copy number gains and losses among 33 individuals. (a) Distribution of absolute copy number gains (copy number >2) and losses (copy number <2) in 33 individuals. (b) Distribution of relative and absolute copy number gains and losses by CNV size. The x and y axes represent size and number of CNV segments, respectively.
There were 20,099 CNV segments ultimately identified in this study. On the average, 670 CNV segments were found in each Asian individual studied, which covered 11.31 Mb of the total DNA sequence and involved 389 RefSeq genes per person (Supplementary Table 4). We randomly selected 116 CNV elements and performed 1,881 quantitative PCR (qPCR) experiments on them. A total of 1,717 of the qPCR experiments were correlated with our aCGH data, resulting in a predictive value of 91% (Supplementary Tables 5 and 6).
To compare CNV segments between individuals, CNV segments from this study were merged into groups, termed ‘CNV elements’ (CNVEs), based on greater than 50% overlap between segments (Supplementary Fig. 6). We obtained absolute copy number states for 5,177 Asian CNVEs, with the group of CNVEs having a median size of 2,667 bp (Supplementary Table 7) and covering 95.40 Mb (3.32%) of the human reference genome. To identify potential Asianspecific CNVEs, we compared these 5,177 CNVEs with 4,978 CNVEs recently identified by Conrad et al.2. Although those researchers found 56 Asian-specific CNVEs, we identified 3,547 putative Asian-specific CNVEs that were not included in their dataset (Fig. 4a and ref. 2).
Figure 4.

Putative Asian population–specific copy number variants. (a) Venn diagram showing validated putative Asian-specific CNVEs. The lower part of the figure (blue) indicates the ethnic distribution of 4,959 CNVEs that were discovered by a 42M NimbleGen aCGH platform and validated with a genotyping microarray in the same study2. The upper part of the figure indicates that 3,547 out of 5,177 CNVEs found among the 30 Asian individuals in this study do not reach a 1-bp overlap with CNVEs recently found by the Genome Structural Variation Consortium2. The Genome Structural Variation Consortium reported that they found 4,978 validated CNVEs, but we show only 4,959 of them in this Venn diagram because 19 were nonpolymorphic. (b) Distribution of gene ontology categories for genes in which coding sequences overlap with common copy number– gain regions (outer circle) and copy number–loss regions (inner circle) identified from 30 Asian subjects. (c) CNVE location and number of Asian individuals involved (bar graph, right). Red, copy number gain; green, copy number loss. Selected genes and miRNAs are also shown on the left.
The CNVEs identified in our study overlapped with 2,913 RefSeq genes and 1,483 genes present in the OMIM database (Supplementary Tables 7 and 8). These CNVEs were also responsible for copy number changes of 29 microRNA (miRNA) genes (Supplementary Table 9) as well as 35 potential gene fusions (Supplementary Table 10).
We categorized genes overlapping the common CNVEs found in our study using the PANTHER gene ontology (see URLs). Copy number gains had an increased bias toward being contained within genes having functions associated with nucleic acid metabolism and developmental processes. Genic copy number losses were enriched for genes involved in cell adhesion. Subsets of genes involved in signal transduction, immunity and sensory perception were found to have both copy number gains and losses, which is consistent with previous findings involving nonsynonymous SNPs and deletion polymorphisms6,7 (Fig. 4b and Supplementary Table 11).
Notably, 2,183 of the 2,913 RefSeq genes that we identified as being copy number variable among the Asian individuals studied did not appear to be copy number variable in the populations studied by Conrad et al.2. For example, CNVs in CLPS, LPA and CEBPB, which have been reported to be involved in type 2 diabetes, myocardial infarction and cancer, respectively, are found at a frequency of ≥10% among Asian populations, which are not found in the Conrad et al. study2,7,8. Some genes, such as LY9, CNTN5 and PIK3CA, which have been reported to be involved in systemic lupus, cardiovascular disease and oncogenesis, respectively9–12, were first found to be copy number variable either in this study or in our previous report of the whole genome sequence of AK1 (ref. 5). Examples of genes with CNVs are shown in Figure 4c and Table 1.
Table 1.
