

Abstract
The consolidation of conditioned fear involves upregulation of genes necessary for long-term memory formation. An important question remains as to whether this results in part from epigenetic regulation and chromatin modulation. We examined whether Homer1a, which is required for memory formation, is necessary for Pavlovian cued fear conditioning, whether it is downstream of BDNF-TrkB activation, and whether this pathway utilizes histone modifications for activity-dependent transcriptional regulation. We initially found that Homer1a knock-out mice exhibited deficits in cued fear conditioning (5 tone–shock presentations with 70 dB, 6 kHz tones and 0.5 s, 0.6 mA footshocks). We then demonstrated that: (1) Homer1a mRNA increases after fear conditioning in vivo within both amygdala and hippocampus of wild-type mice; (2) it increases after BDNF application to primary hippocampal and amygdala cultures in vitro; and (3) these increases are dependent on transcription and MAPK signaling. Furthermore, using chromatin immunoprecipitation we found that both in vitro and in vivo manipulations result in decreases in Homer1 promoter H3K9 methylation in amygdala cells but increases in Homer1 promoter H3 acetylation in hippocampal cells. However, no changes were observed in H4 acetylation or H3K27 dimethylation. Inhibition of histone deacetylation by sodium butyrate enhanced contextual but not cued fear conditioning and enhanced Homer1 H3 acetylation in the hippocampus. These data provide evidence for dynamic epigenetic regulation of Homer1a following BDNF-induced plasticity and during a BDNF-dependent learning process. Furthermore, upregulation of this gene may be regulated through distinct epigenetic modifications in the hippocampus and amygdala.
Introduction
Understanding the molecular mechanisms of fear is crucial for progress in fear-related disorders such as posttraumatic stress disorder. Many genes are dynamically regulated during the consolidation of conditioned fear, including immediate-early genes (Hall et al., 2001; Radwanska et al., 2002; Strekalova et al., 2003; Lonergan et al., 2010), kinases (Ploski et al., 2010), neurotrophic factors (Ressler et al., 2002; Ou and Gean, 2007, Von Hertzen and Gies, 2005), and synaptic structural proteins (Lonergan et al., 2010). Gene expression is dynamically regulated by modification of chromatin structure, which regulates accessibility of transcriptional machinery to promoter regions (Barrett and Wood, 2008; Renthal and Nestler, 2008; Day and Sweatt, 2011). Modifications include acetylation, methylation and phosphorylation of histone tails, which can in turn affect the chromatin structure, and promoter accessibility (Krishnan et al., 2011). Histone modifications occur after contextual fear conditioning in the hippocampus (Chwang et al. 2006), and drugs that increase histone acetylation such as histone deacetylase (HDAC) inhibitors enhance long-term memory for contextual fear (Levenson et al., 2004). This suggests that epigenetic mechanisms are linked extensively to associative learning and gene transcription (Sweatt, 2009; Chwang et al., 2006; Lattal et al., 2007; Miller et al., 2008; Peleg et al., 2010).
Here we examine epigenetic regulation of the gene variant Homer1a (also known as vesl-1S). Homer1a belongs to a family of scaffolding proteins that localize at the postsynaptic density (PSD) (Shirashi-Yamaguchi and Furuich, 2007; Foa and Gasperini, 2009) and play a role in intracellular calcium homeostasis, receptor trafficking, gene transcription, and signal transduction (Shirashi-Yamaguchi and Furuich, 2007; Foa and Gasperini, 2009). The long forms of Homer (Homer 1b-f) are constitutively expressed and contain of two functional domains: (1) the EVH1 binding domain, which targets Shank, mGluR1/5, and ryanodine receptors among others; and (2) a coiled-coil structure that aggregates with other Homer molecules and PSD proteins (Shirashi-Yamaguchi and Furuich, 2007).
The shorter variant of the Homer1 family (Homer1a) lacks the coiled-coil domain and is expressed in an activity-dependent manner (Shirashi-Yamaguchi and Furuich, 2007) (Fig. 1a). This suggests that Homer1a might act to disrupt homer–protein clusters by competitively binding to target proteins (Shirashi-Yamaguchi and Furuich, 2007). Furthermore, it has been shown that Homer1a can disrupt the physical link between mGluR and NMDA, altering the ability of mGluR1/5 to modulate NMDA functioning (Bertaso et al., 2010).
Figure 1.

The Homer1 gene family. a, Functional domains of Homer1a and 1c. The EVH1 binding domain recognizes proline-rich regions of synaptic receptors and scaffolding proteins. The coiled-coil (CC) domain recognizes other CC domains of other homer molecules. Homer1a lacks the CC domain but competitively binds with other receptors with its EVH1 binding domain. b, Schematic of the Homer1a and Homer1c gene variants. Both genes contain similar EVH1 binding domains. Homer1c, however, contains an additional CC domain. Homer1a and 1c contain different transitional domains as well, which were used to design RT-PCR primers for quantifying mRNA expression levels. c, The promoter region of the Homer1 gene family. The promoter region contains several CRE binding sites, suggesting CREB mediation of gene expression. Primers for chromatin immunoprecipitation experiments were designed to recognize this region about 1 kb upstream of the transcription start site.
Most of the current behavioral epigenetic literature focuses on global histone modifications. In contrast, here we examine mechanisms of transcription during fear conditioning and BDNF-induced plasticity of a specific gene, Homer1a, which may regulate synaptic plasticity during memory consolidation. The BDNF-TrkB pathway has been heavily implicated in amygdala and hippocampus during fear conditioning, with BDNF mRNA and protein upregulation during memory consolidation (Rattiner et al., 2004b). Moreover, inhibiting BDNF signaling impairs consolidation of conditioned fear (Rattiner et al., 2004a). Thus, examining BDNF-induced plasticity in vivo as well as in primary cell culture may provide mechanistic insight into regulation of Homer1a in Pavlovian fear conditioning.
Materials and Methods
Animals.
All experiments were performed on adult (6–10 weeks old) wild-type strain C57BL/6J male mice from Jackson Laboratory. All procedures were approved by the Institutional Animal Care and Use Committee of Emory University (Atlanta, GA) and were in compliance with National Institutes of Health guidelines. Separate cohorts of animals were used for each experiment. The Erk inducible knock-out (KO) mice contain loxP sites flanking exon 2 of the Erk2 gene (Samuels et al., 2008). These mice have an intact Erk1 gene, but deletion of the Erk2 exon 2 prevents translation and protein production of the Erk2 protein product in cells expressing Cre recombinase (Samuels et al., 2008).
