

Abstract
Microglia do not constitute a single, uniform cell population, but rather comprise cells with varied phenotypes, some which are beneficial and others that may require active regulatory control. Thus, gaining a better understanding of the heterogeneity of resident microglia responses will contribute to any interpretation regarding the impact of any such response in the brain. Microglia are the primary source of the pro-inflammatory cytokine, tumor necrosis factor (TNF) that can initiate various effects through the activation of membrane receptors. The TNF p55 receptor contains a death domain and activation normally leads to cellular apoptosis; however, under specific conditions, receptor activation can also lead to the activation of NFκB and contribute to cell survival. These divergent outcomes have been linked to receptor localization with receptor internalization leading to cell death and membrane localization supporting cell survival. A second TNF receptor, TNF p75 receptor, is normally linked to cell growth and survival, however, it can cooperate with the p55 receptor and contribute to cell death. Thus, while an elevation in TNFα in the brain is often considered an indicator of microglia activation and neuroinflammation, a number of factors come into play to determine the final outcome. Data is reviewed demonstrating that heterogeneity in morphological response of microglia and the expression of TNFα and TNF receptors are critical in identifying and characterizing neurotoxic events as they relate to neuroinflammation, neuronal damage and in stimulating neuroprotection.
Keywords: microglia, neuroinflammation, hippocampus, trimethyltin, TNFα, TNF receptors
Introduction
Microglia serve as the resident mononuclear phagocytes of the brain and are highly heterogeneous within the healthy CNS. They comprise only 10% of the total cell population of the brain; yet, they have multiple morphological and potential functional profiles depending on their environment (Streit et al., 2004; Streit, 2006). Structurally, microglia display a dynamic and active phenotype with ongoing retraction and extension of processes into the brain parenchyma (Raivich, 2005). This supports the idea of a surveillance function for microglia in the healthy brain and that these cells are poised to rapidly respond to environmental changes. Microglia serve as the first line of defense against pathogens entering the CNS parenchyma and thus, play an important role during injury and infection in the central nervous system (CNS; Streit et al., 1988; Perry et al., 1993; Gehrmann et al., 1993; 1995; Gebicke-Haerter et al., 1996; Kreutzberg, 1996). In the human brain, microglia activation and neuroinflammation have been associated with viral or bacterial infection, autoimmune disease such as multiple sclerosis, head trauma, vascular system damage, neuropsychiatric disorders, and neurodegenerative diseases. In these conditions, microglia serve as the primary resident immune cell of the brain and produce many of the pro-inflammatory cytokines, such as tumor necrosis factor-α and interleukin-1 α.
It has been suggested that microglial responses are tailored in regional and insult-specific manners (Carson et al., 2007). The most recognizable role of microglia in brain defense is as a scavenger role to remove cellular debris by phagocytosis, as occurs in the event of infection, inflammation, trauma, ischemia, and neuronal death (Thomas, 1992; Gonzalez-Scarano and Baltuch, 1999; Carson, 2002; Danton and Dietrich, 2003). However, we now know that, not only do microglia dynamically survey the CNS and clear damaged cellular constituents, but that they are capable of initiating a rapid and specific response to subtle changes in the microenvironment. In addition, microglia are in intimate contact with neurons, for which they serve important developmental support (Kimoto et al., 2009) and maintenance functions, such as clearance of aberrant proteins (Kreutzberg, 1996; Nimmerjahn et al., 2005; Davalos et al., 2005; Raivich, 2005).
With changes in neuronal activity, presence of pathogen, or a mechanical/physical injury, microglia respond with dramatic changes in cell morphology and increased expression of macrophage markers. Due to the observation that microglia activation is likely an early event in all forms of pathology, the presence of activated microglia was initially considered as a sensitive marker to identify sites predestined for imminent tissue destruction (Galea et al., 2007). However, further work has demonstrated that changes in microglia morphology or functional activation do not inevitably lead to neuron loss nor does it only indicate damage.