Summary statistics of selected copy number variants
| CN gain |
CN loss |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reported gene |
Trait | Chr. | Asian | CHB | JPT | KOR | CEPHa | YRIa | Asian | CHB | JPT | KOR | CEPHa | YRIa | PMID |
| ADAMTS14 | Multiple sclerosis | 10 | 3 | 1 | 2 | 0 | – | – | 0 | 0 | 0 | 0 | – | – | 15913795 |
| CCL4 | Type 1 diabetes mellitus | 17 | 4 | 0 | 2 | 2 | – | – | 0 | 0 | 0 | 0 | – | – | 17327452 |
| CES1 | Lipid metabolism | 16 | 10 | 3 | 5 | 2 | 46 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 19332024 |
| CLPS | Type 2 diabetes mellitus | 6 | 10 | 5 | 1 | 4 | – | – | 0 | 0 | 0 | 0 | – | – | 18726866 |
| FOXC1 | Congenital glaucoma and aniridia | 6 | 4 | 1 | 0 | 3 | – | – | 0 | 0 | 0 | 0 | – | – | 18484311 |
| HYLS1 | Hydrolethalus syndrome | 11 | 4 | 1 | 3 | 0 | – | – | 0 | 0 | 0 | 0 | – | – | 18648327 |
| IRF4 | Hair and skin pigmentation, lymphoma | 6 | 11 | 3 | 3 | 5 | – | – | 0 | 0 | 0 | 0 | – | – | 18483556 |
| IRX1 | Myopia, head and neck squamous-cell carcinoma | 5 | 5 | 0 | 2 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 18559491 |
| KRT34 | Urothelial carcinoma | 17 | 3 | 0 | 1 | 2 | 177 | 178 | 0 | 0 | 0 | 0 | 0 | 0 | 16286979 |
| LPA | Myocardial infarction | 6 | 24 | 8 | 6 | 10 | – | – | 0 | 0 | 0 | 0 | – | – | 19509380 |
| NBAS | Neuroblastoma | 2 | 6 | 2 | 2 | 2 | – | – | 0 | 0 | 0 | 0 | – | – | 12706883 |
| NBEA | Autism | 13 | 3 | 1 | 2 | 0 | – | – | 0 | 0 | 0 | 0 | – | – | 12746398 |
| PIK3CA | Small-cell lung cancer, | 3 | 9 | 4 | 3 | 2 | – | – | 0 | 0 | 0 | 0 | – | – | 19394761 |
| PITX1 | Non–small-cell lung cancer | 5 | 5 | 0 | 2 | 3 | – | – | 0 | 0 | 0 | 0 | – | – | 19414376 |
| SKI | Pancreatic cancer | 1 | 3 | 1 | 1 | 1 | – | – | 0 | 0 | 0 | 0 | – | – | 19546161 |
| TPPP | Central nervous system disease | 5 | 1 | 0 | 1 | 0 | – | – | 0 | 0 | 0 | 0 | – | – | 19382230 |
| CFH | Age-related macular degeneration | 1 | 0 | 0 | 0 | 0 | – | – | 6 | 4 | 2 | 0 | – | – | 19692124 |
| CNR2 | Osteoporosis | 1 | 0 | 0 | 0 | 0 | 3 | 0 | 5 | 0 | 3 | 2 | 0 | 0 | 19442614 |
| DAZL | Spermatogenesis | 3 | 0 | 0 | 0 | 0 | – | – | 27 | 7 | 10 | 10 | – | – | 15066460 |
| GHR | Growth | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 3 | 0 | 2 | 82 | 134 | 18793346 |
| GSTT2 | Colorectal cancer | 22 | 0 | 0 | 0 | 0 | 158 | 121 | 4 | 3 | 0 | 1 | 0 | 0 | 17250773 |
| LY9 | Systemic lupus erythematosus | 1 | 0 | 0 | 0 | 0 | – | – | 6 | 4 | 2 | 0 | – | – | 18216865 |
| PGA3,4,5 | Duodenal ulcer, gastric cancer | 11 | 0 | 0 | 0 | 0 | 0 | 0 | 28 | 10 | 9 | 9 | 4 | 0 | 17559360 19196398 |
| PRSS2 | Pancreatitis | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 1 | 0 | 2 | 112 | 48 | 19052022 |
| CEL | Lipid metabolism | 9 | 6 | 2 | 2 | 2 | 6 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 18803939 |
| DMBT1 | Brain tumor | 10 | 6 | 2 | 1 | 3 | 175 | 177 | 9 | 3 | 2 | 4 | 2 | 0 | 19207948 |
| EBF3 | Head and neck squamous-cell carcinoma | 10 | 8 | 1 | 4 | 3 | – | – | 1 | 0 | 1 | 0 | – | – | 18559491 |
| IRS2 | Metabolic syndrome | 13 | 8 | 3 | 2 | 3 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 18802016 |