Homer1a KO breeder mice (Hu et al., 2010) were a gift from P.F.W. J.-H.H. and colleagues generated the Homer1a targeting construct by fusing 2.7 kb of genomic DNA, including intron 4 and exon 5 of the Homer1 gene, with part of the rat Homer1c cDNA (2 kb), containing exons 6–10. The resulting targeted recombinant mouse results in normal levels of Homer1b/c, 2, and 3 protein expression but not that of Homer1a (Hu et al., 2010). We crossed heterozygous Homer1a KO mice to generate wild-type and KO littermates (as seen in Fig. 2a). Six- to ten-week-old male and female Homer1a wild-type and KO littermates were fear conditioned and tested as described in the next paragraph.
Figure 2.
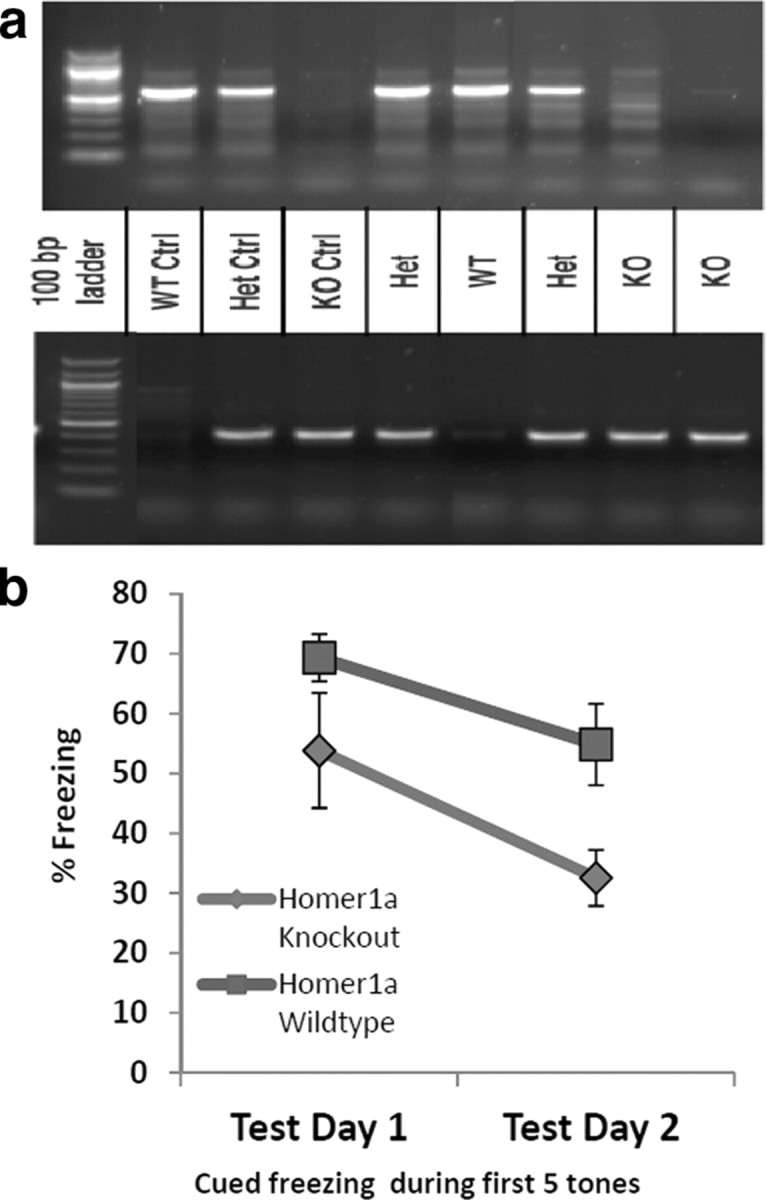
Homer1a knock-out mice have a deficit in Pavlovian fear conditioning. a, Agarose gel showing how the mice were genotyped. Tail DNA from each mouse was extracted, amplified with the respective WT and KO primers (as described previously by Hu et al., 2010), and separated with gel electrophoresis. The top gel was amplified with the WT primer, while the bottom gel was amplified with the KO primer. b, One day after training with five tone–shock pairings, percent freezing was measured with 15 tone-alone trials (Test Day 1). The next day the same animals were tested again in a separate context from training and percent freezing was measured for the 15 tone-alone trials (Test Day 2). The percent freezing during the first five tones is shown, and there is a significant overall difference between freezing levels in WT and KO mice (n = 9 per group).
Behavior.
Fear conditioning was conducted in nonrestrictive acrylic cylinders (SR-LAB startle response system, San Diego Instruments) located in a ventilated, sound-attenuated chamber. The footshock (unconditioned stimulus) was delivered through a stainless steel grid floor. Shock reactivity was defined as the peak activity (measured with a piezoelectric accelerometer) that occurred during the 200 ms after the onset of the unconditioned stimulus. The tone-conditioned stimulus was generated by a Tektronix function generator audio oscillator and delivered through a high-frequency speaker. One day before training, mice were pre-exposed to the tone through a five “tone-alone” presentation program to both habituate them to handling and to the tone but to also obtain baseline fear responses to the tone presentation. Pre-exposure was done in a separate context. During cued fear conditioning, mice received five trials of a conditioned stimulus tone (30 s, 6 kHz, 70 dB) coterminating with an unconditioned stimulus footshock (500 ms, 0.6–1 mA) with a variable intertrial interval between 60 and 180 s. The expression of fear was assessed 24 h after fear conditioning and consisted of 3 min in the same context for which the training occurred (contextual memory) with no stimulus and five conditioned stimulus tone presentations of 30 s each with a 1.5 min intertrial interval in a different context (see Fig. 3 for schematic). Stimulus presentation and data acquisition were controlled and digitized by, and stored in, an interfacing desktop computer using SR-LAB and analyzed with the Freeze View software program (Coulbourn Instruments).
Figure 3.

Pavlovian cued and contextual fear conditioning. a, Schematic diagram of behavioral experiments. On day 1, animals are presented with five tones and baseline freezing is recorded. On day 2, in a novel context the five tones coterminate with a mild footshock. Two hours after training for the molecular experiments, hippocampal and amygdalar tissues were dissected. For behavioral experiments, on day 3 animals are first placed into the day 2 context and contextual freezing is measured. Then they are placed into the context from day 1 and presented with the tone. Cued freezing is then measured. b, Percent freezing during acquisition of fear conditioning. Freezing was measured during presentation of the tone immediately before footshock. c, Animals that received tone–shock pairs had enhanced freezing in response to the final tone during acquisition, enhanced freezing when placed in the same context 24 h later, and enhanced freezing when presented with the cue (tone) test in a novel context (*p < 0.01, n = 10/group).
Cell culture.
