

Abstract
An optical fiber has been developed with a maneuverable mini-probe tip that sparges O2 gas and photo-detaches pheophorbide (sensitizer) molecules. Singlet oxygen is produced at the probe tip surface which reacts with an alkene spacer group releasing sensitizer upon fragmentation of a dioxetane intermediate. Optimal sensitizer photorelease occurred when the probe tip was loaded with 60 nmol sensitizer, where crowding of the pheophorbide molecules and self-quenching were kept to a minimum. The fiber optic tip delivered pheophorbide molecules and singlet oxygen to discrete locations. 60 nmol sensitizer was delivered into petrolatum; however, sensitizer release was less efficient in toluene-d8 (3.6 nmol) where most had remained adsorbed on the probe tip, even after the covalent alkene spacer bond had been broken. The results open the door to a new area of fiber optic-guided sensitizer delivery for the potential photodynamic therapy of hypoxic structures requiring cytotoxic control.
Introduction
Current photodynamic therapy (PDT) methods all employ systemic administration of dyes.1 Instead of injecting a photosensitizer into a patient with irradiation, the PDT of tumors could benefit from a fiber optic that guides the photosensitizer to a specific location. However, at present, no point-source fiber-optic 1O2 generator exists as an alternative PDT method.
The study reported here describes the efficacy of sensitizers to cleave free from the porous silica caps of the fiber optic in Figure 1. The probe tip photocleaves pheophorbide formate ester (3) and leaves behind co-fragment 4. Hydrolysis products include 4-hydroxybenzylic alcohol and formic acid, where the amount of formic acid generated is far less than what is irritating (13 mmol, open skin) or lethal (LD50 in mice is 700 mg/kg).2
Figure 1.

Concept of the singlet oxygen fiber optic: (1) illuminator-to-fiber coupling, (2) compressed oxygen-to-fiber coupling via a flare T valve to a borosilicate fiber optic consisting of a Teflon gas flow tube, (3) porous Vycor glass (PVG) cap-to-fiber coupling, (4) photocleavable sensitizer solid, (5) internally flowing light and oxygen, externally produced 1O2, [2 + 2] cycloaddition at the alkene site, (6) cleavage of sensitizer 3 free from the probe tip via the scission of dioxetane 2, and (7) production of co-fragment 4 and hydrolysis by-products.
Our system uses visible light to cleave the photosensitizer away from a solid surface. Photocleavable groups on solids or biological surfaces typically use UV light, such as the benzoin,3 nitrobenzene,4 phenyacyl,5 and coumarin systems,6 although some have used 2-photon excitation.7 The previous work of Dolphin8 and Breslow9 employed visible light in solution phase photocleaving and drug-delivery systems.
Our hypothesis was that sensitizer molecules will cleave away from the fiber optic probe tip in a “three-phase” experiment: the gas phase was the hollow core of the fiber, the solid phase was the fiber cap, and the “outer” phase was the bulk solution or semi-solid media. O2 gas flowed from a compressed oxygen tank to a T-valve in the custom optical fiber, which was connected to the sensitizer cap 1 via a Teflon inner flow tube. A pheophorbide derivative was selected as the 1O2 sensitizer,10 and a Z-enol ether was selected as the spacer group bridging the sensitizer and the glass tip, which can react with 1O2 and be cleaved apart by way of scission of a dioxetane intermediate.11 Thus, dioxetane cleavage could be conducted for sensitizer release to effectively increase the short diffusion distance of 1O2 away from the probe tip (~150 nm in water and less in vivo12). The previous 1O2 fiber optic we developed lacks a potential PDT utility due to the extremely short diffusion distance of 1O2 away from the probe tip13 in the absence of a photocleavable sensitizer.
The manuscript contains three parts (1) the synthesis of the new heterogeneous sensitizer 1, (2) the optimal loading of sensitizer for maximum visible-light photorelease, and (3) the percent disappearance of starting material and the percent yield of surface-released sensitizer in different media. Our aim was to open the door to a brand new area of fiber-optic guided drug delivery whose origins come from concepts in organic synthesis and molecular photochemistry. Unexpectedly, the probe tip had adsorbtive affinity for sensitizer 3 in toluene, while its quantitative departure took place in the semi-solid petrolatum. Sensitizer 3 localization into the petrolatum indicates a potential for local dye delivery via permeability enhancement.14
Results and Discussion
1. Synthesis of the Heterogeneous Sensitizer
We sought a versatile heterogeneous sensitizer for rapid photorelease, and designed the pheophorbide/alkene conjugate as a new photocleavable sensitizer. The conversion of 4-bromophenol (5) to cis-1,2-bis(4-bromophenoxy)ethene (8) was carried out in 3 steps using a known procedure (Scheme 1).15 Meso-7 and dl-7 were formed, and a separation of the meso was necessary to reach 8, which reacted with n-BuLi and DMF to generate the bis-aldehyde (9) in 60% yield. Bis-aldehyde 9 reacted with sodium borohydride quantitatively to give “spacer group” 10. Spacer group 10 was prepared as a photocleavable group bridging the silica cap and the photosensitizer. Pyropheophorbide-a reacted with 10, EDC, and DMAP yielding pyropheophorbide monoester (11), which was purified and isolated in 60% yield. Sensitizer silane 12 and 3-iodopropyltrimethoxy silane were then covalently bonded to the porous Vycor glass (PVG) (step vii, Scheme 1), which consisted of free, isolated or associated, and hydrogen bonded clusters of silanol groups, Si-OH.16,17 In 1, the ratio of the sensitizer-to-iodosilane-to-silanol sites was typically ~1:3:300, but as will be seen in Section 2, tunable loading amounts were critical for optimal and controllable sensitizer release. Solid 1 was stable in the dark, no sensitizer leaching was observed when the material was (i) repeatedly washed with toluene, THF, chloroform, ether, and hexane solvents, (ii) Soxhlet extracted with chloroform and ethanol, or (iii) immersed in water solution at pH 4–7 for 4 h at room temperature. Because the filtrates of (i)–(iii) showed no photosensitizer activity, we concluded that 1 contained siloxane bonds where the sensitizer was chemically bound to the silica matrix. The FTIR data further bolstered the structural assignment of the saturated carbons of the spacer methylene groups of 1 (Figure 2). The depth that the sensitizer penetrated into PVG was examined using a microscope equipped with a CCD camera. Figure 3 shows a sensitizer-attached PVG sample 1, cut so that the ~0.08 mm depth and localization of the sensitizer on the outer face of the cap could be viewed. Unlike conventional gas-liquid systems that introduce O2 gas into the liquid phase,18 the present system is improved and transmits O2 gas through the pores of the PVG membrane tip where an anaerobic solution becomes oxygen saturated with access to the covalently excited sensitizer sites (Figure 4). The next step was determining the optimal loading of sensitizer for maximum photorelease from the solid surface.
