

Abstract
Primary neoplasms of the skeleton are rare, accounting for 0.2% of overall human tumor burden. Osteosarcoma (OS) accounts for 15–35% of all primary bone tumors, while gnathic osteosarcomas (GOS) represent 4–8% of all osteosarcomas. GOS shows a predilection for men, a peak incidence of 33 years, and affects the mandible more than the maxilla. We review the scientific literature for a better understanding of the clinical, radiographic, and histopathological features of GOS, along with its etiology, staging, treatment protocol, prognosis, and survival. Evidence from molecular research suggests that it is a differentiation disease that disrupts osteoblasts differentiation from mesenchymal stem cells. The classical radiographic finding of a “sunburst” appearance is appreciated only in 50% of GOS. The universally accepted staging system is not commonly used due to the rarity with which they metastasize to the regional lymph nodes. A number of distinct histopathological subtypes have been described, of which osteoblastic GOS are most common. The treatment protocol is multimodal consisting of preoperative chemotherapy followed by surgery and postoperative chemotherapy, and has a 60-70% five-year survival rate. We present two case reports of osteosarcoma involving the maxillary that were initially misdiagnosed as peripheral giant cell granuloma and osteoma of the maxilla, respectively. These case reports demonstrate the diverse clinical, radiographic, and histopathological features that can be encountered in GOS.
Keywords: Bone tumor, gnathic, jaw, maxilla, metastasis, osteogenic sarcoma, osteosarcoma, prognosis, recurrence, staging, treatment
INTRODUCTION
Osteosarcoma (synonym: osteogenic sarcoma) is a primary malignant bone tumor characterized by direct formation of disorganized immature woven bone from mesenchymal tumor cells.[1–6] Osteosarcomas (OS) accounts for 15–35% of all primary bone tumors and is the most common nonhemopoietic primary malignant bone tumor of children and adolescence. Gnathic osteosarcomas (GOS) are relatively rare and represent 4–8% of all the OS.[1,2,4–8]
The age-specific frequencies and incidence rates of OS are bimodal; 75% occur between 15 and 25 years, with the characteristic site for its development being the metaphyseal growth plates of long tubular bones of the lower limbs (femur, tibia, and humerus).[5,6] The second peak is observed in adults above 50 years, with the tumor developing in the axial skeleton and flat bones (vertebrae and pelvic bone).[1,5] More rarely extraskeletal OS arise in soft tissues, commonly the thigh, upper extremity, and retroperitoneum.[9] Men develop OS more frequently than women (ratio 1.5:1)[4–6,10]; 60% of GOS occur in males and the peak incidence is observed in the third to fourth decade (33 years). They show a predilection for mandible; however, some studies found that it affects the mandible and maxilla almost equally.[1,4,5,10,11] Mandibular tumors arise more frequently in the horizontal ramus, while the maxillary lesions are commonly discovered in the alveolar ridge, sinus floor, and palate.[5,12]
Majority of this primary bone malignancy arise de novo, but some apparently develop in association with Paget's disease, fibrous dysplasia, bone infarcts, chronic osteomyelitis, trauma, viral infection, or on exposure to high-dose radiation.[1,6,7,10] It has also been associated with metallic implants, joint prostheses, and in genetic syndromes such as Li–Fraumeni syndrome, hereditary retinoblastoma, and Rothmund–Thomson syndrome.[1,7,9–12] Emerging evidence from molecular research suggests that OS is a differentiation disease caused by genetic and epigenetic changes that disrupt osteoblast differentiation from mesenchymal stem cells. Nearly 70% of OS display a multitude of cytogenetic abnormalities including haploidy in chromosomes 1p11-p13, 1q11-q12, 1q21-q22, 11p14-p15, 14p11-p13, 15p11-p13, 17p, and 19q13; gain of chromosome 1; loss of chromosomes 9, 10, 13, and 17; and amplification in chromosomes 6p12-p21, 17p11, and 12q13-q14.[13]