Primary cultures of postnatal hippocampal neurons were as described previously (Brewer, 1997) with modifications. C57BL/6J mice (21 d postnatal) were decapitated, and the hippocampus and amygdala were removed and immersed in ice-cold dissection buffer consisting of Hibernate-A medium (BrainBits), B27 supplement, and gentamycin (Invitrogen) (12 g/ml) for the preparation of separate hippocampal and amygdalar neuronal cell cultures. The hippocampal and amygdalar tissues were sliced and then enzymatically digested with papain (Worthington) in Hibernate-A medium at 32°C for 30 min. Cells were dissociated by triturating with Pasteur pipettes fired on the tips to narrow openings. Neurons were purified in a density gradient medium including Hibernate-A and OptiPrep (Sigma) by centrifugation. The density gradient media consisted of four layers. The first was 1 ml of dissection buffer containing 35% OptiPrep, the second 1 ml of dissection buffer containing 25% OptiPrep, the third 1 ml of dissection buffer containing 20% OptiPrep, and the fourth 1 ml of dissection buffer containing 15% OptiPrep. They were added on the top of each other carefully, resulting in clear layer separation. Then cells were added on the top of the density gradient medium. After centrifugation, the densest layer with a cream color, located at the middle of the tube, could be seen. This layer of neurons was taken out by using a sterile transfer pipette and put into a new tube. After washing with dissection buffer, neuronal cells were plated onto poly-d-lysine (Sigma)-coated plates or glass coverslips at the density of 2.5 × 105 cells/cm2 in culture media consisting of Neurobasal A medium (Invitrogen) with 2% B27 supplement and 2 mm glutamax and gentamycin (5 g/ml). Thereafter, the cultures were kept in a humidified incubator at 37°C and 5% CO2, and media were changed every 5 d until used for experiments. After 2–3 weeks in vitro, the cells were used for the experiments reported in the present study.
Drugs.
Recombinant human BDNF was purchased from Cell Sciences and reconstituted in sterile PBS as 100 mg/ml stock. The aliquots of stock were stored at −30°C, and final drugs and concentrations for cell culture experiments were as follows: BDNF (100 ng/ml), 7,8-dihydroxyflavone (7,8-DHF; 500 nm), U0126 (10 μm; Tocris Bioscience), and actinomycin D (ActD) (25 μm; Sigma). Mice received intraperitoneal injections of 1.2 g/kg sodium butyrate (NaB; B5887, Sigma-Aldrich) dissolved in distilled water or an equal volume of distilled water alone (vehicle). This dose has been shown previously to enhance contextual fear memories (Levenson et al., 2004). The injections occurred immediately after training. For in vitro studies a concentration of 10 μm dissolved in distilled water was used.
Chromatin immunoprecipitation.
Tissue samples were treated using an EpiQuik tissue chromatin immunoprecipitation (ChIP) kit (Epigentek Group). Brains were extracted using rapid decapitation 2 h after training. Amygdalar and hippocampal tissues were rapidly dissected under a dissecting scope with a scalpel in ice-cold PBS and then frozen immediately on dry ice and stored at −80°C until ready to use. Cells/tissues were harvested and mixed with formaldehyde at a final concentration of 1.0% for 10 min at 37°C to cross-link protein to DNA. Cells/tissue were then suspended in 0.2 ml of SDS lysis buffer and allowed to settle on ice for 10 min. DNA cross-linked with protein was then sonicated into fragments of 200–1000 bp. One-tenth of the sample was set aside as an input control, and the rest was then immunoprecipitated 1.5 h at room temperature with 5 g of primary antibody in the ChIP kit strip wells. As a control, samples were immunoprecipitated with 5 g of nonimmune rabbit IgG. After immunoprecipitation, the DNA–protein complex was eluted and the proteins were digested with DNA release buffer and proteinase K. DNA was dissociated at 65°C for 1.5 h under reverse buffer. The DNA, associated with an antibody of interest (pan-H3 acetylation, pan-H4 acetylation, H3K9 dimethylation, and H3K27 dimethylation; antibodies were obtained from Epigentek) was extracted with binding buffer, precipitated with 70% and 90% ethanol and finally retrieved with elution buffer. Quantitative real-time PCR was performed with primers specific to the Homer1 promoter and for the GAPDH promoter regions.
RNA preparation.
Total RNA was prepared from frozen amygdala and hippocampal dissections in mice. Brains were extracted using rapid decapitation 2 h after training. Amygdalar and hippocampal tissues were rapidly dissected in ice-cold PBS and then frozen immediately on dry ice and stored at −80°C until ready to use. Briefly, tissue samples were homogenized and centrifuged at 13,000 × g for 3 min. RNA was washed with 70% ETOH and purified using RNeasy columns (Qiagen). RNA amount and quality were determined using a NanoDrop spectrophotometer.
Quantitative RT-PCR.
One hundred forty micrograms of total RNA were reverse transcribed using the RT2-First Strand Kit (C-03, SA Biosciences). Quantitative PCR was performed using the Applied Biosystems 7500 Fast. Online detection of reaction products was carried out using the SYBR Green Gene Assay with custom-made primers for Homer1a, Homer1c, and GAPDH. SYBR Green Master Mix was obtained from SA Biosciences, and manufacturer's instructions were followed. Calculated values are presented as mean ± SEM to indicate accuracy of measurement. Homer1a and Homer1c values were normalized for measurements of GADPH. PCR conditions were 2 min at 50°C, 10 min at 95°C, and 40 cycles with 15 s at 95°C and 60 s at 60°C.
Primer design.
Primers were designed and confirmed by Primer-BLAST. The sequences are as follows: Homer1a forward, 5′-GAAGTCGCAGGAGAAGATG-3′; Homer1a reverse, 5′-TGATTGCTGAATTGAATGTGTACC-3′; Homer 1c forward, 5′-ACACCCGATGTGACACAGAACT-3; Homer 1c reverse, 5′-TCAACCTCCCAGTGGTTGCT-3′; Homer1 promoter forward, 5′-GGTGACGTATGTGCGGAGAGGA-3′; Homer1 promoter reverse, 5′-GGTCCGTCGGTCCGTCCCTTT-3′; Primers for GAPDH and GAPDH promoter region were obtained from SA Biosciences.
Statistical analysis.
Statistically significant differences were determined by Student's t test or by between-subjects two-way ANOVA. The results were presented as mean ± SEM. For all chromatin immunoprecipitation and mRNA data, fold changes relative to control were determined using the ΔΔCt method; a mean fold change value along with an SEM value were determined; the ΔΔCt values from each dataset were used in two-tailed paired t tests (adjusted for multiple comparisons) to determine statistical significance (*p < 0.05). All values included in the figure legends represent mean ± SEM. The RT-PCR ChIP data were analyzed identically as the mRNA data using the ΔΔCt method, except that ChIP data were normalized to “input” rather than GAPDH.