Scheme 1a. Synthesis of Sensitizer Functionalized Cap 1.

aReagents and conditions: (i) BrCH2CH2Br, NaOH, 100 °C, 6 h; (ii) NBS, benzoyl peroxide, CCl4, 80 °C, 6 h (meso-7 was carried on to step iii); (iii) NaI, acetone, 25 °C, 2 h; (iv) n-BuLi, DMF, THF, −78 °C, 3.5 h; (v) NaBH4, CH3OH, 25 °C, 14 h; (vi) pyropheophorbide-a, EDC, DMAP, CH2Cl2, 25 °C, 24 h; (vii) (CH3O)3SiCH2CH2CH2I, NaH, THF, under N2, 70 °C, 24 h; (viii) porous Vycor glass (pre-dried at 500 °C), toluene, reflux 110 °C, 24 h.
Figure 2.

The FTIR spectrum of functionalized porous glass 1, in which the C-Hx stretching modes observed at 2851 and 2954 cm−1 were assigned to saturated carbons of the spacer methylene groups indicating that 12 and (CH3O)3SiCH2CH2CH2I (1:3 ratio) were anchored to the PVG surface. The FTIR spectrum of a clean piece of PVG was not identical, no C-Hx stretching modes were observed.
Figure 3.

A low-magnification (10×) cross-sectional optical image. The dark green thin coating shows the depth of 1 accessed into PVG. The image shows ~0.08 mm penetration depth on the outer face of the cylinder-shaped PVG cap 1 (diameter 5.0 mm × length 8.0 mm).
Figure 4.

Schematic of the sensitizer functionalized porous cap 1 where oxygen and light come internally from the hollow optical fiber. Typically 0.3 μmol or 0.33% silane 12 was loaded per g PVG. The sensitizer may adopt various conformations on the isotropic PVG material.
2. Optimal Loading of Sensitizer for Maximum Photorelease
0.06 to 1.1 μmol quantities of silane 12 were loaded onto PVG per g resulting in sensitizer sites separated by 8.9 to 38.4 nm (Table 1). A likely spatial distance between the sensitizer molecules can be estimated with eqs 1–4 (Experimental Section), in which a simple surface geometry was assumed. The loading of 0.3-μmol (0.33%) sensitizer onto the fiber caps resulted in maximal photocleavage of 3. Higher or lower sensitizer loading reduced the photocleavage efficiency and was attributed to less available sensitizer and self-quenching, respectively (Figure 5). For example, a 3-fold mole increased loading of 12 resulted in an 11-fold decrease in sensitizer photocleavage (cf. entries 2 and 5, Table 1). This suggests that dye molecules sufficiently isolated from each other (>~17 nm) efficiently cleave with minimal dipole-dipole energy transfer due to congestion (Förster pheophorbide radius = 6.2 Å). 19 The stability of photocleaved 3 in methanol-water solution (9:1) at pH 2–8 was also investigated by LCMS. As expected, after several minutes it remained unchanged, but at high or low pH, the disappearance of 3 was mostly due to the hydrolysis of the formate ester bond (path A), and then later there was the appearance of 14 (path B) (Scheme 2). Each pair of 3 and 4 can potentially liberate 2 equivalents of formic acid, for a maximum of 120 nmol formic acid arising from an 0.33% sensitizer-loaded 0.2 g fiber cap, along with 60 nmol 4-hydroxybenzylic alcohol.
Table 1.
Loading of Pheophorbide Silane 12 onto Porous Vycor Glass (PVG)a
| entry | loaded, % | sens:SiOH ratio | ave. sens-sens distance (nm) | photocleavage, %c |
|---|---|---|---|---|
| 1 | 0.068 | 1:1470 | 38.4 | ~2.1 |
| 2 | 0.33 | 1:290 | 17.1 | 6.06 |
| 3 | 0.48 | 1:210 | 14.5 | 3.32 |
| 4 | 0.60 | 1:170 | 12.9 | 1.42 |
| 5 | 0.99 | 1:100 | 10.0 | 0.53 |
| 6 | 1.24 | 1:80 | 8.9 | 0.39 |
The sensitizer is thinly coated, it reaches a maximum depth of 0.08 mm into PVG. The percent of sensitizer loaded onto PVG was defined as the number of sensitizer molecules attached vs the number of silanol groups at a depth of 0.08 mm.
The sensitizer loadings were varied based on the ratio of 12 to (CH3O)3SiCH2CH2CH2I added. The range was 0.06 to 1.1 μmol sensitizer silane 12 with concomitant decreases of iodosilane from 0.84 to 0.32 μmol. Thus, the 12 to iodosilane ratio ranged from 1:14 to 1:0.3.
Experimental Section describes the calculation of the sens-sens distance.
White light from a Rayonet reactor was used to photocleave the sensitizer.
Figure 5.

Percent of 3 photoreleased into toluene-d8 solution from the porous cap 1. Silane 12 was loaded in 0.06 to 1.1 μmol amounts (0.068 to 1.24%) onto porous Vycor glass per gram and was exposed to white light.
Scheme 2.

Stability of Photosensitizer 3 in Acid and Base Solution
3. The Percent Disappearance of Starting Material and the Percent Yield of Surface-Released Sensitizer in Different Media
We have carried out a systematic study of the sensitizer photorelease in different media (Figures 6–8, Table 2). Figure 6 shows the amount of sensitizer 3 photoreleased into toluene-d8 solution, as a plot of sensitizer release, i.e. OD vs. time. The increase in OD indicates the release into solution, which gave a maximum of 7300 nM 3. Significant quantities of sensitizer 3 remained adsorbed on the probe tip even though the alkene bond bridging the sensitizer and glass was broken (trace c, Figure 7). The sensitizer photorelease chemistry was worse in D2O, as indicated by the observation that 3 was not detected in D2O solution (entry 8, Table 2). An effort to separate 3 from the glass surface was successful with Soxhlet extraction, where photolyzed 1 resulted in the dissociation of the nonpolar 3 adsorbate into the surrounding solution.