Swelling, gnawing pain, and general discomfort are the usual nonspecific clinical findings.[2,5,10,12] Radiographic findings vary from dense sclerotic to mixed (moth-eaten or cumulus cloud) to radiolucent lesion.[1,5,10] The classical radiographic finding is that of a “sunray” or “sunburst” appearance, due to rapid osteophytic bone formation creating small radiating streaks from the bone surface. But this appearance is appreciated in only 50% of GOS and is best observed in occlusal radiographs and computerized tomography (CT) scans.[2,5,7,10,12] Codman's triangle may be identified, formed due to elevation of periosteum over the expanding tumor mass in a tent-like fashion.[10] Ortho pantograph (OPG) may show Garrington's sign that represents widening of the periodontal ligament space around the affected teeth due to tumor infiltration, along with tapered resorption of the root apex.[2,5,7,10]
Staging a tumor helps estimate the prognosis of the patient, and it incorporates the degree of differentiation and distant metastasis.[1] The universally accepted TNM staging system is not commonly used for sarcomas because of the rarity with which they metastasize to the regional lymph nodes. The Musculoskeletal Tumor Society Staging System [Table 1] and the American Joint Committee on Cancer (AJCC) Staging System [Table 2] have gained acceptance for OS staging.[1,6,14] AJCC 2006 staging system is based on tumor size (T), regional lymph node (N), distant metastasis (M), and histopathological grade (G). The anatomic extent of the tumor is subdivided into intracompartmental (A) and extracompartmental (B), depending on whether the tumor is confined within the cortex or is invading beyond the cortex.[1,6,14]
Table 1.
Musculoskeletal Tumor Society staging system

Table 2.
American Joint Committee on Cancer Staging System (2006)
OS show a broad spectrum of histopathological features, with common characteristics of sarcomatous stroma having atypical neoplastic osteoblasts that produce tumor osteoid arranged in a disorderly irregular fashion, sometimes in solid sheets, along with varying degree of anaplastic fibroblast, cartilage, and myxomatous tissues.[1,4,5,10,12] A number of distinct histopathological subtypes of OS have been described [Table 3].[1,6,10,12] Osteoblastic OS is the most common subtype reported in the long bones of children. Nearly 60% of GOS are osteoblastic, 34% fibroblastic, and less than 10% chondroblastic.[1,5,7,10,12] Most authorities currently believe that even if a malignant bone tumor is chiefly composed of malignant cartilage, it should be designated as OS if significant malignant tumor osteoid can be identified, because the clinical course of the lesion will be that of OS rather than that of a chondrosarcoma that has a better prognosis.[4,5,10,12]
Table 3.
Histopathological subtypes of osteosarcomas

The modern treatment protocol for OS is multimodal, consisting of preoperative chemotherapy followed by extensive surgery and postoperative chemotherapy.[3,12,15] Drugs commonly used for chemotherapy are high-dose combinations of methotrexate, cisplatin, adriamycin, doxorubicin, and ifosfamide. Unfortunately, 40% develop multidrug resistance, and drugs such as vincristine, bleomycin, and dactinomycin are found to be ineffective.[6,12] The tumor is radio-resistant at standard doses, and radiotherapy plays no significant role.[6,10] Due to the high frequency of local micro-metastases, the survival rate of OS patients is 12–20% with surgery alone, while it improves to 60–70% when combined with chemotherapy. Recurrence rate is as high as 25% if adequate clear surgical margins are not achieved.[6]
The prognosis is more favorable for mandibular OS in comparison to those arising in the maxilla, with the maxillary antral tumors having the worst prognosis. Chondroblastic subtype is associated with a better prognosis than other histopathological variants.[5,10,15] Patients with nonmetastatic OS have 70% five-year survival rate, while patients with metastasis or recurrence have a poorer prognosis with only 20% surviving at 5 years.[3,12,15] The average survival after recurrence is less than 1 year.[12] The most common site for metastasis is the lungs where they appear as small pulmonary nodules.[6,12] Achieving complete surgical resection of metastatic pulmonary nodules improves 5-year survival to 45%.[6]
We present two diverse case reports of OS involving the maxillary jaw in a 54-year male and a 43-year female who were clinically and radiographically misdiagnosed. They were reported histopathologically after wide surgical excision as chondroblastic OS and osteoblastic OS, respectively.