| MCTP2 | Schizophrenia | 15 | 1 | 0 | 1 | 0 | 59 | 23 | 17 | 6 | 5 | 6 | 0 | 0 | 19223264 |
| MGAM | Starch absorption | 7 | 2 | 0 | 1 | 1 | 93 | 102 | 4 | 1 | 3 | 0 | 16 | 0 | 17485087 |
| MUC4 | Gastric cancer | 3 | 4 | 0 | 2 | 2 | – | – | 1 | 0 | 1 | 0 | – | – | 18781152 |
| MUC20 | IgA nephropathy | 3 | 5 | 1 | 2 | 2 | – | – | 1 | 0 | 1 | 0 | – | – | 16029633 |
| NAIP | Spinal muscular atrophy | 5 | 3 | 1 | 1 | 1 | – | – | 2 | 1 | 1 | 0 | – | – | 17932457 |
| PRKRA | Dystonia | 2 | 19 | 6 | 6 | 7 | 104 | 35 | 1 | 0 | 0 | 1 | 0 | 0 | 18420150 |
| RHD | Rh blood type | 1 | 2 | 0 | 0 | 2 | 148 | 174 | 3 | 2 | 0 | 1 | 0 | 0 | 10938938 |
Chr., chromosome; CN, copy number; PMID, PubMed unique identifier.
CN gains and losses in CEU and YRI data (n = 180 in each) from Conrad et al.2
Because we used a new approach to call absolute copy number states, some of our data showed different genetic variation frequencies than previous reports. For example, we identified copy number losses (specifically, a one-copy loss) in 3 out of 30 Asian subjects in RHD, whereas Conrad et al.2 found copy number gains in all of the 88 Asian individuals genotyped. A deletion in the gene encoding RhD, which is one of the Rhesus blood antigens, results in an RhD-negative blood type when both copies of the gene are deleted13. The incidence of the homozygous deletion resulting in an RhD-negative blood type is between 0.3% and 0.5% in Asians, which is consistent with our observed 10.0% (3 individuals out of 30 total) one-copy loss rate14. Examples of other discrepancies with data obtained from the study conducted by Conrad et al.2 are listed in Supplementary Table 12. Further studies comparing and validating different absolute calling methods, including the one used here, will be required to design the most efficient and accurate method for determining absolute copy number genotypes.
Because the method proposed in this study uses read-depth sequence information from NA10851, which is one of the most commonly used control samples in aCGH, it should be amenable to other aCGH studies that use NA10851 as the control sample. One example in which absolute copy number states could be determined from data generated from other aCGH platforms is shown in Supplementary Figure 7. Alternatively, if an individual other than NA10851 was used as a control sample in aCGH experiments, read-depth sequence information for that individual could subsequently be used to generate an absolute copy number calling algorithm for those studies.
We also developed a custom 180k CNV genotyping array and used this array to simultaneously examine 17,760 CNVs in a large Asian family comprising 13 individuals across three generations. Using relative copy number information, Mendelian inconsistencies for copy number deletions were estimated at a rate of 6.42%. Using absolute copy number information, the Mendelian inconsistencies for copy number deletions dropped to 2.59% (Supplementary Fig. 8), which is probably closer to the true rate of de novo formation of CNVs at this resolution.