Results
Homer1a knock-out mice exhibit a deficit in Pavlovian cued fear conditioning
Heterozygous Homer1a KO mice were crossed to generate wild-type and KO littermates and genotyped as described previously (Hu et al., 2010; Fig. 2a). Six- to ten-week-old male and female Homer1a wild-type and KO littermates were then fear conditioned and tested to determine whether Homer1a was required for normal fear learning. When we examined shock reactivity, we found that there was no difference in activity response to a 0.6 mA footshock, suggesting that mice of both genetic backgrounds have similar pain sensitivity and unconditioned footshock responses. Following a single session of five trials of 0.6 mA footshock paired with a 6 kHz tone and pretraining saline injection stress, mice were tested for fear expression to the tone 24 and 48 h later. During fear testing, we found a significant decrease (F(1,16) = 4.96, p < 0.05, N = 9/group) in the expression of cued fear across both testing sessions (Fig. 2b). These data suggest that Homer1a expression, even in the presence of normal Homer1b, 1c, 2, and 3, is required for normal cued fear learning.
Pavlovian fear conditioning results in an increase in Homer1a mRNA
Homer1a was dynamically regulated during consolidation of Pavlovian fear conditioning. Homer1a contains a unique stop site at the end of exon 5 that makes its sequence unique from the longer gene variants of homer such as Homer1c. Primers for RT-PCR were designed based on this sequence to differentiate between Homer1a and Homer1c expression (Fig. 1b). For all of the experiments described, amygdalar and hippocampal tissues were extracted during consolidation of fear 2 h after training (Fig. 3a). As illustrated in Figure 3a, all animals were measured for baseline freezing to presentation of a tone in one context (Context A). One day later, animals were presented with five tone–shock pairings (30 s 6 kHz tone coterminating with 0.5 s, 1 mA shock) or five tones without any shock in a novel context (Context B). On the third day, animals were tested in Context B without any tones or shocks for 3 min as an assessment of contextual fear conditioning and then immediately placed into context A, where freezing in response to five tone-alone trials was assessed. Using this procedure, we found that one training session achieved both retention of contextual and cued fear conditioning (Fig. 3b,c) (b: t(18) = 4.20; p < 0.05, n = 10/group; c: t(18) = 2.69, p < 0.05, n = 10). In a separate cohort of animals, quantitative PCR demonstrated an increase in Homer1a mRNA in the hippocampus (Figs. 4a,b) (t(18) = 3.35, p < 0.05, n = 10) and in the amygdala (Fig. 4c,d) (t(18) = 2.39, p < 0.05, n = 10) 2 h after fear conditioning. RNA for Homer1c (a longer gene variant of the Homer1 gene family) was not increased during Pavlovian fear conditioning in either brain region (Fig. 4a,c) (a: hippocampus; c: amygdala). No changes in Homer1a mRNA levels were seen in the striatum.
Figure 4.

Expressional analysis of homer1a and homer1c after Pavlovian fear conditioning in the hippocampus and amygdala. The mRNA levels of Homer1a and 1c in the amygdala and hippocampus were measured by QT-PCR in fear conditioned and non-fear conditioned mice. a, Homer1a mRNA but not Homer1c mRNA in the hippocampus, was increased in the fear-conditioned group 2 h after training. b, Average QT-PCR spectra of Homer1a and GAPDH levels in the hippocampus 2 h after fear conditioning. c, Homer1a mRNA but not Homer1c mRNA was upregulated in the amygdala 2 h after Pavlovian fear conditioning. d, Average QT-PCR spectra of Homer1a and GAPDH levels in the amygdala 2 h after fear conditioning. (*p < 0.01, n = 10/group).
BDNF causes an upregulation of Homer 1a in primary amygdala and hippocampal cultures that is transcription dependent, MEK dependent, and ERK dependent
To assess differential regulation of Homer1a through TrkB signaling, primary hippocampal and amygdala cell cultures were used. Much like in fear conditioning, BDNF-induced plasticity increased Homer1a mRNA levels in both hippocampal and amygdala cell culture (Fig. 5a,d) (hippocampus t(10) = 3.25, p < 0.05, n = 6/group; amygdala t(10) = 2.67, p < 0.05, n = 6), but not Homer1c levels. The trkB-specific agonist (7,8-DHF) (Jang et al., 2010; Andero et al., 2011) upregulated Homer1a in cell culture (hippocampus t(10) = 2.25, p < 0.05, n = 6/group; amygdala t(10) = 4.86, p < 0.05, n = 6), and also modestly increased Homer1c levels. Figure 5, b and e, are representative pictures of immunostaining for CaMKII in both hippocampal (b) and amygdala (e) primary neuronal cultures. Figure 5, c and f, are representative pictures of immunostaining for parvalbumin in both hippocampal (c) and amygdala (f) primary neuronal cell cultures.
Figure 5.

Homer1a is upregulated through the BDNF-trkB signaling pathway in cultured amygdala and hippocampal neurons. a, d, BDNF application to primary hippocampal (a) and amygdala (d) cells resulted in an increase in Homer1a (a, *p < 0.01; d, *p < 0.05, n = 6/group) but not in homer1c. b, e, Hippocampal (b) and amygdala (e) neurons expressed CaMKII. c, f, Hippocampal (c) and amygdala (f) neurons expressed parvalbumin.
Blocking transcription with ActD inhibited BDNF-induced upregulation of Homer1a in both hippocampal and amygdalar cell cultures (Fig. 6a,d) (a: F(3,20) = 27.34, p < 0.05, n = 6/group; d: F(3,20) = 258.90, p < 0.05, n = 6/group). In addition, MEK inhibition by U0126 blocked BDNF-induced increases in Homer1a in both hippocampal and amygdalar cells (Fig. 6b,e) (b: F(3,20) = 7.039, p < 0.05, n = 6/group; e: F(3,20) = 14.57, p < 0.05, n = 6/group). We next used primary cell culture from floxed-ERK knock-out mice in which we transfected cells with a Cre recombinase-expressing lentivirus to delete the ERK gene. We found that genetically deleting ERK impaired BDNF-induced upregulation of Homer1a in both hippocampal and amygdalar cells as well (Fig. 6c,f) (c: F(3,20) = 23.42, p < 0.01, n = 6/group, f: F(3,20) = 89.61, p < 0.01, n = 6/group). Thus, BDNF appears to upregulate Homer1a in a transcriptionally dependent manner and through MEK and ERK signaling mechanisms. None of these manipulations had any effect on Homer1c mRNA levels (Table 1). Genetic deletion of ERK was demonstrated through QT-PCR in amygdala (t(10) = 2.28, p < 0.05, n = 6/group) and hippocampal cells (t(10) = 2.37; p < 0.05, n = 6).
