

Abstract
Complement factor C3 is the central component of the complement system and a key inflammatory protein activated in Alzheimer's disease (AD). Previous studies demonstrated that inhibition of C3 by overexpression of soluble complement receptor-related protein y in an AD mouse model led to reduced microgliosis, increased amyloid β (Aβ) plaque burden, and neurodegeneration. To further address the role of C3 in AD pathology, we generated a complement C3-deficient amyloid precursor protein (APP) transgenic AD mouse model (APP;C3−/−). Brains were analyzed at 8, 12, and 17 months of age by immunohistochemical and biochemical methods and compared with age-matched APP transgenic mice. At younger ages (8–12 months), no significant neuropathological differences were observed between the two transgenic lines. In contrast, at 17 months of age, APP;C3−/− mice showed significant changes of up to twofold increased total Aβ and fibrillar amyloid plaque burden in midfrontal cortex and hippocampus, which correlated with (1) significantly increased Tris-buffered saline (TBS)-insoluble Aβ42 levels and reduced TBS-soluble Aβ42 and Aβ40 levels in brain homogenates, (2) a trend for increased Aβ levels in the plasma, (3) a significant loss of neuronal-specific nuclear protein-positive neurons in the hippocampus, and (4) differential activation of microglia toward a more alternative phenotype (e.g., significantly increased CD45-positive microglia, increased brain levels of interleukins 4 and 10, and reduced levels of CD68, F4/80, inducible nitric oxide synthase, and tumor necrosis factor). Our results suggest a beneficial role for complement C3 in plaque clearance and neuronal health as well as in modulation of the microglia phenotype.
Keywords: Alzheimer's disease, amyloid β, Aβ peptide, complement, C3, neurodegeneration
Introduction
The complement system is essential for immune-mediated defense against pathogens. It may be activated by three pathways: first, via the “classical” activation route through activation of the C1q complex by Ig/antigen immune complexes or non-immune molecules; second, via the immune-complex-independent alternative activation pathway leading to deposition of C3 fragments on target cells; and, third, via the lectin route by binding mannose-binding lectin to pathogen-associated molecular patterns (PAMPs) (for review, see Beek et al., 2003). Activation leads to the formation of multimolecular enzymes that cleave and activate the central component C3. This may lead to the formation of the cytolytic membrane attack complex (MAC), resulting in direct lysis of pathogens. Alternatively, coating of complement activating structures (e.g., immune complexes or PAMPs) with complement C1q activates C3 byproducts C3b and iC3b that promote phagocytosis by cells (such as microglia) expressing the corresponding complement receptors (CRs) such as CR1 (CD35), CR3 (CD11b/CD18), or CR4 (CD11c/CD18). Furthermore, the complement system is involved in the production of anaphylatoxins (C3a and C5a), modulating inflammation and neuroprotective pathways, clearance of immune complexes, and the regulation of adaptive immunity (van Beek et al., 2003).
In mammals, the liver is the major source of complement proteins, but glial cells as well as neurons express complement proteins during stimulation by inflammatory cytokines (Lévi-Strauss and Mallat, 1987; Gasque et al., 1995; Thomas et al., 2000). Complement activation plays an essential role in the inflammatory reactions in the nervous system, including in chronic neurodegenerative disorders such as Alzheimer's disease (AD). Complement is fully activated, and complement components, with their corresponding mRNA levels, are upregulated in AD brains (Eikelenboom et al., 1989; McGeer et al., 1989; Yasojima et al., 1999). Complement components are observed in pyramidal neurons (Shen et al., 1997) and in neurofibrillary tangles and senile plaques in AD brain (Terai et al., 1997; Stoltzner et al., 2000). Dystrophic neurites are immunopositive for MAC (McGeer and McGeer, 2002) suggesting that MAC may contribute to neuritic dystrophy and neuronal loss in AD.
Complement may have also a protective role in AD and normal brains, but its role remains controversial. For example, the C5-derived anaphylatoxin, C5a was reported to protect against excitotoxicity in vitro and in vivo (Pasinetti et al., 1996; Osaka et al., 1999). Overproduction of TGF-β1 in amyloid precursor protein (APP) transgenic (Tg) mice results in elevated C3 brain levels, increased microglial activation, and reduced amyloid β (Aβ) accumulation (Wyss-Coray et al., 2001). Inhibition of complement activation by transgenic overexpression of the soluble complement receptor-related protein y (sCrry) in APP mice resulted in increased Aβ accumulation and neuronal degeneration and reduced microglial activation but did not alter total C3 levels in the brain (Wyss-Coray et al., 2002). However, C1q was found to colocalize with fibrillar Aβ in a presenilin 1 (PS1)/APP mouse model (Matsuoka et al., 2001). APP transgenic mice lacking C1q had no change in Aβ plaque burden compared with C1q-sufficient controls but showed decreased glial activation surrounding plaques and a slowing of neuronal pathogenesis, suggesting a detrimental effect of C1q on neuronal integrity (Fonseca et al., 2004).
Here, we generated an AD mouse model (APP) deficient for C3 (C3−/−) to study the in vivo role of complement C3 on AD pathogenesis. Our results suggest a beneficial role of complement C3 in AD, particularly with advanced aging and pathogenesis.
Materials and Methods
Animals.
Hemizygous APP transgenic mice (Mucke et al., 2000) [line J20, harboring the (PDGF promoter)-human APPsw (K670N, M671L), IN (V717F) transgene; C57BL/6 background] from our breeding colony were bred with homozygous C3-deficient mice (C3−/−) (Wessels et al., 1995) initially obtained from The Jackson Laboratory (line B6.129S4-C3tm1Crr/J; C57BL/6 background) or with C57BL/6 (The Jackson Laboratory). To generate APP;C3−/− animals, C3−/− mice were bred initially with APP mice, then with APP;C3+/−, and later with APP;C3−/− mice. Animals were genotyped by PCR with the following primers: 5′-CTTGGGTGGAGAGGCTATTC-3′ and 5′-GGTTGCAGCAGTCTATGAAGG-3′ for C3 wild-type allele in the same reaction with 5′-AGGTGAGATGACAGGAGATC-3′ and 5′-ATCTTGAGTGCACCA-AGCC-3′ for mutated C3 allele; 5′-GGTGAGTTTGTAAGTGATGCC-3′ and 5′-TCTTCTTCTTCCACCTCAGC-3′ for hAPP allele together with 5′-GCGCGCTCGTGCACACTTATCACA-3′ and 5′-CTGCCCCTGACTTCCTGGAAGCAC-3′ for DNA (GFAP) control. Groups at the different ages were gender balanced and exactly age matched, and the maximal age difference of individual animals within each group was <1 month. All animal use was approved by the Harvard Standing Committee for Animal Use and was in compliance with all state and federal regulations.