CASE REPORTS
Case 1
Clinical details
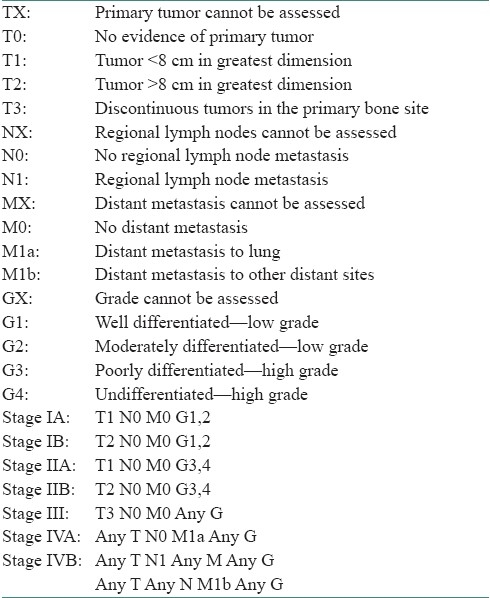
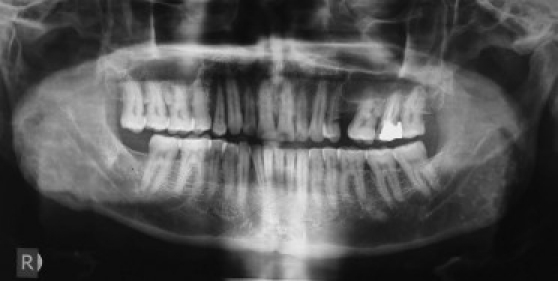
A 54-year-old otherwise healthy businessman reported to a dental practitioner with mobility of upper right teeth and pain on chewing since 2 weeks. On intraoral examination, he was clinically diagnosed to have localized periodontitis with gingival swelling and grade II mobility of 14 (right maxillary first premolar). OPG showed localized alveolar bone loss and widening of the periodontal ligament space in relation to 14 [Figure 1a]. Extraction of 14 along with excision of the gingival swelling was done under local anesthesia and antibiotic coverage. Excised gingival lesion on histopathological examination was reported as peripheral giant cell granuloma. Recurrence of the lesion occurred at the same site within 10–12 days, and he was advised reexcision. After 1 month, he reported to our primary health care center for surgical excision of the said gingival lesion. Clinical examination revealed a 4 × 5 cm well-defined firm sessile asymptomatic gingival swelling in 13–15 regions (right maxillary canine to second premolar), overhanging the 14 extraction socket and covering the cervical third of 13 and 15. The surface was smooth, interspersed with erosive areas, having grayish white to pale pink mucosa [Figure 1b]; 13 and 15 were caries free and had grade I mobility. Regional lymph nodes were not palpable.
Figure 1a.
Ortho pantograph
Figure 1b.
Clinical appearance
Biopsy
A wide surgical excision with extraction of 13 and 15 along with interdental alveolar bone septal osteotomy was performed under general anesthesia. No perforation to maxillary sinus or erosion of palatal bone was noted. Primary closure of surgical site was done using buccal flap.
Histopathological report
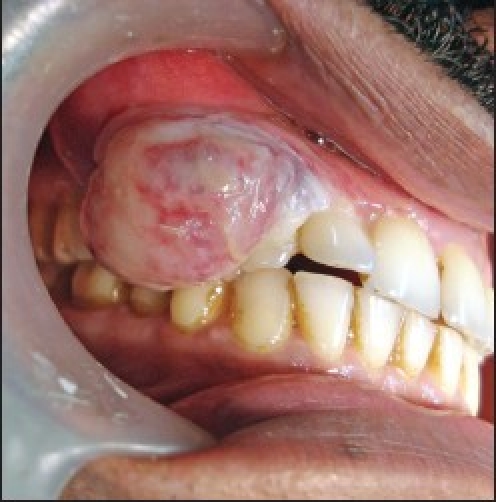
The hematoxylin and eosin (H and E) sections showed lobular areas of chondroblastic differentiation with atypical chondrocytes and chondroblasts, and actively proliferating fibroblast showing nuclear and cellular pleomorphism [Figure 1c]. Peripheral areas of few of these lobular areas showed formation of disorganized woven immature bone [Figure 1d]. Focal areas of atypical haphazardly arranged spindle-shaped cells with myxomatous areas and few diffuse bizarre multinucleated giant cells were seen. Based on these varied histopathological findings, a diagnosis of chondroblastic OS was given. Previous histopathological slides could not be reviewed.