Figure 6.

Time course of photorelease of 3 into toluene-d8 solution arising from photooxidative cleavage and departure from the fiber optic device tip. The absorption spectra show the fourth Q-band of 3 and were normalized at 640 nm: (a) orange 0.0 h, (b) turquoise 0.5 h, (c) blue 1.0 h, (d) green 1.5 h, (e) red 2.0 h, and (f) black 4.0 h.
Figure 8.

Time course of the diffusion of 3 away from the probe tip into petrolatum at 65 °C.
Table 2.
Photorelease of Sensitizer 3a
| entry | medium | irrad. time (h) | photocleaved state
|
comments | ||
|---|---|---|---|---|---|---|
| free 3 (%) | free 3 (nmol) | free 3 (nM) | ||||
| 1 | toluene-d8 | 0 | 0 | 0 | 0 | in dark |
| 2 | toluene-d8 | 0.5 | 0.8 | 0.5 | 960 | a,b |
| 3 | toluene-d8 | 1.0 | 2.6 | 1.6 | 3,110 | a,b |
| 4 | toluene-d8 | 1.5 | 3.7 | 2.2 | 4,400 | a,b |
| 5 | toluene-d8 | 2.0 | 4.9 | 2.9 | 5,860 | a,b |
| 6 | toluene-d8 | 4.0 | 6.1 | 3.6 | 7,300 | a,b |
| 7 | toluene-d8 | 1.5 | ~70 | ~250 | ~280,00 | c |
| 8 | D2O | 3.0 | 0 | 0 | 0 | a,b |
| 9 | petrolatum | 0 | 0 | 0 | 0 | in dark |
| 10 | petrolatum | 0.5 | 37 | 11.1 | 27,750 | a,b,d |
| 11 | petrolatum | 1.0 | 46 | 13.8 | 34,500 | a,b,d |
| 12 | petrolatum | 4.0 | 100 | 30.0 | 75,000 | a,b,d |
Irradiation source: internal irradiation of tip via fiber optic with an output energy density of 444 mJ/cm2.
The 0.2 g PVG cap was loaded with 60 nmol 12 (0.33% surface coverage). PVG fiber tip dimensions: cylinder shape with a length of 8.0 mm, diameter of 5.0 mm, and hole (2.0 length × 3.0 mm diameter).
External Rayonet reactor irradiation of 1.0 g PVG loaded with 360 nmol 12 (0.33% surface coverage) in 0.9 mL toluene-d8 followed by Soxhlet extraction to dissociate the adsorbate 3 into the surrounding solution.
Absorption spectroscopy was used for the quantitation of 3 into surrounding toluene-d8 (0.5 mL). An epifluorescence microscope was used to 3 in petrolatum at 65 °C (0.4 mL).
Figure 7.

Reaction profile of fiber optic delivering light and oxygen to the probe tip (a) percent sensitizer bioconjugate 1, (b) percent surface-bound dioxetane 2, (c) percent 3 photoreleased into toluene-d8 at room temperature, and (d) percent 3 photoreleased into petrolatum at 65 °C.
An interesting result was this in contrast to toluene-d8 and D2O, the sensitizer payload release was efficient in petrolatum (soft paraffin, mixture of hydrocarbons) at 65 °C. We have considered petrolatum as an adequate semi-solid medium, that is, for physical organic studies with relevance to lipophilic biological media. After 30 min, the diffusion distance of the sensitizer away from the fiber-optic tip was 1.03 mm, and the geometry of the spot was approximately circular (Figure 8). After 4 h, quantitative sensitizer departure occurred in petrolatum. No significant adsorption of the sensitizer occurred with the petrolatum, the sensitizer remained in the surrounding semi-solid phase (trace d, Figure 7). Thus, it is evident that the surrounding medium influences the photorelease efficiency at the probe tip. By monitoring the course of the reaction, the progress of the photorelease could be scrutinized. With the fiber optic delivering visible light and oxygen to the sensitizer tip, a rapid 47% photodegradation of the alkene bonds was observed in 30 min. (trace a, Figure 7). After about 2 h, 92% of the alkene bonds were cleaved, whereas full photodegradation of the alkene bonds required 4 h to complete. Because the covalently attached sensitizer reached a 0.08 mm depth, the observed fast and slow alkene photodegradation components can be attributed to the depth in which the sensitizer is linked and the geometric isolation of the latter.
Indirect evidence for the intermediacy of a surface-bound 1,2-dioxetane species shown in Scheme 3 is also reported. Singlet oxygen reacted with the alkene bond of glass 1 (0.6 g) producing dioxetane 2 which deoxygenated to an epoxide (16), likely via a phosphorane (15),20 by trimethylphosphite (0.6 M). The amount of trimethylphosphate detected was 0.57±0.09% of the number of alkene sites after ~70% conversion in toluene-d8. A homogeneous photolysis experiment of diol 10 in toluene-d8 was conducted in a similar manner, but showed a 6-fold increase in dioxetane trapping than could be achieved with the heterogeneous system 2. We propose the sensitivity of dioxetane 2 comes by virtue of its proximity to the silanol and silanoxy anion surface groups (Scheme 4). Peroxide bond decomposition by nucleophiles has been discussed before.21 In some cases anionic nucleophiles or oxyanion substituents can promote O-O bond homolysis by an electron transfer reaction.22
Scheme 3.

Scheme 4.

Shown in Schemes 4 and 5 is a proposed mechanism for the photooxidation and cleavage of the alkene group covalently bonded to the porous glass surface. The mechanism involves 3 steps: (i) visible light and O2 gas emerge from the opposite face of the attached sensitizer molecules for a triplet-triplet energy transfer reaction producing 1O2. (ii) Of the 3O2 molecules coming through the tip, 1O2 is located near the surface in a free or adsorbed state, which reacts with the alkene group with inefficient diffusion to the surrounding solution. There is a loss in mobility of 1O2 into the solution phase. Namely, the concentration of 1O2 drops off rapidly in the solution phase with increasing distance away from the probe tip due to 1O2 uptake at the alkene sites and the intrinsic short diffusion distance of 1O2. (iii) Silanol or silanoxy anion nucleophilic attack on a C atom of the dioxetane group is proposed to take place where the heterogeneous catalyst causes a gain in the initial yield of 3 from 1 by a mechanism other than higher loading of the sensitizer silane. Shown in Scheme 6 are two different pathways that can be visualized for the dioxetane cleavage reaction. The first involves an adsorption of 3 and the lack of photorelease due to the resistance to 3 dissolution as revealed by the toluene-d8 and D2O media at the cap/outer-phase boundary. The second pathway involves the release of 3, which effectively increases the diffusion distance of singlet oxygen. In this case, the amount of 3 released into petrolatum would probably relate to local drug delivery via permeabilization,23 which is an important finding to distinguish a new application in PDT. Overall, we ascertained single digit to tens of nmol of sensitizer released, which appears to surpass the objective of 0.25 nmol sensitizer/mL needed for PDT,24 although the quantities needed can depend on the type of tumor targeted.25
Scheme 5.