Plasma and tissue collection.
Mice were killed by CO2 inhalation, and blood was collected by cardiac puncture followed by transcardial perfusion with 20–30 ml of PBS as described previously (Maier et al., 2005). The brain was removed and divided sagittally. One hemibrain was fixed for 2 h in 10% buffered Formalin, whereas the other hemibrain was snap frozen in liquid nitrogen for biochemical analysis. One hemibrain, one liver, and one kidney were embedded in paraffin as described previously (Lemere et al., 2003).
Brain homogenates and Aβ ELISA.
For Tris-buffered saline (TBS) brain homogenates, frozen hemibrains (without cerebellum and brainstem) were homogenized with a Dounce homogenizer in 5 vol of TBS with a protease inhibitor cocktail (Roche Applied Science). The samples were centrifuged at 175,000 × g for 30 min at 4°C. The supernatant (TBS-soluble homogenate) was collected and stored at −20°C. The pellets were resuspended in the same volume of TBS-T (TBS/1% Triton X-100 plus protease inhibitor cocktail) buffer, sonicated for 5 min in 4°C water bath, homogenized, and centrifuged at 175,000 × g for 30 min at 4°C. The supernatant (TBS-T-soluble homogenate), containing membrane-bound Aβ, was collected and stored at −20°C. The pellets were extracted a third time as described previously (Johnson-Wood et al., 1997) using ice-cold guanidine buffer (5 m guanidine-HCl/50 mm Tris, pH 8.0) (herein referred to as TBS-insoluble or guanidine-soluble homogenate). AβX-40 (Aβ40) and AβX-42 (Aβ42) levels were determined in TBS, TBS-T and guanidine brain homogenates, and total Aβ in EDTA plasma samples of tail blood. ELISAs specific for human Aβx-40, Aβx-42, and total Aβ were performed (using antibodies kindly supplied by Elan Pharmaceuticals) according to Johnson-Wood et al. (1997).
C3 and cytokine ELISAs.
Plates were coated overnight at 4°C with goat anti mouse C3 (1:500; MP Biomedicals) in 50 mm carbonate buffer, pH 9.6, and then blocked for 2 h in blocking buffer (PBS/1% BSA). Serum or TBS–brain homogenates were diluted (1:32,000 for serum and straight for brain homogenates) in wash buffer (PBS/0.1% BSA/0.05% Tween 20), added to the plate for 2 h, hand washed three times, incubated with biotinylated goat anti-mouse C3 antibody (1:1000; MP Biomedicals) in blocking buffer for 1 h, washed, incubated with streptavidin–HRP in blocking buffer for 30 min, washed, and developed with TMB peroxidase substrate for 2–5 min. Stop reagent was added, and optical density was measured at 450 nm. Matching antibody pairs, composed of capture and detection antibodies for murine interleukin-4 (IL-4), IL-10, and tumor necrosis factor α (TNFα) (BD PharMingen), were used according to the instructions of the manufacturer. Serial dilutions of exogenous cytokine were added as standards. TBS and TBS-T brain homogenates were added to the plate without dilution.
APP, secreted αAPP, synaptophysin, C3, inducible nitric oxide synthase, CD68, and F4/80 Western blot.
Western blot was performed as reported previously (Peng et al., 2006). TBS–brain homogenates were used to detect secreted αAPP (αAPPs), C3, and inducible nitric oxide synthase (iNOS) levels, whereas TBS-T brain homogenates were used to examine total APP, CD68, F4/80, and synaptophysin (SYN) levels. Briefly, samples were denatured in SDS sample buffer and separated on 10–20% Tricine gels (Invitrogen). Polyclonal antibody R1736, which recognizes residues 595–611 of APP695 (1:1000; gift from D. Selkoe, Center for Neurological Disorders, Boston, MA) was used to specifically detect the α-secretase-generated ectodomain fragment of APP. Monoclonal antibody 8E5 (1:1000), which recognizes residues 444–591 of human APP, was used to detect the full-length APP (a gift from Elan Pharmaceuticals). Polyclonal anti-C3 (1:500; MP Biomedicals), anti-synaptophysin (1:1000; Sigma), anti-iNOS (1:200; BD Bioscience), anti-CD68 (1:200; Serotec), anti-F4/80 (1:200; Serotec), and anti-β-actin (1:10,000; Sigma) were used to probe the membrane. Horseradish peroxidase-coupled anti-rabbit or anti-mouse IgG were used as secondary antibodies. The signal was detected with an enhanced chemiluminescence kit (Pierce). Densitometric evaluation was performed using an imaging system and the corresponding analyzing software (FluorchemTMIS-8800 software; Alpha Innotech). Protein levels were normalized to β-actin.
Immunohistochemistry.
Serial 10 μm sagittal paraffin sections of mouse brain were mounted on glass slides, and immunohistochemistry was performed as reported previously (Lemere et al., 2002) using Vector ELITE ABC kits (Vector Laboratories). The following antibodies were used for neuropathological analysis: anti-CD45 (1:5000; Serotec), anti-Iba1 (ionized-calcium binding adapter molecule 1) (1:500; Wako Bioproducts), anti-GFAP (1:500; DakoCytomation), anti-neuronal-specific nuclear protein (NeuN) (1:250; Millipore Bioscience Research Reagents), anti-APP antibody 22C11 (1:200; Millipore Bioscience Research Reagents), rabbit polyclonal anti-Aβ R1282 (1:1000; gift from D. Selkoe), and anti-Aβ40 and anti-Aβ42 (1:500; BioSource International). Computer-assisted image analysis was used to quantify the percentage area of thioflavin S staining and immunoreactivity as described previously (Maier et al., 2006). Briefly, the percentage area occupied by immunoreactivity was calculated for three to four equidistant sections per animal of total hippocampus area or for one visual field of the whole cortex dorsal to the hippocampus (midfrontal cortex, ∼1.5 mm2). The threshold for detection of immunoreactivity was kept constant for the analysis of an entire series of sections for each antibody. Presynaptic terminals were labeled with anti-SYN (1:200; Sigma), neuronal cell bodies and dendrites with anti-MAP2 (1:200; Sigma), and newly generated neurons with anti-doublecortin (C-18, 1:500; Santa Cruz Biotechnology), followed by Alexa488, Rhodamine Red-X (Invitrogen), or cyanine 3 (The Jackson Laboratory) coupled secondary antibodies. To quantify immunoreactivity, acquisition of images was performed in a single session using a SPOT camera with individual red–green–blue channels set for fluorescence (Diagnostic Instruments) and analyzed by NIH ImageJ software.