Because the Conrad et al.2 dataset used a different aCGH platform than was used here, we considered whether the unique CNVEs found in our study were platform specific. There are some differences between the Agilent aCGH platform used in this study and the NimbleGen array used in the previous study by Conrad et al.2. The Agilent platform we used excluded areas of repetitive sequence using a homology filter, whereas 43% of the NimbleGen probes are in repetitive regions (Supplementary Fig. 1). The use of the Agilent platform in this study resulted in an effectively smaller portion of the genome being assayed for CNVs and resulted in a lower false positive rate. The Agilent and NimbleGen platforms identified 722 and 1,829 CNVs, respectively, in AK1 (Supplementary Fig. 7). Four hundred and fifty CNVs were common to both platforms. Of the 1,282 CNVs specific to the NimbleGen array, 655 were in moderately to highly repetitive regions, whereas 424 had log2 ratios that did not meet the more stringent filter criteria established for the Agilent array. Consequently, only 203 NimbleGen-specific CNVs were relevant in this comparison. Further comparison of CNVs identified in NA12878 (from CEU) and NA19240 (from YRI) revealed that ~60% of the calls on the Agilent 24M array were common to the Conrad et al. study, whereas ~40% of the calls were specific to our platform only (Fig. 5 and Supplementary Fig. 9). Taken together, these comparisons indicate that ~40% of the Agilent 24M CNV calls may be platform dependent and would not be captured by the NimbleGen 42M array. This suggests that about 60% out of the 3,547 potential Asian-specific CNVEs used here are truly specific to the Asian population and were missed in Conrad et al.2 due to the lack of samples from Asian individuals used in their CNV discovery phase.
Figure 5.
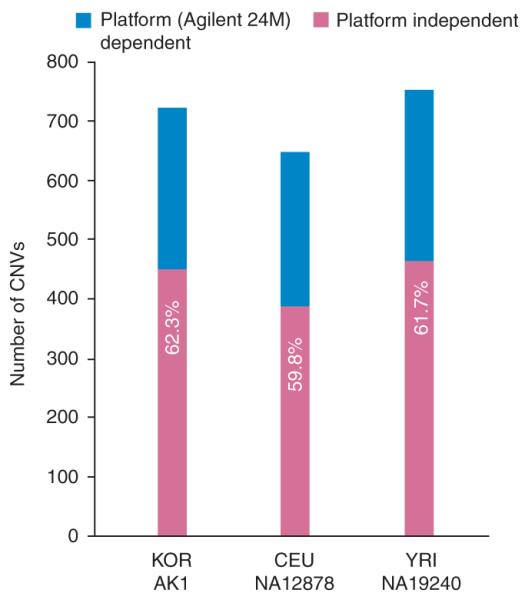
Effect of aCGH platforms in the CNV discovery. Absolute CNVs found from two HapMap individuals (NA12878 and NA19240) in this study using the Agilent 24M aCGH array were compared with CNVs found by genotyping microarray in the Genomic Structural Variation Consortium data. We also compared CNVs from AK1 obtained by Agilent 24M and Nimblegen 42M microarrays.
In summary, we have comprehensively identified common CNVs (minor allele frequency > 1.7%) among individuals in Asian populations at a resolution sufficient to detect CNVs as small as 438 bp using an integrated high-resolution aCGH approach combined with next-generation sequencing data. The discovery of many more new CNVs in our study, despite the large number of previous studies in this area, is most likely due to the increased resolution and comprehensive nature of our aCGH platform design, which targets smaller variants, combined with a focus on Asian populations, which to date have been relatively neglected in CNV studies15–18. We also provide a paradigm for large-scale genome sequencing initiatives, such as the 1000 Genomes Project (see URLs), to combine DNA sequencing data with high-resolution CNV mapping via aCGH for more accurate CNV identification and characterization in individual genome sequences. To more accurately apply CNV research to personalized medicine, copy number genotyping must not rely on relative copy number data, but should be able to identify the absolute copy number state in any given individual. Our results also provide guidance for future studies in genomic medicine in the Asian population, especially in those that identify ethnic differences in predisposition to disease and drug response19–23.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge R. Govindaraju for editing this manuscript. This work has been supported in part by Macrogen Inc. (MG2009009), Psoma Therapeutics Inc., the Korean Ministry of Education, Science and Technology (grant M10305030000), Green Cross Therapeutics (0411-20080023), the Department of Pathology at Brigham and Women’s Hospital (to C.L.) and a US National Institutes of Health Grant (HG004221 to C.L.).