A reviewer rightfully pointed out that an alternative system could be designed, where 60 nmols of the sensitizer in an appropriate solvent is delivered by the same capillary under the pressure of oxygen without the use of a sensitizer tethered to the porous tip. Because such pump technology in this fiber optic with slow oxygen sparging is not yet available, we have not sought this avenue for the potential facile sensitizer delivery into biological matrices.
Conclusion
There is now a photosensitizer/fiber optic system usable as a point-source 1O2 generator that is unlike current PDT methods, which employ the systemic administration of dyes. The optical fiber was developed for the site-specific delivery of photosensitizer molecules. Three areas were discussed: (1) A porous fiber optic cap with photo-detachable pheophorbide molecules was synthesized, no leaching of the sensitizer was observed in the dark. The fiber optic was configured so that visible light and O2 gas were co-administered through the porous fiber optic cap, and was capable of O2 sparging to hypoxic sites. (2) Sensitizer loading was optimized, the maximal photocleavage arose when the surface distance between sensitizers was ~17 nm. Lower sensitizer loadings reduced the photocleavage efficiency due to less available sensitizer. Higher loadings were inefficient likely due to the crowding of sensitizer molecules and sensitizer-sensitizer self quenching. (3) Pigmentation of semi-solid petrolatum was observed where 100% (60 nmol) sensitizer detached from the probe tip. However, photorelease was much less efficient in toluene-d8 and D2O, because 3 remained adsorbed on the probe tip even though the covalent alkene bond bridging the sensitizer and glass had been broken.
The results reported here provide knowledge of the factors influencing sensitizer photorelease for the development of the new area of fiber optic-guided sensitizer delivery. The potential of this fiber optic for cancer cell killing in discrete locations remains to be determined.
Experimental Section
Materials and Instrumentation
Corning 7930 porous Vycor glass was purchased from Advanced Glass and Ceramics, Holden, MA. Pyropheophorbide-a was purchased from Frontier Scientific and was used as received. Spectrophotometric grade toluene-d8, deuterium oxide-d2, chloroform-d1 were purchased from Sigma Aldrich or Cambridge Isotope Laboratories and were used as received. Acetonitrile-d3 was purchased from Isotec, Inc. Deionized water was purified using a U.S. Filter Corporation deionization system. Reagents and solvents such as NaOH, 4-bromophenol, 1,2-dibromoethane, CH2Cl2, CCl4, benzoyl peroxide, acetone, NaI, sodium thiosulfate, ethanol, n-BuLi, DMF, NH4Cl, anhydrous Na2SO4, MeOH, NBS, NaBH4, DMAP, EDC, NaH, 3-iodopropyltrimethoxysilane, tetramethoxy benzene, toluene, and THF were purchased from Sigma Aldrich and used without further purification. Column chromatography was carried out on silica gel 40–60 Å particles. TLC was conducted on silica gel 60F 254 TLC-plates. The radiant power of the visible light exiting the fiber was measured with a Nova energy meter from Ophir Optronics, Logan, UT. Dissolved oxygen was measured with a Hach sens-ION6 dissolved oxygen meter. Proton NMR spectra were acquired at 400 MHz and 13C NMR spectra were acquired at 100.6 MHz on a Bruker DPX400 MHz instrument. HRMS data were obtained on Agilent 6220-TOF coupled with 1200 series LC. GC/MS data were acquired on Agilent 6890N coupled with 5973 MSD. HPLC data were obtained on a Perkin-Elmer 200 series instrument equipped with bondclone 10 C18 column at 254 nm and FTIR spectra were collected on a Perkin-Elmer Spotlight Imaging System. The melting points were obtained on a MEL-TEMP apparatus. UV-visible spectra were collected on a Hitachi UV-VIS U-2001 instrument. Fluorescence spectra were collected on a Nikon Eclipse TE 200 inverted epifluorescence microscope. Optical images of the glass samples were collected on an Olympus SZX10 stereo microscope. Some samples were irradiated with a Rayonet photoreactor fitted with Sylvania visible light bulbs through a 0.05 M NaNO2 filter solution.
Optical Fiber
The apparatus consisted of a 250-W quartz-halogen illuminator (Fiberoptic Systems, Inc., Simi Valley, CA), a custom-made fiber-optic cable, a compressed oxygen gas tank, and PVG tip 1. A 3 ft 0.55 numerical aperture borosilicate fiber optic was used, which had an external diameter of 1.4 mm including the jacket black polyvinyl chloride. The fiber optic contained a 1.1 mm diameter Teflon gas flow tube running from the distal end to the T-valve, which was surrounded by ~350 excitation fibers (Figure 9). An integral dichroic reflector was used to focus the light of the illuminator into the proximal end of the fiber, and 28 mW was delivered out of the end of the fiber (beam area = 0.126 cm2, energy density of output = 444 mJ/cm2). Some experiments employed the use of a cutoff filter (<400 nm). The T value was connected to a 200 PSI compressed oxygen tank with the gas regulator set at 2 or 10 PSI (flow rate through the cap was 0.2–0.3 ppm/min). PVG caps were shaped into cylindrical pieces with a Buehler IsoMet Low Speed Saw (Model 11-1280-160), a Buehler ultrasonic disc cutter (Model 170), and a Buehler variable speed grinder-polisher. A hole (3.0 mm diameter × 2.0 mm length) was drilled into the PVG cylinders with a drill dremel (Model 200) to accommodate the fiber optic, which was glued in place with ethyl cyanoacrylate. Regarding the propagation of light, most was lost out the end of the tip rather than scattered evenly within the tip. The PVG cap received the excitation light and oxygen gas, but did not heat up, it remained at room temperature throughout the course of the experiments.
Figure 9.