Stereological neuron counts were performed in three 10 μm NeuN-stained sagittal brain sections per animal spaced 200 μm apart using the optical dissector technique (Irizarry et al., 1997). The average number of neurons per section in the CA3 region of hippocampus was estimated using ∼12 optical dissectors and the Bioquant Image Analysis System according to the principle of Cavalieri (West and Gundersen, 1990). Each optical dissector included a 50 × 50 μm sampling box. Using a 100× oil-immersion lens, neurons with a visible NeuN cell body were counted if they were not visible in the initial plane of focus but came into focus as the optical plane moved through the tissue. The average number of hippocampal CA3 neurons per section was determined by calculating the mean of the neuron counts from three sections per animal.
Statistical analyses.
The Mann–Whitney U test was used for statistical analysis of the percentage area of immunoreactivity in brain sections and Aβ levels in brain homogenates. The critical α level was set to 0.05 for all statistical analyses. All values reported are average ± SEM. A two-tailed Spearman's correlation was used for correlation of plaque load and NeuN counts. SPSS software (version 11.5) was used for statistical analysis
Results
Expression of complement C3 protein in the CNS and periphery is absent in C3-deficient APP transgenic (APP;C3−/−) mice
To determine the role of central complement component C3 in Aβ accumulation and neurodegeneration, we generated APP transgenic mice lacking complement C3 by breeding APP (J20 line) (Mucke et al., 2000) to complement C3-deficient mice (Wessels et al., 1995). Complement C3 gene deletion was confirmed by PCR using primers showing the presence of the mutated allele and the absence of the wild-type C3 allele PCR product in APP;C3−/− mice (Fig. 1A). Expression of complement C3 protein was demonstrated by Western blot and C3 ELISA with an anti-C3 antibody in brain homogenates (Fig. 1B,C) and blood plasma (Fig. 1D) in 12- and 17-month-old APP mice. In contrast, C3 protein was undetectable in brain and blood plasma in APP;C3−/− mice at the same ages (Fig. 1). C3 levels in blood as well as brain were slightly higher in 17-month-old compared with 12-month-old APP animals (p = 0.14; n = 5 mice each at 12 and 17 months) and approximately four orders of magnitude higher in plasma than in brain tissue.
Figure 1.

Complement C3 protein is expressed in the brain and in the plasma of 12- and 17-month-old APP mice but is undetectable in APP;C3−/− mice. A, Complement C3 gene deletion was determined by PCR by the absence of the wild-type C3 allele PCR product (top band at ∼350 bp) and the presence of the mutated allele (bottom band at ∼280 bp). B–D, Expression of complement C3 protein in the brain (B, C) and plasma (D) of 12- and 17-month-old APP mice (n = 5 per group) was shown by Western blot (B, C) and C3 ELISA (D). C3 protein was absent in the corresponding APP;C3−/− mice. The α/β dimer of C3 protein (∼185 kDa) in brain is indicated by the arrow in B.
Plaque load and gliosis are unaltered in 12-month-old complement C3-deficient APP Tg mice
To study the role of C3 deficiency on the onset of plaque pathology, we analyzed total Aβ immunoreactive plaque load in the hippocampi of 8-month-old APP and APP;C3−/− mice (n = 5 mice per group). Quantitative image analysis showed no significant difference in plaque burden between the groups (data not shown). Semiquantitative analysis of CD45-specific immunoreactivity as a marker for activated microglia and GFAP-specific immunoreactivity as a measure to quantify astrogliosis in the hippocampus revealed no major differences between the groups (data not shown).
At 12 months of age, quantitative image analysis of brain sections stained with Aβ42-, Aβ40-, or pan-Aβ-specific R1282 antibodies revealed no significant differences in plaque load in hippocampus or midfrontal cortex in APP;C3−/− mice (n = 8) compared with their age-matched APP controls (n = 6) (Fig. 2A,B) (11.8 ± 2.0 vs 10.2 ± 0.9% area of hippocampus for R1282; p > 0.05). At this age, thioflavin S-positive plaques were mainly detected in the hippocampus, and, again, quantitative image analysis showed no significant differences between the two groups of mice (Fig. 2C). In parallel, no significant differences were found for CD45- and GFAP-specific immunoreactivity (Fig. 2D,E). Quantification of Aβ42 and Aβ40 by ELISA in TBS, TBS-T, and guanidine-HCl brain extracts also showed no significant differences in Aβ levels (Fig. 2F,G). Total Aβ levels in plasma were increased by 31%, but the difference did not reach significance because of variability (Fig. 2H) (n = 6 per group; p = 0.11).
Figure 2.

Neuropathological and biochemical analysis at 12 months of age showed no significant differences in plaque load, microgliosis, and astrocytosis in APP;C3−/− (filled bars; n = 8) compared with age-matched APP control mice (open bars; n = 6). A, B, Quantitative image analysis of Aβ42- (A) and Aβ40- (B) specific immunoreactivity did not show any significant differences in hippocampus or midfrontal cortex. C–E, Image analysis of thioflavin S-positive plaque load (C) as well as CD45- and GFAP-specific immunoreactivity in the hippocampus (D, E) revealed no significant differences between the APP and APP;C3−/− groups. F–H, Quantification of Aβ42 and Aβ40 by ELISA in TBS, TBS-T, and guanidine extracts of brain homogenates (F, G) or in plasma (H) did not reveal any significant difference between the groups. % ROI, Percentage of immunoreactivity within the region of interest.
Aβ deposition and insoluble Aβ42 brain levels are significantly elevated, whereas TBS-soluble Aβ levels are decreased in 17-month-old complement C3-deficient APP Tg mice
In contrast to 12-month-old mice, total Aβ plaque load at 17 months of age was approximately doubled in both the hippocampus and midfrontal cortex in APP;C3−/− mice compared with APP mice as shown by Aβ42- and Aβ40-specific immunoreactivity (Fig. 3A,B), general Aβ immunolabeling (Fig. 4A,B,G,H, R1282 antibody), and thioflavin S-positive staining of fibrillar amyloid deposits (Figs. 3C, 4M,N) (p < 0.05; n = 5 mice per group). Guanidine-soluble (TBS-T-insoluble) Aβ42 and Aβ40 levels were higher in APP;C3−/− mice compared with APP mice (Fig. 3D,E) at 17 months of age, although only the difference in Aβ42 levels reached significance (p < 0.01). These data are in agreement with the corresponding Aβ42 and Aβ40 plaque loads. In contrast, TBS-soluble Aβ42 and Aβ40 levels were significantly lower in APP;C3−/− mice compared with APP mice (Fig. 3D,E) (p < 0.05). TBS-T soluble Aβ42 and Aβ40 levels were nonsignificantly reduced in the APP;C3−/− mice compared with APP mice. Total Aβ levels in plasma were increased by 51% in APP;C3−/− mice compared with APP mice (Fig. 3F) (nonsignificant trend, p = 0.06). These results indicate that a deficiency of the key complement factor C3 results in greater accumulation of insoluble Aβ in the brain and a trend for more soluble Aβ in the plasma with aging in APP Tg mice.