Figure 6.

Homer1a upregulation by BDNF is transcription dependent, MEK dependent, and ERK dependent. a, d, BDNF-induced upregulation of Homer1a was blocked by transcriptional inhibitor ActD in hippocampal or amygdala primary cells. b, e, BDNF-induced upregulation of Homer1a was blocked by MEK inhibitor, U0126, in hippocampal and amygdala primary cell culture. c, f, BDNF-induced upregulation of Homer1a was blocked by genetic deletion of ERK in hippocampal and amygdala primary cell culture.
Table 1.
Homer1c values during BDNF-induced plasticity
| Brain region | Experiment | Treatment | ΔCT | Statistics |
|---|---|---|---|---|
| Hippocampus | 78DHF | 78DHF | 10.89 | p = 0.70 |
| Control | 11.21 | |||
| Actinomycin D (ActD) | BDNF | 12.44 | BDNF: F(1,20) = 1.09 | |
| BDNF + ActD | 13.43 | ActD: F(1,20) = 0.083 | ||
| ActD | 13.26 | BDNF × ActD: F(1,20) = 0.22 | ||
| Control | 12.93 | |||
| U0126 | BDNF | 11.99 | BDNF: F(1,20) = 0.13 | |
| BDNF + U0126 | 15.87 | U0126: F(1,20) = 0.065 | ||
| U0126 | 15.94 | BDNF × U0126: F(1,20) = 0.0031 | ||
| Control | 15.72 | |||
| ERK | BDNF | −5.383 | BDNF: F(1,20) = 11.13 | |
| BDNF + ERK −/− | −1.91 | ERK: F(1,20) = 16.94 | ||
| ERK −/− | −1.52 | BDNF × ERK: F(1,20) = 0.087 | ||
| WT | 7.87 | |||
| Amygdala | 78DHF | 78DHF | 11.08 | p < 0.05 |
| Control | 13.82 | |||
| Actinomycin D | BDNF | 11.26 | BDNF: F(1,20) = 0.99 | |
| BDNF + ActD | 12.16 | ActD: F(1,20) = 2.83 | ||
| ActD | 13.67 | BDNF × ActD: F(1,20) = 0.017 | ||
| Control | 11.99 | |||
| U0126 | BDNF | 12.79 | BDNF: F(1,20) = 0.25 | |
| BDNF + U0126 | 14.89 | U0126: F(1,20) = 0.13 | ||
| U0126 | 16.94 | BDNF × U0126: F(1,20) = 0.093 | ||
| Control | 15.60 | |||
| ERK | BDNF | −4.35 | BDNF: F(1,20) = 0.018 | |
| BDNF + ERK −/− | −1.54 | ERK: F(1,20) = 0.43 | ||
| ERK −/− | −3.81 | BDNF × ERK: F(1,20) = 0.096 | ||
| WT | −1.92 |
BDNF application onto amygdala cells results in histone modifications along the Homer1 promoter
To determine the epigenetic role of BDNF signaling on homer1a expression, we examined histone modifications around the Homer1a promoter region after BDNF-induced plasticity. ChIP assays were performed to measure the levels of several histone modifications around the Homer1 promoter after BDNF-induced plasticity. Levels of promoter enrichment were quantified by QT-PCR. We found that BDNF application had distinct effects in hippocampal and amygdala primary cell culture. In the hippocampal cell cultures, there was a significant increase in H3 acetylation (t(10) = 6.80, p < 0.05, n = 6/group) following BDNF application, but no changes were apparent in H4 acetylation, H3K9 methylation, or H3K27 methylation (Fig. 7a). In amygdala cell cultures, however, there appears to be a decrease in H3K9 methylation (t(10) = 2.44, p < 0.05, n = 6/group) following BDNF application, but no changes in H3 acetylation, H4 acetylation, or H3K27 methylation (Fig. 7c). Significant changes in acetylation or methylation were not detected at the GAPDH promoter region.
Figure 7.

BDNF-induced and fear conditioning-induced histone modifications around the Homer1 promoter. ChIP assays were performed to measure the levels of several histone modifications around the Homer1 promoter after fear conditioning and BDNF-induced plasticity. Levels of promoter enrichment were quantified by QT-PCR. a, Histone H3 acetylation, an enhancive marker of transcription, was strongly increased at the Homer1 promoter after BDNF-induced plasticity in hippocampal cells (*p < 0.01, n = 6/group) b, Histone H3 acetylation was also increased at the Homer1 promoter in the hippocampus after fear conditioning (*p < 0.05, n = 10/group). Histone H4 acetylation, H3K9 methylation, and H3K27 methylation were not altered after BDNF-induced plasticity or fear conditioning. c, Histone H3K9 methylation, a repressive marker of transcription, was strongly decreased after BDNF-induced plasticity at the Homer1 promoter in amygdala cells (*p < 0.05, n = 6/group). d, Histone H3K9 methylation was also decreased after fear conditioning in the amygdala (*p < 0.01, n = 6/group). Significant changes in acetylation or methylation were not detected at the GAPDH promoter region (data not shown).
Pavlovian fear conditioning induces epigenetic modifications of histones along the Homer1 promoter
We next examined the effect of Pavlovian fear conditioning on histone modifications around the Homer1 promoter region. In the hippocampus there was a significant increase in H3 acetylation (t(18) = 2.37, p < 0.05, n = 10/group), but no difference in H4 acetylation, H3K9 methylation, or H3K27 methylation (Fig. 7b). In the amygdala, however, there was a significant decrease in H3K9 methylation (t(18) = 3.14, p < 0.05, n = 10/group), but no changes in H3 acetylation, H4 acetylation, or H3K27 methylation (Fig. 7d). Significant changes in acetylation or methylation were not detected at the GAPDH promoter region. Notably, these in vivo results parallel the histone modification-specific findings seen in amygdala and hippocampal primary cell culture.
HDAC inhibition enhances fear conditioning, Homer1a expression, and modifications of the Homer1 promoter
HDAC inhibitors have been shown to enhance contextual fear conditioning (Levenson et al., 2004). In this experiment, we examined the effect of the HDAC inhibitor, sodium butyrate, on fear conditioning. Note that the overall level of fear retrieval in controls (Fig. 8a) was less than that in our initial study (Fig. 3c), which is likely due to injection stress. We showed that intraparietal administration of NaB can induce increases in contextual fear memories (t(18) = 2.10, p < 0.05, n = 10/group) but did not appear to cause an increase in cued fear conditioning (Fig. 8a). NaB also appeared to enhance hippocampal Homer1a mRNA expression (Fig. 8b; F(3,36) = 5.01, p < 0.05, n = 10/group) but seemed to reverse the mRNA increase in amygdalar tissue (Fig. 8c; F(3,36) = 5.45, p < 0.05, n = 10/group). Sodium butyrate enhanced H3 acetylation in hippocampal tissue (Fig. 8d; F(3,36) = 9.54, p < 0.01, n = 10/group) but reversed fear conditioning-induced decreases in H3K9 methylation in amygdalar tissue (Fig. 8e; F(3,36) = 4.58, p < 0.05, n = 10/group).