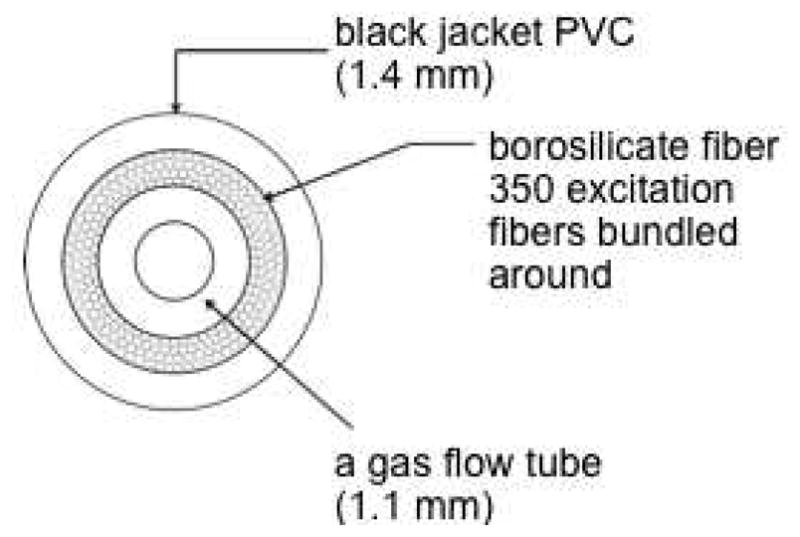
A schematic cross-section picture of the hollow-core fiber optic, in which ~350 excitation fibers surround the Teflon inner flow tube coaxially.
Pheophorbide-Modified Glass (1)
Covalent bonding of the sensitizer to the PVG was achieved by adding pheophorbide monoester 11 (10 mg, 0.0126 mmol) to 3-iodopropyltrimethoxysilane (0.250 mmol) and NaH (0.302 mg, 0.0126 mmol) in 5 mL dry THF, and refluxing the mixture at 70 °C for 24 h. THF was evaporated under N2 leaving the pyropheophorbide–3-iodopropyltrimethoxysilane 12 residue, which was then added to 100 mL dry toluene and sixteen 0.2 g PVG caps (pre-dried at 500 °C in a Fisher Scientific Isotemp muffle furnace for 24 hours) and refluxed at 110 °C for 24 h. The colorless PVG tips were converted to a deep green color when silane 12 was anchored to the glass. Control experiments were carried out in order to establish that the sensitizer molecules derived from 12 were anchored and not adsorbed to the silanol groups of PVG. Any silanes that were not covalently attached to the PVG surface were washed away with toluene, THF, chloroform, ether, and hexane, followed by Soxhlet extraction with methanol for 24 h. In the absence of propyltrimethoxysilane and NaH, a weak non-covalent interaction existed between 11 molecules and PVG that were readily carried off the surface by solvent washing and Soxhlet extraction. Regarding the dark stability of 1, no leaching of the photosensitizer was observed under the following conditions: (a) 1.5 mL of 0.01 M HCl solution (pH 2) was added to 156 mg of 1 at rt for 4 h; (b) 1.5 mL 0.01 M dilute NaOH solution (pH 12) was added to 130 mg of 1 at rt for 4 h. In both cases [(a) and (b)], the aqueous solution or chloroform extract did not show any trace of organics in solution by absorption spectroscopy (detection limit of pheophorbide-a = 10−8 M)). The heterogeneous sensitizer 1 was recovered by drying under vacuum at rt for 12 h. UV (air) λ: 507, 538, 610, and 666 nm; FT-IR: 2851 and 2954 cm−1. In 1, the ratio of sensitizer to iodosilane to silanol groups was ~1:3:300.
Photocleaved 4-(Formyloxy)benzyl-pyropheophorbide Ester Sensitizer (3)
One gram of pheophorbide-modified glass 1 (3 × 10−7 mol/g) was taken into 1 mL of toluene-d8 solution. The solution was purged with oxygen for 10 min. The heterogeneous solution was irradiated with the Rayonet reactor for 2 h. The photocleaved compound 3 was analyzed by following LC-MS condition: Solvent A (water containing 0.1% formic acid and 5 mM ammonium formate) and Solvent B (MeOH containing 0.1% formic acid and 5 mM ammonium formate) were used for isocratic elution. 90% B was delivered through the 30 mm C-18 column for 6 min wherein compound 3 was found to elute at 2.3 min. For the heterogeneous experiments, Soxhlet extraction was conducted to quantitate the pheophorbide formate ester (3) that detached from the silica matrix HRMS [+ESI] calcd C41H40N4O5 (M+) 688.2999, found 688.2997.
(Z)-4,4’-(Ethene-1,2-diylbis(oxy))dibenzaldehyde (9)
Yield 0.30 g (60%). To 0.5 g of arylbromide 8 in 5 mL THF, 1.6 mL of (0.003 mol) n-BuLi was added and the solution was stirred at −78 °C for 0.5 h. DMF (0.0058 mol, 0.5 mL) was added to the reaction mixture and stirred for 20 min at −78 °C. The solution was warmed to room temperature and was stirred for an additional 3.5 h. The reaction mixture was quenched with 20 mL cold saturated aqueous NH4Cl and then 25 mL of CH2Cl2 was added to the solution. The organic layer was washed with saturated sodium chloride solution and dried with Na2SO4. The crude liquid was purified by column chromatography (3:1 hexane and ethyl acetate) to yield 9 (mp 108–110°C). Rf = 0.15; 1H NMR (400 MHz, CDCl3) δ 9.93 (s, 2H), 7.88 (d, J = 8.8 Hz, 4H), 7.20 (d, J = 8.8 Hz, 4H), 6.34 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 116.2, 128.4, 131.8, 131.9, 161.5, 190.5. HRMS [+ESI] calcd for C16H12O4 (M+) 268.0736, found: 268.0732.