Figure 3.
A–E, Quantitative neuropathological analysis (A–C) and biochemical analysis of brain homogenates by quantitative Aβ42- and Aβ40-specific ELISAs (D, E) of 17-month-old mice shows significantly higher Aβ plaque load and significantly higher Aβ42 levels in guanidine brain extract in APP;C3−/− mice (filled bars; n = 5) compared with age-matched APP controls (open bars; n = 5). A–C, Aβ42- and Aβ40- specific immunoreactivity (A, B), as well as thioflavin S-positive staining (C), were significantly increased in hippocampus and midfrontal cortex of APP;C3−/− mice (A–C; p < 0.05). D, E, Aβ42 and Aβ40 levels in TBS-soluble extracts of brain homogenates were significantly reduced, whereas Aβ42 was significantly elevated and Aβ40 nonsignificantly increased in TBS-insoluble guanidine extracts in APP;C3−/− mice. Aβ42 and Aβ40 levels in the membrane-bound TBS-T extracts were nonsignificantly reduced. F, A trend was evident for higher total Aβ levels in plasma samples from APP;C3−/− mice compared with APP mice (51% increase; p = 0.06). *p < 0.05, **p < 0.01. % ROI, Percentage of immunoreactivity within the region of interest.
Figure 4.

A–N, Immunohistochemical analysis of hippocampus of representative sections of 17-month-old APP (left column; 2 sets of serial sections: A, C, E and G, I, K, M) and APP;C3−/− (right column; 2 sets of serial sections: B, D, F and H, J, L, N) mice. A, B, Relative to complement-sufficient APP Tg mice (A), the total plaque load demonstrated using the pan-specific Aβ antibody R1282 was increased in the complement-deficient APP;C3−/− mice (B). C, D, In parallel, the number of CD45-positive microglia was also increased in APP;C3−/− mice, and the microglia were mostly associated with compacted plaques (arrows). E, F, GFAP immunoreactivity, a marker for astrogliosis, was comparable between both groups, although modest increases were observed in APP;C3−/− mice. G–N, In parallel to the increase of total (G, H; R1282 antibody) and fibrillar, compact plaque load (M, N; thioflavin S), the number of Iba1-immunoreactive microglia/macrophage (I, J) and dystrophic neurites (K, L) observed in APP;C3−/− mice (right column) was increased compared with that of APP mice (left column). Scale bars, 200 μm.
Microglia/macrophage are differentially activated in the brain of complement C3-deficient APP Tg mice toward a more alternative M2 activation phenotype
Activation of microglia, measured by CD45-specific immunoreactivity, was significantly increased in hippocampus (p < 0.01) and cortex (p < 0.05) of APP;C3−/− mice (Figs. 4C,D, 5A). Because CD45 staining was associated mainly with compacted plaques and correlated well with thioflavin S-positive plaque load (r = 0.75; p < 0.05; n = 5 mice per group), we calculated the CD45/thioflavin S ratio as an indicator for activation of microglia corrected by the difference in plaque load (as performed by Wilcock et al., 2006). A trend for an increased CD45/thioflavin S ratio was observed in the hippocampus of APP;C3−/− mice compared with APP mice (p = 0.10) (Fig. 5B) even after correction for plaque load.
Figure 5.

Quantitative neuropathological and biochemical analysis of gliosis at 17 months of age. A, B, Total CD45 immunoreactivity was significantly increased in APP;C3−/− mice (A; *p < 0.05, **p < 0.01), whereas the CD45/thioflavin S ratio (B) only showed a trend to be elevated in APP;C3−/− mice (p = 0.10). C, Iba1 immunoreactivity was modestly and nonsignificantly elevated in APP;C3−/− compared with APP mice. D, GFAP immunoreactivity was only slightly higher in APP;C3−/− compared with APP mice, but the difference was nonsignificant. E, F, In contrast to CD45 and Iba1, the levels of microglial markers CD68 and F4/80 by Western blot were nonsignificantly lower in TBS-soluble brain homogenates of APP;C3−/− compared with APP mice. The levels in the APP;C3−/− mice are presented as a percentage of the levels in the APP transgenic mice. β-Actin was used as a protein loading control.
To further characterize the microglial phenotype, we used anti-Iba1 immunoreactivity and Western blot levels of CD68 and F4/80 as additional markers of microglia/macrophage activity. Western blot was used because immunohistochemistry with anti-CD68 and anti-F4/80 was unsuccessful on paraffin sections. In hippocampus and cortex, Iba1 immunoreactivity was detected mainly in microglia/macrophages associated with compacted plaques, but it also stained cells not directly associated with plaques, particularly in the cortex. The number of Iba1-positive cells was somewhat higher in APP;C3−/− compared with APP mice (Fig. 4I,J), but quantitative image analysis revealed no significant differences in the percentage area of Iba-1 immunoreactivity between the two groups (Fig. 5C). In contrast to CD45 and Iba1, quantification of CD68 and F4/80 microglia/macrophage markers in TBS-T total brain homogenate by Western blot revealed reduced levels of each in APP;C3−/− compared with APP mice, suggesting a differential regulation of microglia, but the difference was not statistically significant. No differences in CD68 and F4/80 levels were observed in 12-month-old APP;C3−/− mice compared with APP mice (data not shown).
Next, iNOS, TNF, IL-4, and IL-10 levels, as indicators of microglia/macrophage phenotype, were examined. iNOS levels in TBS-soluble brain homogenates were significantly lower by Western blot in APP;C3−/− mice compared with APP mice (Fig. 6A) (p < 0.05). TBS-T-soluble, membrane-bound TNF levels were significantly reduced (p < 0.05) by ELISA, whereas TBS-soluble TNF levels showed a strong trend for reduction (p = 0.05) (Fig. 6B). A significant increase in IL-4 was observed by ELISA in TBS-soluble brain homogenates in APP;C3−/− compared with APP mice (Fig. 6C) (p < 0.05). IL-10 levels in both TBS-soluble and TBS-T-soluble brain homogenates were elevated in APP;C3−/− mice compared with APP mice (Fig. 6D), although the differences did not reach statistical differences because of a high level of variability among the mice in each group. Together, these results are indicative of a shift of the microglia/macrophage toward a more alternative M2 activation phenotype in APP;C3−/− mice compared with APP mice (Mantovani et al., 2004; Morgan et al., 2005).
Figure 6.