Phenotypes of Microglia
Microglia share phenotypic characteristics and innate immunological functions with other mononuclear phagocytes such as, monocytes, macrophages, and dendritic cells (Flaris et al., 1993). The classical bone-marrow-derived microglial cells reside in the gray matter and are ramified (highly branched) with a small amount of perinuclear cytoplasm and a small, dense and heterochromatic nucleus. Under normal conditions, microglia interact with their surroundings and provide neurotrophic factors. Microglia can be transformed to a reactive or activated state by localized changes in the environment, influences from infiltrating blood-borne cells, local blood-brain-barrier disruption (Nimmerjahn et al., 2005), or the presence of endangered neurons. In exchange, the transformed microglia can influence other neural, vasculature, and blood-borne cells by a number of secreted factors including pro-inflammatory cytokines and chemokines, as well as, nitric oxide and reactive oxygen intermediates. Chemokines and cytokines produced by microglia can serve to signal lymphocytes from the vascular system allowing for transendothelial migration into the brain and the production of interferon gamma, which then propagates microglia activation.
The functional changes of activated microglia are often accompanied by a morphological transformation leading from cells with thin, ramified processes to cells with larger somata and shorter and coarser cytoplasmic processes. Microglia can display a ramified, hyper-ramified, or activated/amoeboid morphology depending upon the type of ongoing response/injury. For example, increased staining of ramified microglia indicative of thickened processes is evident in the CA1 pyramidal cell layer and molecular layer of the hippocampus within a few hours following an acute injection of lipopolysaccride (LPS) and in the contralateral hippocampus following a direct injection of kainic acid. In each case, these morphological changes occur in the absence of neuronal death (Cunningham et al., 2009). However, microglia localized to areas of neuronal death assume a more amoeboid shape with dense cell bodies and a reduced number of shortened, thick processes (Cunningham et al., 2009) progressing to a structural morphology similar to macrophages. While morphology may not reflect functional activation states of microglia (Schwartz et al., 2006), ongoing efforts to characterize functional differences between the various microglia structural phenotypes continue. Such characterization and identification of relationship to other cells in the brain are critical in determining the contribution of these dynamic cells in acute and chronic brain insults.
Sources of Brain Macrophages
The brain has two indigenous sources of brain macrophages (a general term encompassing all phagocytic cells, including activated microglia resident to the brain and blood-derived monocytes entering the brain upon vascular injury). Once in the brain, the macrophages are morphologically indistinguishable and identification requires either separation by flow cytometry on the basis of CD11b/CD45 expression levels (Sedgwick et al., 1991; Renno et al., 1995; Carson et al. 1998) or passive immune transfer with bone marrow chimera mice (Ajami et al., 2007). While the entry into the brain is limited and delayed as compared to peripheral tissue (Andersson et al., 1992), blood-derived macrophages may enter brain tissue, react upon it, and then return to the circulation in the presence or absence of injury (Matsumoto and Fujiwara, 1987; Lassmann et al., 1993). Alternatively, macrophages may differentiate to microglial morphology and remain in the brain tissue for an extended period of time. In this state they can persist until destroyed by senescence or prompted to move back into the circulation. Peripheral macrophages provide an enriched source of cytokine and inflammatory factors as compared to resident microglia. In addition, the impact of these two different sources of microglia can be very different on neurons. This may be due to innate features of the two cells yet to be identified or simply related to the magnitude of microglia versus macrophage secreted factors, such as pro-inflammatory cytokines, chemokines, nitric oxide, and production of reactive oxygen species.
TNFα and TNF receptor signaling
One such pro-inflammatory cytokine produced by macrophages is tumor necrosis factor alpha (TNFα). TNFα has been considered as a possible master inflammatory regulator that can induce further cytokine production, gliosis, blood-brain-barrier damage, demyelination, inflammation, cell adhesion, and immune reactivity. In the CNS, microglia are the primary source of TNFα (Gregersen et al., 2000; Hanisch, 2002) and the release of TNFα by microglia is implicated in neurotoxicity (Badie et al., 2000; Taylor et al., 2005). Although TNFα has not been demonstrated to cause neuronal death in healthy brain tissue or normal neurons (Gendelman and Folks, 1999) and normal cellular architecture is maintained in mice deficient in pro-inflammatory cytokines or receptors, one can not rule out that a localized activation could initiate neuronal death.