Figure 1c.
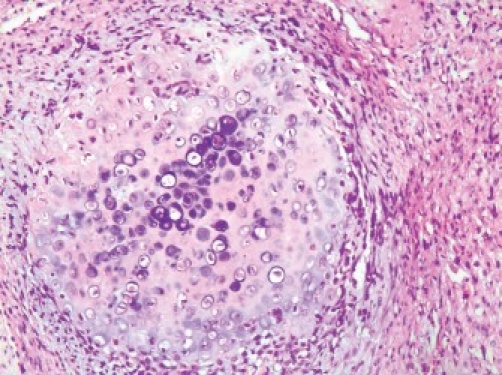
Photomicrograph (×10) showing lobular areas of neoplastic cartilage (H and E section)
Figure 1d.
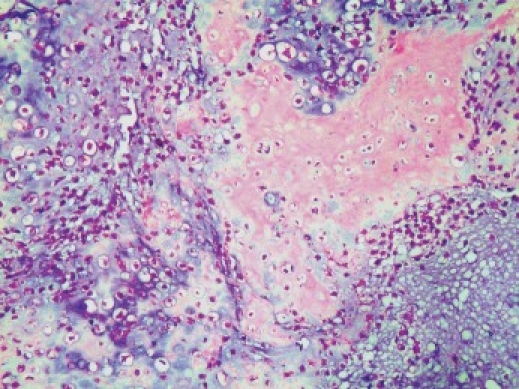
Photomicrograph (10×) showing tumor osteoid in neoplastic cartilage (H and E section)
Treatment
No characteristic bone changes could be detected on CT examination. Chest X-ray was reviewed for metastatic lesions in the lungs and none was detected. Case was referred to higher oncology center where partial maxillectomy and adjuvant chemotherapy were performed. After 1.6 years, the patient is doing well with no reported recurrence.
Case 2
Clinical details
A 43-year-old otherwise healthy housewife reported to a head and neck surgeon with a chief complaint of right nasal obstruction since 1 month. Routine paranasal sinus x-ray showed a dense sclerotic radiopaque mass obliterating the right maxillary sinus with deviated nasal septum. On clinical reevaluation at our primary health care center, a mild facial swelling along with mild proptosis of right eye with no apparent restriction of eye movements or vision was noted. Intraoral examination showed obliteration of the entire right buccal sulcus by a diffuse bony hard mass covered by a normal appearing oral mucosa; 16 (right maxillary first molar) had deep mesial class II carious lesion, with extrusion of 16 and 18 (right maxillary third molar). Grade I painless mobility of 16 and 15 was noted; 46 and 48 (right mandibular first and third molar) were missing with a history of extraction after carious exposure 6–8 years ago. Regional lymph nodes were not palpable. OPG showed dense to mixed sclerotic radiopaque mass of the right maxillary sinus along with widening of the periodontal ligament space of 15–18 [Figure 2a]. CT scan showed mixed sclerotic mass involving the right maxilla and nasal cavity with involvement of infraorbital floor, nasal cavity, frontal, sphenoidal, and ethemoidal sinuses and was reported as osteoma of maxilla [Figure 2b].
Figure 2a.
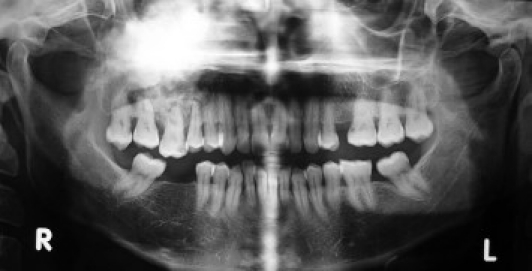
Ortho pantograph
Figure 2b.