Differential activation of microglia/macrophage in the brains of APP;C3−/− mice toward a more alternative activation/M2 phenotype. A, iNOS levels were significantly reduced in TBS-soluble, but not TBS-T-soluble, membrane-bound brain homogenates by Western blot in APP;C3−/− compared with APP mice (*p < 0.05). B, TBS-T-soluble, membrane-bound and TBS-soluble TNF levels by ELISA were lower in APP;C3−/− compared with APP mice, but only the difference in membrane-bound TNF reached significance (*p < 0.05). C, D, IL-4 (C) and IL-10 (D) levels in TBS and TBS-T brain homogenates by ELISA were elevated in APP;C3−/− compared with APP mice, but only the difference in IL-4 in TBS-soluble brain homogenates reached significance (*p < 0.05).
GFAP immunoreactivity of astrocytes in the hippocampus (Fig. 4E,F) and cortex was nonsignificantly increased in APP;C3−/− compared with APP mice (Fig. 5D).
APP processing is not altered in complement C3-deficient APP Tg mice
To determine whether altered plaque pathology is attributable to altered expression or processing of APP protein, total APP protein levels and αAPPs fragments were quantified in brain homogenates by Western blot and normalized to β-actin levels (Fig. 7) (average of two experiments, n = 4 mice per group). Total APP levels were slightly increased in APP;C3−/− mice at 12 months of age (Fig. 7A), but, in general, APP levels were not significantly different between the two lines of mice at either 12 or 17 months of age. No differences were observed in αAPPs levels between the two groups at either 12 or 17 months, indicating that there was no obvious effect of C3 deficiency on APP processing (Fig. 7C,D).
Figure 7.

APP processing was not altered in C3-deficient APP mice (filled bars) at either 12 or 17 months of age. Quantification of β-actin normalized total APP protein (A, B; average of 2 experiments, n = 4 mice per group) and αAPPs fragment (C, D; average of 2 experiments, n = 4 mice per group) in brain homogenates by Western blot revealed no difference in protein levels (p > 0.05).
NeuN-positive neurons are reduced in the CA3 region of hippocampus in 17-month-old complement C3-deficient APP Tg mice
Anti-NeuN is a neuronal marker that labels most differentiated neurons in the adult neocortex (Mullen et al., 1992), but degenerating neurons seem to lose this marker (Larsson et al., 2001). Consequently, the amount of NeuN-positive neurons was used as a marker to determine the role of complement C3 on neuronal survival. The average number of NeuN-positive neurons per section (based on three 10 μm sections) in the CA3 region of hippocampus, determined by stereological counting, was similar between 12-month-old APP and APP;C3−/− mice (Fig. 8C) (221 ± 8 vs 225 ± 5; n = 6 mice per group). In contrast, at 17 months of age, the average number of NeuN-positive neurons per section in the CA3 region of hippocampus was significantly reduced in APP;C3−/− mice (201 ± 8) compared with APP mice (225 ± 8; p < 0.05) (Fig. 8A–C). Furthermore, the average number of NeuN-positive neurons per section was significantly reduced in 17-month-old versus 12-month-old APP;C3−/− mice (p = 0.015) but not in C3-sufficient APP mice. The average number of NeuN-positive neurons per section within individual mice was inversely correlated with the thioflavin S plaque load (Fig. 8D) (r = −0.636; p < 0.05; n = 5 mice per group). There was a more pronounced loss of neurons in the CA3 compared with the CA1 region of the hippocampus (data not shown). This may correlate with the observation of an increased expression of the hAPP protein (Fig. 4K,L) and increased Aβ plaque load (Fig. 4A,B,G,H) in CA3 compared with CA1 in the hippocampus of this mouse model.
Figure 8.
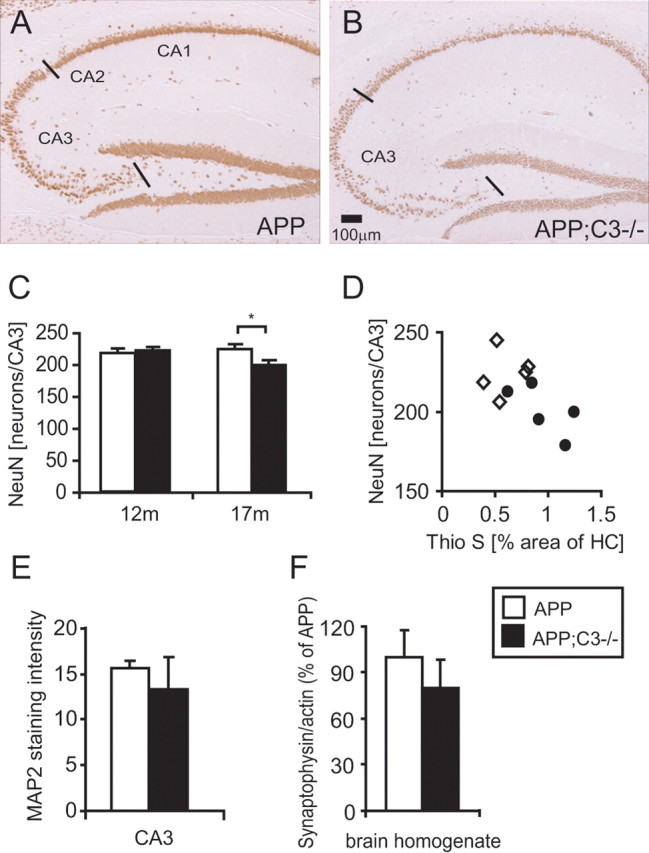
At 17 months of age, C3-deficient APP transgenic mice showed a reduction in the number of NeuN-positive neurons that correlated with the thioflavin S plaque load. A, B, Seventeen-month-old APP;C3−/− mice (B; n = 5) had fewer NeuN-positive neurons in the CA3 region of the hippocampus compared with the APP control group (A; n = 5). Neuronal counting was performed in CA3 between the black lines indicated in A and B. C, Quantification of NeuN-positive neurons in the CA3 region of the hippocampus by stereological counting showed a significant reduction in APP;C3−/− mice (filled bars) compared with APP mice (open bars) (*p < 0.05). No difference in neuronal counts was detected between APP and APP;C3−/− mice at 12 months of age. A significant reduction in neurons was observed in APP;C3−/− mice, but not APP mice, from 12 to 17 months of age (p = 0.015). D, The number of NeuN-positive neurons of individual mice correlated significantly with their thioflavin S plaque load (r = −0.636; p < 0.05; n = 5 mice per group; open diamonds indicate APP mice, and black circles indicate APP;C3−/− mice). E, MAP2 immunoreactivity was nonsignificantly reduced in complement C3-deficient APP mice compared with complement-sufficient APP mice. F, Synaptophysin levels in TBS-T extracts of brain homogenate were nonsignificantly reduced in APP;C3−/− mice by Western blot compared with APP mice. Scale bar, 100 μm.