Footnotes
URLs. Sequence data for AK1, AK2 and NA10851, http://www.gmi.ac.kr; PANTHER gene ontology, http://www.pantherdb.org/; 1000 Genomes Project, http://www.1000genomes.org.
METHODS Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
Accession codes. Massively parallel sequencing data of AK1, AK2 and NA10851 have been deposited in the NCBI short read archive under accession number SRA008370, SRA010321 and SRA010320, respectively. Array CGH data have been deposited in the NCBI GEO (gene expression omnibus) under accession number GSE19651.
Note: Supplementary information is available on the Nature Genetics website.
AUTHOR CONTRIBUTIONS J.-S.S. and C.L. planned and managed the project. H.P., J.-I.K., Y.S.J., O.G., R.E.M., Y.J.Y., J.-Y.S., J.-S.H., W.C., G.-R.H. and K.D. executed and analyzed aCGH experiments. J.-I.K., Y.S.J., S.K., D.H., H.-J.K. and D.H. executed sequencing of the genome and analyzed sequence data. D.S., S.L., M.Y., Y.W.C., HyeRan Kim, S.J.Y., K.-S.Y. and Hyungtae Kim performed validation experiments; M.E.H., S.W.S., N.P.C. and C.T.-S. assisted in data analyses; J.-S.S., C.L., H.P., J.-I.K., Y.S.J. and H.P.K. wrote the manuscript.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 2.Conrad DF, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2009 Oct 7; doi: 10.1038/nature08516. advance online publication, doi:10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perry GH, et al. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008;82:685–695. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JI, et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–1015. doi: 10.1038/nature08211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nat. Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- 7.Lindner I, et al. Putative association between a new polymorphism in exon 3 (Arg109Cys) of the pancreatic colipase gene and type 2 diabetes mellitus in two independent Caucasian study populations. Mol. Nutr. Food Res. 2005;49:972–976. doi: 10.1002/mnfr.200500087. [DOI] [PubMed] [Google Scholar]
- 8.Shiffman D, et al. Analysis of 17,576 potentially functional SNPs in three case-control studies of myocardial infarction. PLoS One. 2008;3:e2895. doi: 10.1371/journal.pone.0002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunninghame Graham DS, et al. Association of LY9 in UK and Canadian SLE families. Genes Immun. 2008;9:93–102. doi: 10.1038/sj.gene.6364453. [DOI] [PubMed] [Google Scholar]
- 10.Larson MG, et al. Framingham Heart Study 100K project: genome-wide associations for cardiovascular disease outcomes. BMC Med. Genet. 2007;8(suppl 1):S5. doi: 10.1186/1471-2350-8-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 12.Lee JW, et al. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24:1477–1480. doi: 10.1038/sj.onc.1208304. [DOI] [PubMed] [Google Scholar]
- 13.Colin Y, et al. Genetic basis of the RhD-positive and RhD-negative blood group polymorphism as determined by Southern analysis. Blood. 1991;78:2747–2752. [PubMed] [Google Scholar]
- 14.Wang YH, et al. Detection of RhD(el) in RhD-negative persons in clinical laboratory. J. Lab. Clin. Med. 2005;146:321–325. doi: 10.1016/j.lab.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohmueller KE, et al. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuzun E, et al. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 19.Aitman TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 20.Burchard EG, et al. The importance of race and ethnic background in biomedical research and clinical practice. N. Engl. J. Med. 2003;348:1170–1175. doi: 10.1056/NEJMsb025007. [DOI] [PubMed] [Google Scholar]
- 21.Horowitz RE. Gastric cancer in Japan. N. Engl. J. Med. 2008;359:2393–2394. doi: 10.1056/NEJMc081797. author reply 2394–2395. [DOI] [PubMed] [Google Scholar]
- 22.Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world—a growing challenge. N. Engl. J. Med. 2007;356:213–215. doi: 10.1056/NEJMp068177. [DOI] [PubMed] [Google Scholar]
- 23.Jee SH, et al. Body-mass index and mortality in Korean men and women. N. Engl. J. Med. 2006;355:779–787. doi: 10.1056/NEJMoa054017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.