Figure 8.

Effects of histone deacetylase inhibition on fear conditioning, Homer1a mRNA levels, and histone modifications around the Homer1 promoter. a, Sodium butyrate, a histone deacetylase inhibitor, had no effect on baseline freezing, acquisition, or cued fear memory expression. However, NaB significantly enhanced contextual freezing (*p < 0.05, n = 10/group). b, NaB enhanced homer1a mRNA in the hippocampus (*p < 0.05, n = 10). C, NaB appeared to inhibit fear-induced upregulation of Homer1a mRNA in the amygdala (*p < 0.05, n = 10). d, NaB enhanced H3 acetylation in the hippocampus around the Homer1 promoter after fear conditioning. e, While NaB treatment in the absence of fear significantly reduced H3K9 methylation in the amygdala, as did fear in the absence of NaB, NaB and fear combined showed no changes in H3K9 methylation relative to control.
Discussion
In this paper we demonstrate that the Homer1a knock-out mice have a deficit in cued fear conditioning and that Homer1a mRNA is increased during the consolidation of Pavlovian cued fear conditioning in the hippocampus and amygdala, as well as during BDNF-induced plasticity in amygdala and hippocampal primary cell culture. As mentioned previously, BDNF has been shown to play a critical role in fear conditioning in both the hippocampus and amygdala. We chose to use a primary cell culture model as a way to more directly address the molecular mechanisms of Homer1a gene transcription and potentially tease apart any differences in hippocampal and amygdala neurons separate from their functional connectivity seen in the brain. We fail to rule out the possibility that other receptor–ligand systems play a role in Homer1a transcription. There are several potential signaling pathways that could result in upregulation of Homer1a during Pavlovian fear conditioning, including NMDA activation (Ango et al., 2000; Sato et al., 2001). However, we have demonstrated that BDNF-induced plasticity in cell culture is one mechanism for Homer1a upregulation. Given the role of BDNF in the amygdala and in the hippocampus during consolidation of fear conditioning (Rattiner et al., 2004a,b; Heldt et al., 2007; Musumeci et al., 2009), it is plausible that an increase in BDNF in the hippocampus and amygdala might result in an increase in Homer1a signaling during the consolidation of Pavlovian fear conditioning.
Posttranslational histone modifications regulating Homer1a transcription in vivo and in vitro
We also demonstrate that Pavlovian fear conditioning results in an increase in H3 acetylation around the Homer1 promoter in the hippocampus and a decrease in H3K9 methylation around the Homer1 promoter in the amygdala. However, we saw no differences in H4 acetylation or H3K27 methylation. Given that there is no existing evidence for histone posttranslational modifications regulating Homer1a and very limited data for specific modifications regulating the expression of other genes during Pavlovian fear conditioning (Fuchikami et al., 2010; Gupta et al., 2010) we began this work using a representative sample of well characterized histone-specific antibodies. There is evidence for H3K9 global methylation and H3 and H4 global acetylation during Pavlovian fear conditioning (Gupta et al., 2010). In addition, there are data demonstrating an increase in H3 acetylation specifically around the promoter region of BDNF (Fuchikami et al., 2010; Gupta et al., 2010; Takei et al., 2011), although no study to date has examined histone modifications around the Homer1 promoter or any other genes during Pavlovian fear conditioning. For future studies, it may be informative to look at a wider array of posttranslational modifications involved in homer1a during Pavlovian fear conditioning. However, with our very limited selection, we were able to find distinct epigenetic regulation of Homer1a in hippocampal and amygdalar tissue during Pavlovian fear conditioning and in cells during BDNF-induced plasticity.
BDNF-induced upregulation of Homer1a
Interestingly, during BDNF-induced plasticity we also demonstrate an increase in H3 acetylation around the Homer1 promoter in hippocampal primary cell culture and a decrease in H3K9 methylation in amygdala primary cell culture. Despite the fact that homer1a mRNA was upregulated in both the hippocampus and amygdala, histone modifications around the Homer1 promoter were distinct between the two brain regions. The hippocampus primarily exhibited increases in histone H3 acetylation, which is associated with enhanced gene transcription, within the Homer1 promoter region. The amygdala, however, primarily exhibited decreases in histone H3K9 methylation, a repressive marker of transcription, in the Homer1 promoter region. Notably, although the specific histone regulation was different in these two brain regions and in primary cell culture from these regions, both histone tail modifications would result in enhanced transcription. These differences were seen in vivo after fear conditioning and in vitro after BDNF-induced plasticity, suggesting that these differences are not caused by the unique functional connectivity of the hippocampus and amygdala but are due to intrinsic molecular properties of the neurons themselves.
It is important to note that we have not directly demonstrated, in vivo, the connection between BDNF-dependent ERK activation and epigenetic regulation of the Homer1 promoter. Rather, our data combine convergent studies demonstrating correlational relationships in vivo and some causal relationships in vitro. Future studies should inhibit BDNF at the time of learning, perhaps via BDNF knockdown approaches (Choi et al., 2010) or through blocking TrkB activation with genetic approaches (Chhatwal et al., 2006).
Effects of HDAC inhibition on epigenetic modifications around the Homer1 promoter
Moreover, histone deacetylase inhibition with sodium butyrate had differential effects on hippocampal and amygdalar tissues. As expected, NaB enhanced hippocampal H3 acetylation around the Homer1 promoter in fear-conditioned mice but not in control mice. NaB did not induce global increases in acetylation in that the GAPDH promoter showed no enhancement in H3 or H4 acetylation, nor did NaB induce an increase in histone H4 acetylation around the Homer1 promoter in the hippocampus. This result is straightforward in that NaB prevents the removal of acetyl groups from histone tails. In order for there to be a resulting increase in acetylation, there would already have to be an initial addition of acetyl groups, which only would occur around already activated genes. If histone H3 but not H4 is increasingly acetylated around the Homer1 promoter, then NaB should only enhance H3 acetylation. This may also explain why NaB did not enhance acetylation of H3/H4 tails around the Homer1 promoter in amygdalar tissues. NaB did however reduce H3K9 methylation around the Homer1 promoter in control mice and reverse the decrease in H3K9 methylation seen in fear-conditioned mice. While it has been demonstrated that HDAC inhibition can decrease H3K9 methylation (Gupta et al., 2010), the mechanism is less clear and the mechanism underlying NaB effects on H3K9 methylation in the amygdala is not so obvious.