MAP2 immunoreactivity, a marker for neuronal dendrites and cell bodies, was slightly reduced in APP;C3−/− mice compared with APP mice, but the difference was not significant (Fig. 8E). Synaptophysin is used as a marker to assess presynaptic terminals and neuronal integrity. Similar to MAP2, no significant difference was observed in synaptophysin staining intensity in the CA1 and CA3 region of hippocampus in brain sections between the two groups (data not shown). Synaptophysin levels, quantified by Western blot of TBS-T brain homogenates, were similar between APP and APP;C3−/− mice at 12 months of age (data not shown) but showed a tendency to be lower in APP;C3−/− compared with APP mice at 17 months of age; however, this difference was not significant (Fig. 8F).
A reduction of neuronal numbers might possibly be attributable to a reduction in neurogenesis, especially because it was reported recently that complement C3 and its receptor C3aR play a role in basal and ischemia induced neurogenesis (Rahpeymai et al., 2006). Neurogenesis usually takes place in the subventricular zone (SVZ) and in the granular cell layer (GCL) of the dentate gyrus (for review, see Lledo et al., 2006). Therefore, we used Doublecortin (DCX) as a marker for newly generated neurons and quantified DCX staining in the SVZ and GCL. Quantification of DCX-positive cells in the GCL, the potential source of new neurons in the hippocampus, was not possible as the labeling of DCX-positive cells in the GCL of 12- and 17-month-old APP or APP;C3−/− mice was below the limit of detection unlike the clusters of DCX-positive cells in the SVZ. This finding is in agreement with previous studies characterizing neurogenesis in this animal model (Jin et al., 2004), showing very low levels of neurogenesis in the GCL.
Discussion
To experimentally address the role of complement C3 in the pathogenesis of AD, we generated C3-deficient APP transgenic mice and analyzed them neuropathologically and biochemically at 8, 12, and 17 months of age, comparing them with age-matched APP transgenic mice. At younger ages (8 and 12 months), no significant differences were observed between APP and APP;C3−/− mice in plaque load, biochemical levels of Aβ, or any of the neuronal or glial markers examined. In contrast, at 17 months of age, the APP;C3−/− mice showed a significant approximate twofold increase in total Aβ and fibrillar amyloid plaque burden that correlated with significantly increased guanidine-soluble Aβ42 levels and reduced TBS-soluble Aβ42 and Aβ40 in brain homogenates, a nonsignificant trend for increased plasma Aβ levels, a significant reduction of NeuN-positive neurons in the CA3 region of the hippocampus, and a shift of microglia/macrophage toward a more alternative activation/M2 phenotype (according to Mantovani et al., 2004; Morgan et al., 2005). Our findings demonstrate that the complete absence of the central complement component, C3, accelerates AD-like plaque pathology with aging once plaque pathogenesis is underway. Although many of the differences between the APP;C3−/− and APP mice reported here were statistically significant, many comparisons resulted in a nonsignificant trend attributable to the relatively small number of mice studied and the high variability observed in APP transgenic mice, in general. However, these trends provide additional support to the overall findings.
The rather late effect of C3 deficiency may be related to an attempt of APP mice to protect the brain by upregulating complement C3 once AD-like pathogenesis is underway. Similar to a previous report by Wyss-Coray et al. (2002), we observed an increase in C3 protein levels with aging and AD pathogenesis in APP mice. In their study, Wyss-Coray and colleagues demonstrated that C3 mRNA levels increased with age in APP mice and that C3 protein levels were elevated when TGF-β was overexpressed in APP mice (Wyss-Coray et al., 2002). Inhibition of complement C3 convertase (to block activation of C3) by overexpression of sCrry in APP mice resulted in increased plaque burden and neurodegeneration, even in the presence of C3 protein (Wyss-Coray, 2002).
Consistent with an important role of complement in late-stage AD-like pathogenesis is a study (Matsuoka et al., 2001) showing the colocalization of complement-activating C1q and an upregulation of C1q with increased formation of fibrillar Aβ plaques in a PS1/APP transgenic mouse model. In addition, we reported previously prominent C1q and C3 immunoreactivity in highly compacted Aβ42-positive neuritic plaques associated with microgliosis in the cortex of middle-aged and older individuals with Down syndrome (Stoltzner et al., 2000). Whether the elevation of complement proteins in AD brain is an attempt to protect the brain or a consequence of neuronal damage is unclear, but our results, along with those of Wyss-Coray et al. (2002), suggest that complement C3 may play a protective role in the brain.
Amyloid fibrils have been detected in microglia in human AD brain, suggesting that microglia are involved in the clearing of Aβ protein deposits (Wegiel and Wisniewski, 1990). Furthermore, it has also been shown in multiple studies that different forms of aggregated Aβ activate and become bound by complement opsonins such as C3b (Webster et al., 1997; Bradt et al., 1998), which facilitates CR3-mediated phagocytosis of Aβ (Ehlers, 2000). Recent mouse studies report a reduction in the microglial markers F4/80 and major histocompatibility complex class II alloantigens, I-A/I-E, in old C1q-deficient Tg2576 (APP) transgenic mice (Fonseca et al., 2004). In addition, F4/80-positive microglia were reduced in APP/sCrry mice (Wyss-Coray et al., 2002). By Western blot, we, too, found a nonsignificant trend for reduced F4/80 as well as CD68 levels in our 17-month-old APP;C3−/− mice. Therefore, it is possible that the increased fibrillar plaque load observed in our C3-deficient APP Tg mice may be attributable to less efficient phagocytosis of Aβ fibrils in the absence of C3 attributable to lack of C3b- or iC3b-mediated opsonization.
Interestingly, although F4/80 and CD68 levels were reduced in our 17-month-old APP;C3−/− mice compared with age-matched APP mice, we found increases in other microglia/macrophage markers of activation, including CD45 (significant) and Iba1 (nonsignificant trend), correlating with increased Aβ plaque burden and neurodegeneration. C3 deficiency in 17-month-old APP mice resulted in increased IL-4 (p < 0.05 in TBS-soluble fraction) and IL-10 (nonsignificant trend) and reduced iNOS (p < 0.05) and TNF (p < 0.05 in TBS-T membrane-bound fraction) brain levels. Others have demonstrated that anti-inflammatory cytokines, such as IL-4 and IL-10, increased fibrillar Aβ phagocytosis by murine primary microglia in vitro (Koenigsknecht-Talboo and Landreth, 2005). Although anti-inflammatory cytokine levels were elevated in the APP;C3−/− mice in our study, Aβ deposition was increased as well, indicating that Aβ phagocytosis by microglia was ineffective at removing Aβ deposits. Thus, it is possible that the presence of complement C3 may be necessary for anti-inflammatory cytokines to stimulate microglial phagocytosis of aggregated Aβ.