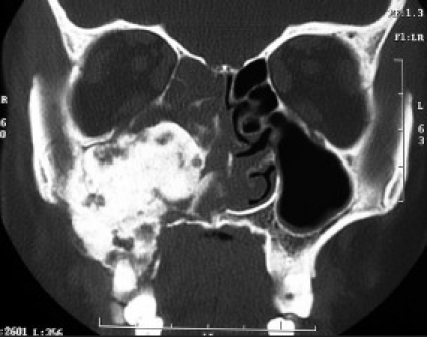
Computerized tomography
Biopsy
The patient underwent excision and curettage of the mass through a Fergusons incision under general anesthesia. One single large mass of bone within the maxillary sinus and multiple bits of bony tissue were curetted piecemeal. The inferior orbital floor was involved and was reconstructed using mandibular cortical bone graft to prevent eyeball intrusion into surgical defect.
Histopathological report
Given (H and E) sections showed sheets of disorganized woven immature bone and round cells having large vesicular nucleus and scanty cytoplasm [Figure 2c]. Focal areas of few bizarre multinucleated giant cells were seen. Few areas of irregular calcified spicules along with many areas of disorganized darkly stained immature bone with irregular margins having an acid-etched-glass appearance were present [Figure 2d]. A diagnosis of osteoblastic OS was given.
Figure 2c.
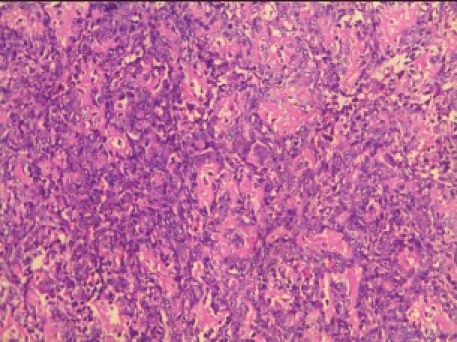
Photomicrograph (10×) showing disorganized woven bone and neoplastic osteoblasts
Figure 2d.
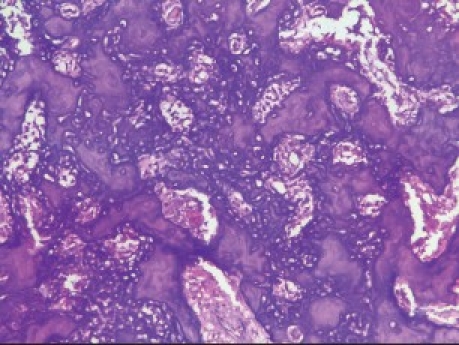
Photomicrograph (10×) showing immature bone having an acid-etched-glass appearance
Treatment
No metastatic lesions were identified on a chest x-ray. Case was referred to higher oncology center where extensive surgery with adjuvant chemotherapy was done. After 1 year, the patient is doing reasonably well (under treatment for severe mental depression) and no recurrence or distant metastasis has been identified.
CONCLUSION
OS is the second most common malignant bone tumor after multiple myeloma.[1,2] These case reports demonstrate the diverse clinical, radiographic, and histopathological features of GOS and the difficulties encountered during diagnosis. The reasons for the first case being initially misdiagnosed as peripheral giant cell granuloma could be rarity of OS clinically presenting as a well-defined sessile gingival swelling, OPG findings being ignored as no classical features were observed, and sampling error during histopathological examination. The second case reported with a chief complaint of nasal obstruction, which on CT imaging was suggestive of osteoma of maxilla possibly because of large solitary radiopaque mass with no classic OS features.
Identifying the genes and signal transduction pathways responsible for the development of OS through more molecular research may help in the development of newer diagnostic markers and help improve therapeutics, leading to better prognosis and patient survival in the future.