(Z)-(4,4’-(Ethene-1,2-diylbis(oxy))bis(1,4-phenylene))dimethanol (10)
Yield 97 mg (100%). To 100 mg (0.37 mmol) 9 in 2.5 mL MeOH, 0.140g (1.86 mmol) NaBH4 was added in small proportions and the reaction mixture was stirred for 14 h. Methanol solvent was then evaporated and the reaction was quenched with saturated aqueous NH4Cl solution. Ethyl acetate was then added to the solution mixture and the organic layer was separated and dried over Na2SO4. Evaporation of solvent gave solid 10 (mp: 150–153 °C). The acid stability of the alkene linkage of bis-alcohol 10 was examined: 0.02 M 10 was added to 0.02 M HCl (pH 2) with 1.1 × 10−4 M 1-pentanol as the internal standard with stirring in 2 mL CDCl3. The solution was stirred and monitored by 1H NMR after 0, 60, and 120 min with no degradation or loss of the alkene peaks. 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 8.4 Hz, 4H), 7.09 (d, J = 8.4 Hz, 4H), 6.15 (s, 2H), 4.66 (d, J = 3.9 Hz, 4H). 13C NMR (100 MHz, CDCl3) δ 64.3, 116.1, 128.3, 128.4, 135.5, 156.8. HRMS [+ESI] calcd for C16H16O4 (M+) 272.1049, found: 272.1047.
(Z)-4-[2-(4-Hydroxymethyl-phenoxy)-vinyloxy]benzyl-pyropheophorbide (11)
Yield 6 mg (60%). 10 mg (0.018 mmol) of commercially available pyropheophorbide-a, 5.0 mg (0.018 mmol) bis-alcohol 10, 2.28 mg (0.018 mmol) DMAP, and 5.37 mg (6.9 mmol) EDC were added into 3 mL DCM. After stirring overnight at room temperature, DCM was evaporated from the reaction mixture and the crude product was purified by column chromatography (1% methanol in CHCl3) yielding a black solid. Rf = 0.48. 1H NMR (400 MHz, CDCl3 δ 9.51 (s, 1H), 9.40 (s, 1H), 8.54 (s, 1H), 8.01 (dd, J = 17.8, 11.5 Hz, 1H), 7.29 (d, J = 8.5 Hz, 2H), 7.14 (d, J = 8.5 Hz, 2H), 7.03 (d, J = 8.5 Hz, 2H), 6.64 (d, J = 8.5 Hz, 2H), 6.28 (d, J = 17.8 Hz, 1H), 6.17 (d, J = 11.5 Hz, 1H), 6.08 (dd, J = 17.0, 3.4 Hz, 2H), 5.24 (d, J = 19.8 Hz, 1H), 5.06 (d, J = 19.8 Hz, 1H), 4.98 (d, J = 16.0 Hz, 1H), 4.90 (d, J = 12.0 Hz, 1H), 4.61 (s, 2H), 4.46 (d, J = 7.08 Hz, 1H), 4.28 (d, J = 8.3 Hz, 1H), 3.70 (m, 4H), 3.40 (s, 3H), 3.24 (s, 3H), 2.61 (m, 2H), 2.31 (m, 2H), 1.78 (d, J = 7.2 Hz, 3H), 1.70 (t, J = 7.6 Hz, 3H), −1.68 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 11.2, 12.0, 17.4, 19.5, 23.1, 29.7, 29.8, 31.1, 48.0, 49.9, 51.6, 64.8, 65.9, 93.0, 97.2, 104.1, 106.1, 116.1, 116.3, 122.5, 128.1, 128.3, 128.5, 128.6, 129.2, 130.0, 130.1, 130.5, 130.6, 131.5, 135.4, 135.9, 136.1, 136.2, 137.9, 141.5, 145.0, 149.0, 150.8, 155.2, 156.9, 157.3, 160.2, 171.3, 172.8, 196.1. HRMS [+ESI] calcd for C49H48 N4O6 (M+) 788.3574, found: 788.3562.
4-Hydroxybenzyl-pyropheophorbide Ester Sensitizer (13)
Pheophorbide ester 3 (2 × 10−5 M) was placed in methanol-water mixtures (9:1) where the pHs ranged from 2–8. The pH of the solution was adjusted by adding 0.01 M NH4OH and 0.01 M HCOOH. Compounds 13 and 14 were monitored by LCMS: Solvent A (water containing 0.1% formic acid and 5 mM ammonium formate) and Solvent B (MeOH containing 0.1% formic acid and 5 mM ammonium formate) were used for isocratic elution. 90% B was run through a 30 mm C-18 column for 6 min. HRMS [+ESI] calcd for C40H40N4O4 (M+) 640.3050, found. 640.3048.
Oxygen Flow Measurements
The rate of oxygen flow through the porous vycor cap was measured using a Hach sensION6 Dissolved Oxygen Meter. The oxygen meter and the fiber optic tip were placed simultaneously into 30 mL of distilled H2O. Oxygen flowed through the fiber optic cable at 2 and 10 PSI. Measurements were taken from the oxygen meter at one-minute intervals for a total of 30 minutes.
Sensitizer Coverage Measurements
Two methods were conducted to determine the amount of sensitizer bonded to the PVG tip. (i) The sensitizer was liberated from the PVG surface by dipping 1 into a 30% (V/W) hydrofluoric acid solution for 12 h using a previously established method.26 The free sensitizer in solution was extracted with chloroform and its concentration determined by UV-visible spectroscopy. (ii) A calculation was performed to determine the amount of 11 or 12 remaining in toluene solution after PVG was removed in 12 h with a correction for any adsorbed sensitizer by Soxhlet extraction. Even though mmol amounts of silane could be loaded due to the available silanol groups (1.66 mmol SiOH/g PVG), the range of sensitizer silane 12 loaded was 0.06 to 1.1 μmol.
Photocleavage Procedure
The 0.2 g fiber optic cap 1 (160 nmol 12) was placed into 0.5 mL toluene-d8 or 1.0 mL D2O, 0.4 mL petrolatum at 65 °C. The amount of photocleaved 3 was determined by absorption spectroscopy for the toluene-d8 and D2O samples, and by fluorescence spectroscopy for the petrolatum samples. The fiber tip was secured and then the cap was irradiated via the fiber with white light for 4 hours. A TXRed optical filter was used and images were created with Metafluor imaging software. Concentrations of dye were calculated by measuring the fluorescence intensity and comparing values to those found in the photorelease of dye from neat glass caps. The diffusion of pyropheophorbide 3 away from the probe tip into petrolatum was viewed using the epifluorescence microscope and was measured with the Adobe Photoshop CS5 ruler tool. The experiment was performed for a total of two hours and images were taken every 30 minutes at 4× and 10× magnification using an epifluorescence microscope.