Understanding the molecular mechanisms of Pavlovian fear conditioning will give us an understanding of the mechanism and potential treatments for posttraumatic stress disorder (PTSD) and other fear-related disorders. Histone-modifying drugs have been shown to enhance fear conditioning as well as fear extinction (Kaplan and Moore, 2011). Our data suggest a distinct epigenetic signature for Homer1a gene expression in hippocampal and amygdala cells/tissues separate from the two regions' functional connectivity. These differences may explain why HDAC inhibitors such as sodium butyrate only enhance hippocampal-dependent contextual memories and mainly enhance hippocampal histone modifications.
Transient increases in Homer1a and synaptic plasticity
At first glance, an increase in Homer1a may seem contradictory relative to other findings with LTP and plasticity in fear conditioning. For example, it has been shown that Homer1a is upregulated during increases in network activity that scale down the expression of synaptic AMPA receptors (Sala et al., 2003; Inoue et al., 2007; Hu et al., 2010). On the other hand, fear conditioning results in LTP of synaptic transmission from auditory thalamus and cortex to the lateral amygdala and increased synaptic GluR1 subunit of AMPA receptors (Clugnet and LeDoux, 1990; McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997). One possible explanation is that Homer1a increases may be occurring only transiently following fear conditioning. We have recently found that β-catenin (Maguschak and Ressler, 2008, 2012), possibly through Wnt-mediated signaling (Maguschak and Ressler, 2011), is associated with transient decreases followed by increases in synaptic stability during fear consolidation. Similarly, Homer1a increases may serve to transiently destabilize synapses, thus allowing for strengthening of the most currently plastic/active synapses as new learning occurs. Further studies should help to clarify its functional role and how this function is differentially modulated via transcriptional regulation.
While we know that fear learning requires long term potentiation and synaptic plasticity, very little direct evidence links the increases in gene transcription with increases in LTP seen in fear conditioning. Homer1a, a known regulator of both Pavlovian fear conditioning and physiological plasticity (Inoue et al., 2009; Tronson et al., 2010), is epigenetically regulated after learning and may provide a useful connection between gene expression and physiological synaptic plasticity. Furthermore, studying the role of Homer1a transcription and activity in the consolidation of fear may help better understand the connection between epigenetics, gene transcription, LTP, and the consolidation of memory formation. In addition, it may provide insight into future drug targets for the enhancement of extinction learning as well as behavioral therapy for PTSD and other fear-related disorders.
Footnotes
Support was provided by NIH Grants DA019624 and P30 NS055077, the Burroughs Wellcome Fund, the Center for Behavioral Neuroscience, a Science and Technology Center program of the National Science Foundation under Agreement No. IBN-9876754, National Primate Research Center Base Grant No. RR-00165, and the Animal Resource Program at NIH.
References
- Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry. 2011;168:163–172. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ango F, Pin JP, Tu JC, Xiao B, Worley PF, Bockaert J, Fagni L. Dendritic and axonal targeting of type 5 metabotropic glutamate receptor is regulated by homer1 proteins and neuronal excitation. J Neurosci. 2000;20:8710–8716. doi: 10.1523/JNEUROSCI.20-23-08710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RM, Wood MA. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem. 2008;15:460–467. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertaso F, Roussignol G, Worley P, Bockaert J, Fagni L, Ango F. Homer1a-dependent crosstalk between NMDA and metabotropic glutamate receptors in mouse neurons. PLoS One. 2010;5:e9755. doi: 10.1371/journal.pone.0009755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods. 1997;71:143–155. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci. 2006;9:870–872. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DC, Maguschak KA, Ye K, Jang SW, Myers KM, Ressler KJ. Prelimbic cortical BDNF is required for memory of learned fear but not extinction or innate fear. Proc Natl Acad Sci U S A. 2010;107:2675–2680. doi: 10.1073/pnas.0909359107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwang WB, O'Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal Histone phosphorylation following contextual fear conditioning. Learn Mem. 2006;13:322–328. doi: 10.1101/lm.152906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clugnet MC, LeDoux JE. Synaptic plasticity in fear conditioning circuits: induction of LTP in the lateral nucleus of the amygdala by stimulation of the medial geniculate body. J Neurosci. 1990;10:2818–2824. doi: 10.1523/JNEUROSCI.10-08-02818.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD. Cognitive neuroepigenetics: a role for epigenetic mechanisms in learning and memory. Neurobiol Learn Mem. 2011;96:2–12. doi: 10.1016/j.nlm.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foa L, Gasperini R. Developmental roles for Homer: more than just a pretty scaffold. J Neurochem. 2009;108:1–10. doi: 10.1111/j.1471-4159.2008.05726.x. [DOI] [PubMed] [Google Scholar]
- Fuchikami M, Yamamoto S, Morinobu S, Takei S, Yamawaki S. Epigenetic regulation of BDNF gene in response to stress. Psychiatry Investig. 2010;7:251–256. doi: 10.4306/pi.2010.7.4.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD. Histone methylation regulates memory formation. J Neurosci. 2010;30:3589–3599. doi: 10.1523/JNEUROSCI.3732-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ. Fear memory retrieval induces CREB phosphorylation and Fos expression within the amygdala. Eur J Neurosci. 2001;13:1453–1458. doi: 10.1046/j.0953-816x.2001.01531.x. [DOI] [PubMed] [Google Scholar]
- Heldt SA, Stanek L, Chhatwal JP, Ressler KJ. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007;12:656–670. doi: 10.1038/sj.mp.4001957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JH, Park JM, Park S, Xiao B, Dehoff MH, Kim S, Hayashi T, Schwarz MK, Huganir RL, Seeburg PH, Linden DJ, Worley PF. Homeostatic scaling requires group 1 mGluR activation mediated by Homer1a. Neuron. 2010;68:1128–1142. doi: 10.1016/j.neuron.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue N, Nakao H, Migishima R, Hino T, Matsui M, Hayashi F, Nakao K, Manabe T, Aiba A, Inokuchi K. Requirement of the immediate early gene vesl-1S/homer-1a for fear memory formation. Mol Brain. 2009;2:7. doi: 10.1186/1756-6606-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue Y, Udo H, Inokuchi K, Sugiyama H. Homer1a regulates the activity-induced remodeling of synaptic structures in cultured hippocampal neurons. Neuroscience. 2007;150:841–852. doi: 10.1016/j.neuroscience.2007.09.081. [DOI] [PubMed] [Google Scholar]
- Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A. 2010;107:2687–2692. doi: 10.1073/pnas.0913572107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan GB, Moore KA. The use of cognitive enhancers in animal models of fear extinction. Pharmacol Biochem Behav. 2011;99:217–228. doi: 10.1016/j.pbb.2011.01.009. [DOI] [PubMed] [Google Scholar]
- Krishnan S, Horowitz S, Trievel RC. Structure and function of histone H3 lysine 9 methyltransferases and demethylases. Chembiochem. 2011;12:254–263. doi: 10.1002/cbic.201000545. [DOI] [PubMed] [Google Scholar]
- Lattal KM, Barrett RM, Wood MA. Systemic or intrahippocampal delivery of Histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci. 2007;121:1125–1131. doi: 10.1037/0735-7044.121.5.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Lonergan ME, Gafford GM, Jarome TJ, Helmstetter FJ. Time-dependent expression of Arc and zif268 after acquisition of fear conditioning. Neural Plast. 2010;2010:139891. doi: 10.1155/2010/139891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguschak KA, Ressler KJ. Beta-catenin is required for memory consolidation. Nat Neurosci. 2008;11:1319–1326. doi: 10.1038/nn.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguschak KA, Ressler KJ. Wnt signaling in amygdala-dependent learning and memory. J Neurosci. 2011;31:13057–13067. doi: 10.1523/JNEUROSCI.3248-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguschak KA, Ressler KJ. The dynamic role of beta-catenin in synaptic plasticity. Neuropharmacology. 2012;62:78–88. doi: 10.1016/j.neuropharm.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–611. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- Miller CA, Campbell SL, Sweatt JD. DNA methylation and Histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem. 2008;89:599–603. doi: 10.1016/j.nlm.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musumeci G, Sciarretta C, Rodríguez-Moreno A, Al Banchaabouchi M, Negrete-Díaz V, Costanzi M, Berno V, Egorov AV, von Bohlen und Halbach O, Cestari V, Delgado-García JM, Minichiello L. TrkB modulates fear learning and amygdalar synaptic plasticity by specific docking sites. J Neurosci. 2009;29:10131–10143. doi: 10.1523/JNEUROSCI.1707-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou LC, Gean PW. Transcriptional regulation of brain-derived neurotrophic factor in the amygdala during consolidation of fear memory. Mol Pharmacol. 2007;72:350–358. doi: 10.1124/mol.107.034934. [DOI] [PubMed] [Google Scholar]
- Peleg S, Sananbenesi F, Zovoilis A, Burkhardt S, Bahari-Javan S, Agis-Balboa RC, Cota P, Wittnam JL, Gogol-Doering A, Opitz L, Salinas-Riester G, Dettenhofer M, Kang H, Farinelli L, Chen W, Fischer A. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328:753–756. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- Ploski JE, Park KW, Ping J, Monsey MS, Schafe GE. Identification of plasticity-associated genes regulated by Pavlovian fear conditioning in the lateral amygdala. J Neurochem. 2010;112:636–650. doi: 10.1111/j.1471-4159.2009.06491.x. [DOI] [PubMed] [Google Scholar]
- Radwanska K, Nikolaev E, Knapska E, Kaczmarek L. Differential response of two subdivisions of lateral amygdala to aversive conditioning as revealed by c-Fos and P-ERK mapping. Neuroreport. 2002;13:2241–2246. doi: 10.1097/00001756-200212030-00015. [DOI] [PubMed] [Google Scholar]
- Rattiner LM, Davis M, French CT, Ressler KJ. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci. 2004a;24:4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattiner LM, Davis M, Ressler KJ. Differential regulation of brain-derived neurotrophic factor transcripts during the consolidation of fear learning. Learn Mem. 2004b;11:727–731. doi: 10.1101/lm.83304. [DOI] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ. Epigenetic mechanisms in drug addiction. Trends Mol Med. 2008;14:341–350. doi: 10.1016/j.molmed.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressler KJ, Paschall G, Zhou XL, Davis M. Regulation of synaptic plasticity genes during consolidation of fear conditioning. J Neurosci. 2002;22:7892–7902. doi: 10.1523/JNEUROSCI.22-18-07892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Stäubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [Erratum (1998) 391:818] [DOI] [PubMed] [Google Scholar]
- Sala C, Futai K, Yamamoto K, Worley PF, Hayashi Y, Sheng M. Inhibition of dendritic spine morphogenesis and synaptic transmission by activity-inducible protein Homer1a. J Neurosci. 2003;23:6327–6337. doi: 10.1523/JNEUROSCI.23-15-06327.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its keyroles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suzuki K, Nakanishi S. NMDA receptor stimulation and brain-derived neurotrophic factor upregulate homer 1a mRNA via the mitogen-activated protein kinase cascade in cultured cerebellar granule cells. J Neurosci. 2001;21:3797–3805. doi: 10.1523/JNEUROSCI.21-11-03797.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S, Takishima K. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi-Yamaguchi Y, Furuichi T. The Homer family proteins. Genome Biol. 2007;8:206. doi: 10.1186/gb-2007-8-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson T, Doyère V, Cain CK, LeDoux JE. Long-term potentiation in the amygdala: a cellular mechanisms of fear learning and memory. Neuropharmacology. 2007;52:215–227. doi: 10.1016/j.neuropharm.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Strekalova T, Zörner B, Zacher C, Sadovska G, Herdegen T, Gass P. Memory retrieval after contextual fear conditioning induces c-Fos and JunB expression in CA1 hippocampus. Genes Brain Behav. 2003;2:3–10. doi: 10.1034/j.1601-183x.2003.00001.x. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Experience-dependent epigenetic modifications in the central nervous system. Biol Psychiatry. 2009;65:191–197. doi: 10.1016/j.biopsych.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei S, Morinobu S, Yamamoto S, Fuchikami M, Matsumoto T, Yamawaki S. Enhanced hippocampal BDNF/TrkB signaling in response to fear conditioning in an animal model of posttraumatic stress disorder. J Psychiatr Res. 2011;45:460–468. doi: 10.1016/j.jpsychires.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Tronson NC, Guzman YF, Guedea AL, Huh KH, Gao C, Schwarz MK, Radulovic J. Metabotropic glutamate receptor 5/Homer interactions underlie stress effects on fear. Biol Psychiatry. 2010;68:1007–1015. doi: 10.1016/j.biopsych.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazdarjanova A, McNaughton BL, Barnes CA, Worley PF, Guzowski JF. Experience-dependent coincident expression of the effector immediate-early genes arc and Homer 1a in hippocampal and neocortical neuronal networks. J Neurosci. 2002;22:10067–10071. doi: 10.1523/JNEUROSCI.22-23-10067.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]