ACKNOWLEDGEMENT
Dr. Krishnan Pisharodi, Anesthetist, prepared and managed the patients. Dr. Arun Kumar, ENT Surgeon, assisted in managing the second patient.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Raymond AK, Ayala AG, Knuutila S. Conventional osteosarcoma. In: Fletcher CD, Unni KK, Mertens F, editors. Pathology and Genetics of Tumors of Soft Tissue and Bone. 1st ed. Lyon: IARC Press; 2002. pp. 264–70. [Google Scholar]
- 2.Fernandes R, Nikitakis NG, Pazoki A, Ord RA. Osteogenic Sarcoma of the Jaws: A 10 year experience. J Oral Maxillofac Surg. 2007;65:1286–91. doi: 10.1016/j.joms.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 3.Amaral MB, Buchholz I, Freire-Maia B, Reher P, deSouza PE, Marigo HA, et al. Advanced osteosarcoma of the maxilla: A case report. Med Oral Pathol Oral Cir Bucal. 2008;13:492–5. [PubMed] [Google Scholar]
- 4.Warnock G. Malignant Neoplasms of the Gnathic Bones. In: Thompson LD, Goldblum JR, editors. Head and Neck Pathology. 1st ed. Amsterdam: Elsevier; 2006. pp. 492–7. [Google Scholar]
- 5.Angela C. Bone Pathology. In: Neville BW, Damm DD, Allen CM, Bouquot JE, editors. Oral and Maxillofacial Pathology. 3rd ed. Philadelphia: Saunders; 2009. pp. 660–4. [Google Scholar]
- 6.Picci P. Osteosarcoma (Osteogenic sarcoma) Orphanet J Rare Dis. 2007;2:6–10. doi: 10.1186/1750-1172-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogunlewe MO, Ajayi OF, Adeyemo WL, Ladeinde AL, James O. Osteogenic sarcoma of the jaw bones: A single institution experience over a 21-year period. Oral Surg Oral Med Oral Pathol Oral Radiol Endo. 2006;101:76–81. doi: 10.1016/j.tripleo.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 8.August M, Magennis P, Dewitt D. Osteogenic sarcoma of the jaws: Factors influencing prognosis. Int J Oral Maxillofac Surg. 1997;26:198–204. doi: 10.1016/s0901-5027(97)80819-3. [DOI] [PubMed] [Google Scholar]
- 9.Bane BL, Evans HL, Ro JY, Carrasco CH, Grignon DJ, Benjamin RS, et al. Extraskeletal osteosarcoma. Cancer. 1990;66:2762–70. doi: 10.1002/1097-0142(19900615)65:12<2762::aid-cncr2820651226>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 10.Rajendran R. Benign and malignant tumors of the oral cavity. In: Rajendran R, Sivapathasundaram B, editors. Shafer's Textbook of Oral Pathology. 6th ed. Amsterdam: Elsevier; 2009. pp. 169–73. [Google Scholar]
- 11.McGuff HS, Heim-Hall J, Holsinger FC, Jones AA, O’Dell DS, Hafemeister AC. Maxillary osteosarcoma associated with a dental implant. Am Dent Assoc. 2008;139:1052–9. doi: 10.14219/jada.archive.2008.0307. [DOI] [PubMed] [Google Scholar]
- 12.Zarbo RJ, Carlson ER. Malignancies of the Jaws. In: Regezi JA, Sciubba JJ, Jordan RK, editors. Oral Pathology. 5th ed. St. Louis: Saunders; 2008. pp. 315–21. [Google Scholar]
- 13.Tang N, Song W, Luo J, Haydon RC, He TC. Osteosarcoma development and stem cell differentiation. Clin Orthop Relat Res. 2008;466:2114–30. doi: 10.1007/s11999-008-0335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greene FL, Compton CC, Fritz AG, Shah JP, Winchester DP. New York: Springer; 2006. AJCC Cancer Staging Atlas; pp. 185–9. [Google Scholar]
- 15.Nielsen GP, Rosenberg AE. Update on bone forming tumors of the head and neck. Head Neck Pathol. 2007;1:87–93. doi: 10.1007/s12105-007-0023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]