Sensitizer-Sensitizer Surface Distance Calculation
PVG is a transparent material with ~4 nm diameter pores and a ~250 m2/g surface area, interconnected pores, and the surface has “stalagmite-like” features running ~3 nm in length and height.16 It is difficult to accurately calculate the spatial distance between sensitizer molecules in the porous glass matrix; however, a likely distance was estimated. With an average distance between silanol groups of 10 Å, the calculation (4 × 250 m2/g)/100 Å2 = 1 × 1021 Si-OH groups is divided by Avogadro’s number = 1.66 × 10−3 mol Si-OH/g PVG. Equations 1–4 show how a simple surface geometry was assumed. Eq 1 gives the volume of the 1 within the 0.08 mm sensitizer intrusion depth of the glass of radius (r) and height (h). The weight of the sensitizer-intruded portion of glass was calculated multiplying its volume by its density (1.38 g/mL). The area of the sensitizer-intruded glass is given by eq 2. The area occupied by 4 sensitizer molecules at a given loading amount (0.06 to 1.1 μmol 12) is given by eq 3. Dye molecules were assumed to be spread out with an orthogonal orientation in relation to the surface. The sensitizer-sensitizer distance on the PVG surface is given by eq 4.
| (1) |
| (2) |
| (3) |
| (4) |
Supplementary Material
Acknowledgments
We acknowledge support from the National Institute of General Medical Sciences (NIH SC1GM093830). A.G. is thankful to Zhong Wang (Hunter College Bio-Imaging Facility), Terry Dowd (Brooklyn College Chemistry Department), and Mim Nakarmi (Brooklyn College Physics Department) for use of requisite equipment.
Footnotes
Supporting Information Available: Details of the syntheses of known compounds 6–8 and spectroscopic data of 1, 6–11, 13, and 14. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kessel D, Foster TH, editors. Symposium-In-Print: Photodynamic therapy. Photochem Photobiol. 2007;83:995–1282. [Google Scholar]
- 2.Recommendations for occupational safety and health: Compendium of policy documents and statements. Cincinnati, OH: U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control, National Institute for Occupational Safety and Health; DHHS (NIOSH) Publication No. 92-100. [Google Scholar]
- 3.(a) Flickinger ST, Patel M, Binkowski BF, Lowe AM, Li MH, Kim C, Cerrina F, Belshaw PJ. Org Lett. 2006;8:2357–2360. doi: 10.1021/ol060644x. [DOI] [PubMed] [Google Scholar]; (b) Cano M, Ladlow M, Balasubramanian S. J Org Chem. 2002;67:129–135. doi: 10.1021/jo010703e. [DOI] [PubMed] [Google Scholar]; (c) Pirrung MC, Fallon L, Lever DC, Shuey SW. J Org Chem. 1996;61:2129–2136. [Google Scholar]
- 4.(a) Kammari L, Solomek T, Ngoy BP, Heger D, Klán P. J Am Chem Soc. 2010;132:11431–11433. doi: 10.1021/ja1047736. [DOI] [PubMed] [Google Scholar]; (b) Xie Z, Hu X, Chen X, Sun J, Shi Q, Jing X. Biomacromolecules. 2008;9:376–380. doi: 10.1021/bm700906k. [DOI] [PubMed] [Google Scholar]; (c) Kessler M, Glatthar R, Giese B, Bochet CG. Org Lett. 2003;5:1179–1181. doi: 10.1021/ol027454g. [DOI] [PubMed] [Google Scholar]; (d) Singh AK, Khade PK. Bioconjugate Chem. 2002;13:1286–1291. doi: 10.1021/bc020021d. [DOI] [PubMed] [Google Scholar]; (e) Hu J, Zhang J, Liu F, Kittredge K, Whitesell JK, Fox MA. J Am Chem Soc. 2001;123:1464–1470. [Google Scholar]; (f) Smet M, Liao LX, Dehaen W, McGrath DV. Org Lett. 2000;2:511–513. doi: 10.1021/ol991373b. [DOI] [PubMed] [Google Scholar]
- 5.(a) Givens RS, Weber JFW, Conrad PG, Orosz G, Donahue SL, Thayer SA. J Am Chem Soc. 2000;122:2687–2697. [Google Scholar]; (b) Givens R. In: CRC Handbook of Organic Photochemistry and Photobiology. 2. Horspool WM, Lenci F, editors. CRC Press; Boca Raton, FL: 2003. pp. 1–46. [Google Scholar]
- 6.Wijtmans M, Rosenthal SJ, Zwanenburg B, Porter NA. J Am Chem Soc. 2006;128:11720–11726. doi: 10.1021/ja063562c. [DOI] [PubMed] [Google Scholar]
- 7.Lin Q, Huang Q, Li C, Bao C, Liu Z, Li F, Zhu L. J Am Chem Soc. 2010;132:10645–10647. doi: 10.1021/ja103415t. [DOI] [PubMed] [Google Scholar]
- 8.Jiang MY, Dolphin D. J Am Chem Soc. 2008;130:4236–4237. doi: 10.1021/ja800140g. [DOI] [PubMed] [Google Scholar]
- 9.(a) Baugh SDP, Yang Z, Leung DK, Wilson DM, Breslow R. J Am Chem Soc. 2001;123:12488–12494. doi: 10.1021/ja011709o. [DOI] [PubMed] [Google Scholar]; (b) Ruebner A, Yang Z, Leung D, Breslow R. Proc Natl Acad Sci. 1999;96:14692–14693. doi: 10.1073/pnas.96.26.14692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Busch TM, Cengel KA, Finlay JC. Cancer Biol Therapy. 2009;8:540–542. doi: 10.4161/cbt.8.6.8067. [DOI] [PubMed] [Google Scholar]; (b) Hajri A, Wack S, Meyer C, Smith MK, Leberquier C, Kedinger M, Aprahamian M. Photochem Photobiol. 2002;75:140–148. doi: 10.1562/0031-8655(2002)075<0140:ivaive>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 11.(a) Zaklika KA, Thayer AL, Schaap AP. J Am Chem Soc. 1978;100:4916–4918. [Google Scholar]; (b) Adam W, Cheng CC, Cueto O, Erden I, Zinner K. J Am Chem Soc. 1979;101:4735–4736. [Google Scholar]; (c) Baumstark A. Advances in Oxygenated Processes. Vol. 1. JAI Press; Greenwich, CT: 1988. pp. 31–84. [Google Scholar]; (d) Zamadar M, Greer A. In: Handbook of Synthetic Photochemistry. Albini A, Fagnoni M, editors. Wiley-VCH; Weinheim: 2010. pp. 353–386. [Google Scholar]; (e) Turro NJ, Ramamurthy V, Scaiano JC. Modern Molecular Photochemistry of Organic Molecules. University Science Books; Sausalito, CA: 2010. pp. 1001–1040. [Google Scholar]