The rapid increase in cytokine expression following injury requires transcription, posttranscription, translation, and the conversion of latent precursors by proteases to biologically active forms. The short half-life of cytokine mRNA transcripts requires rapid translation and secretion into the extracellular space, resulting in a burst of cytokine release. A microglia response and a rapid and dramatic up-regulation of TNFα protein and mRNA is found in animal models of cerebral ischemia (Botchkina et al., 1997; Saito et al., 1996) with increased TNFα expression preceding the onset of neuronal cell death (Botchkina et al., 1999). Recent studies examining cell death and survival following an ischemic insult (Lambertsen et al., 2009), exposure to MPTP (Sairanen et al., 2006), or induced by a systemic injection of trimethyltin (Harry et al., 2008) suggest that the level of TNFα produced by microglia at a specific site and the neuronal expression pattern of TNF receptors can be determining factors for neuronal death or survival. A critical signaling element in TNFα-induced apoptosis is the robust and prolonged activation of JNK, which occurs when NF-κB is inhibited (Sakon et al., 2003; Kamata et al., 2005), suggesting a dose-response effect of TNFα exposure.
TNF initiates its multiple effects on cell function by binding to two distinct cell surface receptors, a 55 kDa type-1 receptor (TNFp55R) and a 75 kDa type-2 receptor (TNFp75R) (Tartaglia and Goeddel, 1992; Medvedev et al., 1996). TNFp55R contains a cytoplasmic sequence identifying an intracellular death domain essential for the transduction of an apoptotic signal (Micheau and Tschopp, 2003; Thorburn 2004). Receptor activation occurs by oligomerization and requires internalization of the ligand-receptor complex. TNFp55R contains tyrosine residues in its intracellular domain and, in most cells, this allows for the ligand-TNFp55R complex to be rapidly internalized by clathrin-coated pits following receptor triggering, which is critical for mediating the death signaling. Within minutes of internalization, the TNF receptosome recruits TNF receptor-associated death domain (TRADD) (Hsu et al., 1996; Schneider-Brachert et al., 2004). The fate of the cell depends on which proteins associate with TRADD (Hsu et al., 1996). TNFp75R activation primarily initiates trophic/protective actions (Shen et al., 1997; Yang et al., 2002), yet this receptor can also initiate apoptosis (Suvannavejh et al., 2000). While cell death has been reported upon triggering of this receptor (Bigda et al., 1994; Grell et al., 1993; Medvedev et al., 1994; 1996b), activation of the apoptotic program was considered to be due to endogenous TNF and the activation of TNFp55R (Grell et al., 1999; Fiers et al. 1995). Later studies using mice in which astrocyte-specific human tm (transgenic mouse) TNF signals exclusively through a regulated human (hu) TNFp75R transgene, in the absence of the endogenous TNFp75R and TNF genes, demonstrated a proinflammatory role for the TNFp75R when signaling alone in the CNS (Akassoglou et al., 2003). This study demonstrated that physiological levels of the TNFp75R are sufficient to induce inflammation in the CNS when TNF is chronically produced. In this case, however, clinical symptoms are not necessarily associated with the inflammation. With increased levels of the TNFp75R, both neuropathology and clinical signs are evident. Further work demonstrated that cytotoxic and inflammatory effects of TNF in the CNS occurred if these transgenic mice are linked with the TNFp55R. When these transgenic mice are linked with the TNFp75R only, the inflammatory effects are observed. With two functional TNFRs, it was concluded that the severity of TNF-induced neuropathology depends on the levels of TNFR expression (Akassoglou et al., 2003).