Together, our findings indicate a shift of the microglia/macrophage response toward an alternative M2 activation phenotype (Mantovani et al., 2004; Morgan et al., 2005) in the absence of complement C3. M2-type microglia/macrophage are often found in association with apoptotic cells to scavenge debris and promote tissue repair, which is in agreement with the increased neurodegeneration we observed in the 17-month-old APP;C3−/− mice. Upregulation of CD45, which is expressed at high levels in infiltrating microglia/macrophage (Ford et al., 1995), could represent enhanced infiltration of peripheral macrophages attributable to increased fibrillar Aβ deposits and a higher incidence of dying cells in brain. It should be noted that, in the study of Wyss-Coray et al. (2002), inhibition of C3 convertase by sCrry overexpression may have only affected certain microglial functions such as Aβ phagocytosis, whereas the complete absence of C3 may have additional affects on microglial function and/or molecules that normally suppress microglial activation along the phagocytic and cytotoxic pathways. Ours is the first study to fully examine the role of complement C3 on the microglia/macrophage phenotype and cytokines in APP mice.
Alternatively, increased Aβ deposition in brain and Aβ levels in plasma in C3-deficient APP Tg mice may also be attributable to reduced peripheral degradation of Aβ. Indeed, a recent study suggested an important role of complement C3 in the peripheral clearance of Aβ by C3b-dependent adherence to CR1 on erythrocytes in blood of humans (Rogers et al., 2006). A recently described complement receptor, CR1g, was found to be important for C3b-dependent clearance of pathogens from the blood (Helmy et al., 2006) and, thus, may represent an additional pathway for clearance of complement-opsonized Aβ in the periphery. Such a mechanism should be affected in C3-deficient APP mice. Indeed, the absence of C3 resulted in the accumulation of Aβ in the periphery, thereby increasing the amount of peripheral Aβ available for influx into the brain.
In contrast to guanidine-soluble Aβ42 levels, which were increased with the fibrillar Aβ plaque load, TBS-soluble and to some extent also TBS-T-soluble Aβ levels were reduced in the brains of C3-deficient APP mice. Therefore, it is possible that complement C3 also plays a role in suppressing aggregation of Aβ, in which case the lack of C3 would result in reduced soluble Aβ levels in brain and increased deposition of aggregated Aβ into plaques. However, this seems unlikely because C3 levels were elevated in the complement-sufficient APP Tg mice with age as Aβ became more aggregated.
Increased amounts of fibrillar Aβ deposition in 17-month-old APP;C3−/− mice correlated with a reduction of NeuN-positive neurons in hippocampus, suggesting a role for complement C3 in neuronal survival and health. Neuronal loss was highest in CA3 compared with CA1, possibly attributable to increased hAPP transgene expression and Aβ deposition. Neurons express C3; thus, the neurons in C3-deficient APP mice may be more vulnerable to the cytotoxic effects of Aβ and/or microglial activation. However, C3-deficient neurons in 12-month-old APP;C3−/− mice appeared relatively healthy. MAP2 and synaptophysin immunoreactivity were nonsignificantly reduced in APP;C3−/− mice, in agreement with the findings of Wyss-Coray et al. (2002) in which a similar neuronal phenotype was observed during inhibition of C3 convertase. By electron microscopy, they reported an accumulation of degenerating neurons in APP/sCrry mice compared with wild-type mice that correlated with the reduction of NeuN-positive neurons. Our findings agree with other studies that have proposed a neuroprotective effect of complement proteins and complement activation products such as C3a and C5a (van Beek et al., 2003).
A recent study attributes an important role to complement C3 and its receptor C3aR in basal- and ischemia-induced neurogenesis in young adult mice (Rahpeymai et al., 2006). The reduction of NeuN-positive neurons in APP;C3−/− mice may be attributable to a reduction of neurogenesis over the lifespan of C3-deficient mice. However, because the average number of hippocampal CA3 NeuN-positive neurons per section was similar in 12-month-old APP;C3−/− versus APP mice, this seems unlikely. Quantification of DCX-positive cells in the GCL of the dentate gyrus, the source for new hippocampal neurons, was not possible because the DCX staining in GCL was below detectable levels in the 12- and 17-month-old APP and APP;C3−/− mouse brain sections (in contrast to DCX-positive cell clusters in the SVZ), consistent with previous reports in APP mice (Jin et al., 2004).
In addition, C1q and C3 were recently reported to play a role in CNS synapse elimination as shown by the failure of C1q-deficient and C3-deficient mice to fully refine retinogeniculate connections during postnatal development (Stevens et al., 2007). Complement-independent mechanisms of synapse elimination may also exist (as suggested by the authors) and may have allowed synapse elimination during development in our APP;C3−/− mice. Furthermore, initial studies in a glaucoma mouse model by the same group suggest that complement-mediated synapse elimination may become aberrantly reactivated under disease conditions via upregulation of C1q at the synapse, leading to synaptic and neuronal degeneration. The presence and possible upregulation of complement C1q in our APP;C3−/− mice with aging may have contributed to the synapse reduction and neuronal loss we observed at 17 months of age.
In summary, we demonstrate that C3 deficiency in APP mice resulted in increased cerebral Aβ deposition and neuronal loss as well as a shift to a M2 microglial/macrophage response, thereby supporting a beneficial, neuroprotective role of complement C3 in brain. Additional studies are underway to further tease apart the various mechanisms by which complement proteins influence neuronal health at different ages.
Footnotes
This work was supported by National Institutes of Health Grant AG20159 (C.A.L.). We thank Guiquan Chen, Admar Verschoor, Diana Li, Eva Luo, Wei Liu, and Weiming Xia for help with experiments, Dan Frenkel for providing some APP mice, and Lennart Mucke for sharing the APP (J20) transgenic mouse line.