- 12.(a) Kanofsky JR. Photochem Photobiol. 1990;51:299–303. doi: 10.1111/j.1751-1097.1990.tb01714.x. [DOI] [PubMed] [Google Scholar]; (b) Moan J, Berg K. Photochem Photobiol. 1991;53:549–553. doi: 10.1111/j.1751-1097.1991.tb03669.x. [DOI] [PubMed] [Google Scholar]; (c) Moor ACE. J Photochem Photobiol B Biol. 2000;57:1–13. doi: 10.1016/s1011-1344(00)00065-8. [DOI] [PubMed] [Google Scholar]; (d) Niedre M, Patterson MS, Wilson BC. Photochem Photobiol. 2002;75:392–397. doi: 10.1562/0031-8655(2002)075<0382:DNILDO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]; (e) Schweitzer C, Schmidt R. Chem Rev. 2003;103:1685–1758. doi: 10.1021/cr010371d. [DOI] [PubMed] [Google Scholar]; (f) Skovsen E, Snyder JW, Lambert JDC, Ogilby PR. J Phys Chem B. 2005;109:8570–8573. doi: 10.1021/jp051163i. [DOI] [PubMed] [Google Scholar]; (g) Redmond RW, Kochevar IE. Photochem Photobiol. 2006;82:1178–1186. doi: 10.1562/2006-04-14-IR-874. [DOI] [PubMed] [Google Scholar]; (h) Flors C, Nonell S. Acc Chem Res. 2006;39:293–300. doi: 10.1021/ar0402863. [DOI] [PubMed] [Google Scholar]; (i) Greer A. Nature. 2007;447:273–274. doi: 10.1038/447273a. [DOI] [PubMed] [Google Scholar]; (j) Baruah A, Šimková K, Hincha DK, Apel K, Laloi C. Plant J. 2009;60:22–32. doi: 10.1111/j.1365-313X.2009.03935.x. [DOI] [PubMed] [Google Scholar]
- 13.(a) Aebisher D, Zamadar M, Mahendran A, Ghosh G, McEntee C, Greer A. Photochem Photobiol. 2010;86:890–894. doi: 10.1111/j.1751-1097.2010.00748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zamadar M, Aebisher D, Greer A. J Phys Chem B. 2009;113:15803–15806. doi: 10.1021/jp907945c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Ehrenberg B, Anderson JL, Foote CS. Photochem Photobiol. 1998;68:135–140. [PubMed] [Google Scholar]; (b) Berg K, Moan J. Photochem Photobiol. 1997;65:403–409. doi: 10.1111/j.1751-1097.1997.tb08578.x. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Bauld NL. J Org Chem. 1999;64:9251–9253. [Google Scholar]
- 16.Pohl E, Osterholtz F. In: Silane Surfaces and Interfaces. Leyden D, editor. Gordon & Breach; New York: 1986. [Google Scholar]
- 17.For potential enhanced spin-orbit coupling and singlet oxygen production, 1000–2000 fold higher loading of iodosilane would be needed as observed in a sol-gel system: Kim S, Ohulchanskyy TY, Bharali D, Chen Y, Pandey RK, Prasad PN. J Phys Chem C. 2009;113:12641–12644. doi: 10.1021/jp900573s.
- 18.(a) Ogilby PR, editor. Symposium-In-Print: Singlet Oxygen [ Photochem Photobiol. 2006;823:1133–1240. doi: 10.1562/2006-06-19-RA-936. [DOI] [PubMed] [Google Scholar]; (b) Greer A, editor. Symposium-In-Print: Organic Chemistry of Singlet Oxygen. Tetrahedron. 2006;62:10603–10776. [Google Scholar]
- 19.(a) Lovell JF, Chen J, Jarvi MT, Cao WG, Allen AD, Liu Y, Tidwell TT, Wilson BC, Zheng G. J Phys Chem B. 2009;113:3203–3211. doi: 10.1021/jp810324v. [DOI] [PubMed] [Google Scholar]; (b) Campo MA, Gabriel D, Kucera P, Gurny R, Lange N. Photochem Photobiol. 2007;83:958–965. doi: 10.1111/j.1751-1097.2007.00090.x. [DOI] [PubMed] [Google Scholar]; (c) Gabriel D, Campo MA, Gurny R, Lange N. Bioconjugate Chem. 2007;18:1070–1077. doi: 10.1021/bc060321l. [DOI] [PubMed] [Google Scholar]
- 20.Bartlett PD, Baumstark AL, Landis ME, Lerman CL. J Am Chem Soc. 1974;96:5267–5268. [Google Scholar]
- 21.(a) Cramail C, Degueil-Castaing M, Maillard B. J Org Chem. 2001;66:3492–3494. doi: 10.1021/jo015506u. [DOI] [PubMed] [Google Scholar]; (b) Nair V, Sheeba V. J Org Chem. 1999;64:6898–6900. doi: 10.1021/jo9904359. [DOI] [PubMed] [Google Scholar]
- 22.(a) Tanimura M, Watanabe N, Ijuin HK, Matsumoto M. J Org Chem. 2011;76:902–908. doi: 10.1021/jo1021822. [DOI] [PubMed] [Google Scholar]; (b) Adam W, Kazakov DV, Kazakov VP. Chem Rev. 2005;105:3371–3387. doi: 10.1021/cr0300035. [DOI] [PubMed] [Google Scholar]; (c) Adam W, Blancafort L. J Org Chem. 1997;62:1623–1629. [Google Scholar]
- 23.Lovell JF, Liu TWB, Chen J, Zheng G. Chem Rev. 2010;110:2839–2857. doi: 10.1021/cr900236h. [DOI] [PubMed] [Google Scholar]
- 24.Laubach HJ, Chang SK, Lee S, Rizvi I, Zurakowski D, Davis SJ, Taylor CR, Hasan T. J Biomed Opt. 2008;13:050504. doi: 10.1117/1.2981813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hahn SM, Putt ME, Metz J, Shin DB, Rickter E, Menon C, Smith D, Glatstein E, Fraker DL, Busch TM. Clin Cancer Res. 2006;12:5464. doi: 10.1158/1078-0432.CCR-06-0953. [DOI] [PubMed] [Google Scholar]
- 26.Houghten RA, Yu Y. J Am Chem Soc. 2005;127:8582–8583. doi: 10.1021/ja0402396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.