References
- Bradt BM, Kolb WP, Cooper NR. Complement-dependent proinflammatory properties of the Alzheimer's disease beta-peptide. J Exp Med. 1998;188:431–438. doi: 10.1084/jem.188.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MR. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect. 2000;2:289–294. doi: 10.1016/s1286-4579(00)00299-9. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, Hack CE, Rozemuller JM, Stam FC. Complement activation in amyloid plaques in Alzheimer's dementia. Virchows Arch B Cell Pathol Incl Mol Pathol. 1989;56:259–262. doi: 10.1007/BF02890024. [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. J Neurosci. 2004;24:6457–6465. doi: 10.1523/JNEUROSCI.0901-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- Gasque P, Fontaine M, Morgan BP. Complement expression in human brain. Biosynthesis of terminal pathway components and regulators in human glial cells and cell lines. J Immunol. 1995;154:4726–4733. [PubMed] [Google Scholar]
- Helmy KY, Katschke KJ, Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, Scales SJ, Ghilardi N, van Lookeren Campagne M. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 2006;124:915–927. doi: 10.1016/j.cell.2005.12.039. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci. 1997;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, Greenberg DA. Enhanced neurogenesis in Alzheimer's disease transgenic (PDGF-APPSw, Ind) mice. Proc Natl Acad Sci USA. 2004;101:13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Landreth GE. Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J Neurosci. 2005;25:8240–8249. doi: 10.1523/JNEUROSCI.1808-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson E, Lindvall O, Kokaia Z. Stereological assessment of vulnerability of immunocytochemically identified striatal and hippocampal neurons after global cerebral ischemia in rats. Brain Res. 2001;913:117–132. doi: 10.1016/s0006-8993(01)02762-7. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, Leverone JF, Mori C, Clements JD. Intranasal immunotherapy for the treatment of Alzheimer's disease: Escherichia coli LT and LT(R192G) as mucosal adjuvants. Neurobiol Aging. 2002;23:991–1000. doi: 10.1016/s0197-4580(02)00127-6. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, LaFrancois J, Malester B, Mori C, Leverone JF, Matsuoka Y, Taylor JW, DeMattos RB, Holtzman DM, Clements JD, Selkoe DJ, Duff KE. Evidence for peripheral clearance of Aβ following chronic, active Aβ immunization in PSAPP mice. Neurobiol Dis. 2003;14:10–18. doi: 10.1016/s0969-9961(03)00044-5. [DOI] [PubMed] [Google Scholar]
- Lévi-Strauss M, Mallat M. Primary cultures of murine astrocytes produce C3 and factor B, two components of the alternative pathway of complement activation. J Immunol. 1987;139:2361–2366. [PubMed] [Google Scholar]
- Lledo PM, Alonso M, Grubb MS. Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci. 2006;7:179–193. doi: 10.1038/nrn1867. [DOI] [PubMed] [Google Scholar]
- Maier M, Seabrook TJ, Lemere CA. Modulation of the humoral and cellular immune response in Abeta immunotherapy by the adjuvants monophosphoryl lipid A (MPL), cholera toxin B subunit (CTB) and E. coli enterotoxin LT(R192G) Vaccine. 2005;23:5149–5159. doi: 10.1016/j.vaccine.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Maier M, Seabrook TJ, Lazo ND, Jiang L, Das P, Janus C, Lemere CA. Short amyloid-β (Aβ) immunogens reduce cerebral Aβ load and learning deficits in an Alzheimer's disease mouse model in the absence of an Aβ-specific cellular immune response. J Neurosci. 2006;26:4717–4728. doi: 10.1523/JNEUROSCI.0381-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O'Banion MK, Tenner AJ, Lemere CA, Duff K. Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer's disease. Am J Pathol. 2001;158:1345–1354. doi: 10.1016/S0002-9440(10)64085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. The possible role of complement activation in Alzheimer disease. Trends Mol Med. 2002;8:519–523. doi: 10.1016/s1471-4914(02)02422-x. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Akiyama H, Itagaki S, McGeer EG. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett. 1989;107:341–346. doi: 10.1016/0304-3940(89)90843-4. [DOI] [PubMed] [Google Scholar]
- Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer's therapeutics. J Neuropathol Exp Neurol. 2005;64:743–753. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Osaka H, Mukherjee P, Aisen PS, Pasinetti GM. Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. J Cell Biochem. 1999;73:303–311. [PubMed] [Google Scholar]
- Pasinetti GM, Tocco G, Sakhi S, Musleh WD, DeSimoni MG, Mascarucci P, Schreiber S, Baudry M, Finch CE. Hereditary deficiencies in complement C5 are associated with intensified neurodegenerative responses that implicate new roles for the C-system in neuronal and astrocytic functions. Neurobiol Dis. 1996;3:197–204. doi: 10.1006/nbdi.1996.0020. [DOI] [PubMed] [Google Scholar]
- Peng Y, Jiang L, Lee DY, Schachter SC, Ma Z, Lemere CA. Effects of huperzine A on amyloid precursor protein processing and beta-amyloid generation in human embryonic kidney 293 APP Swedish mutant cells. J Neurosci Res. 2006;84:903–911. doi: 10.1002/jnr.20987. [DOI] [PubMed] [Google Scholar]
- Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, Zwirner J, Wetsel RA, Gerard C, Pekny M, Pekna M. Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J. 2006;25:1364–1374. doi: 10.1038/sj.emboj.7601004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Li R, Mastroeni D, Grover A, Leonard B, Ahern G, Cao P, Kolody H, Vedders L, Kolb WP, Sabbagh M. Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol Aging. 2006;27:1733–1739. doi: 10.1016/j.neurobiolaging.2005.09.043. [DOI] [PubMed] [Google Scholar]
- Shen Y, Li R, McGeer EG, McGeer PL. Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res. 1997;769:391–395. doi: 10.1016/s0006-8993(97)00850-0. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Stoltzner SE, Grenfell TJ, Mori C, Wisniewski KE, Wisniewski TM, Selkoe DJ, Lemere CA. Temporal accrual of complement proteins in amyloid plaques in Down's syndrome with Alzheimer's disease. Am J Pathol. 2000;156:489–499. doi: 10.1016/S0002-9440(10)64753-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai K, Walker DG, McGeer EG, McGeer PL. Neurons express proteins of the classical complement pathway in Alzheimer disease. Brain Res. 1997;769:385–390. doi: 10.1016/s0006-8993(97)00849-4. [DOI] [PubMed] [Google Scholar]
- Thomas A, Gasque P, Vaudry D, Gonzalez B, Fontaine M. Expression of a complete and functional complement system by human neuronal cells in vitro. Int Immunol. 2000;12:1015–1023. doi: 10.1093/intimm/12.7.1015. [DOI] [PubMed] [Google Scholar]
- van Beek J, Elward K, Gasque P. Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann NY Acad Sci. 2003;992:56–71. doi: 10.1111/j.1749-6632.2003.tb03138.x. [DOI] [PubMed] [Google Scholar]
- Webster S, Bradt B, Rogers J, Cooper N. Aggregation state-dependent activation of the classical complement pathway by the amyloid beta peptide. J Neurochem. 1997;69:388–398. doi: 10.1046/j.1471-4159.1997.69010388.x. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Wisniewski HM. The complex of microglial cells and amyloid star in three-dimensional reconstruction. Acta Neuropathol. 1990;81:116–124. doi: 10.1007/BF00334499. [DOI] [PubMed] [Google Scholar]
- Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate and essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci USA. 1995;92:490–494. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Gundersen HJ. Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, Ronan V, Symmonds K, Gordon MN, Morgan D. Deglycosylated anti-amyloid-β antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–5346. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci USA. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, McGeer PL. Up-regulated production and activation of the complement system in Alzheimer's disease brain. Am J Pathol. 1999;154:927–936. doi: 10.1016/S0002-9440(10)65340-0. [DOI] [PMC free article] [PubMed] [Google Scholar]