A number of studies have supported cooperation between TNF receptors in cytotoxicity (Declercq et al., 1998; Vandenabeele et al., 1995; Grell et al., 1995; Meager et al., 1993; Weiss et al., 1997; Akassoglou et al., 2003). It is thought that the cytoplasmic domain of the TNFp75R has signaling activity for TNFp75R-mediated cytotoxicity. Activation of TNFp75R can enhance TNFp55R-induced cell death (Chan and Lenardo, 2000; Declercq et al., 1998; Vandenabeele et al., 1995; Weiss et al., 1997). Activation of TNFp55R results in activation of NF-κB and induction of associated anti-apoptotic factors (Wajant et al., 1999; 2001; Yang et al., 2001). Thus, TNFp55R-induced apoptosis usually occurs only following down-regulation of the anti-apoptotic NF-κB response. TNFp75R triggering can modify TNFp55R-induced apoptosis via inhibition of NF-kB-dependent production of anti-apoptotic factors and by blocking the action of anti-apoptotic factors at the post-transcriptional level (Fotin-Mleczek et al., 2002).
TNF receptors are expressed by both neurons and glia (Kinouchi et al., 1991; Tchelingerian et al., 1996; Dopp et al., 1997). With injury, the expression of receptors and their distribution on specific cell types can vary depending upon whether the activation is due to apoptosis or inflammatory regulation. For example, macrophages within an ischemic infarct express a high level of TNF receptors while within the prenumbra or with a nerve transection injury, neurons rather than reactive/ramified microglia may be the prominent cell expressing TNF receptors (Botchkina et al., 1997; Sairanen et al., 2001; Lambertsen et al., 2007; Harry et al., 2008). While TNF signals via membrane receptors, TNF exposure also activates a selective extracellular proteolytic cleavage of TNFR resulting in the release of soluble TNFR. In this form, the receptors can serve in a protective fashion by binding TNF and preventing further receptor signaling. It is also known that both the tissue distribution of the receptor and the differentiation state of the target cell influence the cellular response to cytokines. The contradictory effects of TNFα may depend on timing i.e., early proinflammatory functions may be followed by a later stage of immunosuppressive activity (Liu et al., 1998; Korner et al., 1997), such as the inhibition of T-cell proliferation (Lu et al., 2007) or apoptosis (Weishaupt et al., 2000).
For the TNF family of receptors, membrane receptor mechanisms of apoptosis are implicated in neuronal death involving intracellular death-inducing signaling complexes (Martin-Villalba et al., 2001; Henshall and Simon, 2005) activation of the AP-1 and NF-κβ transcription factors (Baud and Karin, 2001; Qiu et al., 2002), as well as signal transduction and activation of caspases (Krammer, 2000; Rosenbaum et al., 2000). It has been proposed that TNFp55R activation provides a molecular mechanism for the rapid apoptosis of injured or sick neurons through a caspase 3-mediated pathway (Yang et al., 2002). However, studies to elucidate the role of individual TNF receptors in the brain using TNF receptor-deficient mice have provided a less than consistent pattern with contradictory results as to the influence of each receptor on the severity of an injury (Sullivan et al., 1999; Suvannavejh et al., 2000; Raivich et al., 2002; Bohatschek et al., 2004; Quintana et al., 2005). Many of these studies however have employed injury models that disrupt the blood brain barrier and thus serum factors or infiltrating cells may serve as a major confounder in the final interpretation. In addition, as a whole, these studies were not designed in a manner to examine the contribution of receptor cooperation.
Parkinson's Disease
One of the more prominent views of a role of microglia and neuroinflammation in neurodegeneration comes in the arena of Parkinson's Disease (Orr et al., 2002; Whitton, 2007). TNF-immunoreactive glial cells have been detected in the substantia nigra and immunoreactivity for TNF receptors was found in cell bodies and processes of most dopaminergic neurons of Parkinsonian patients (Boka et al., 1994). The general assumption has been that, given the elevated vulnerability of dopaminergic neurons to oxidative stress and the increased number of microglia within the substantia nigra (SN), microglia and neuroinflammation significantly contribute to the disease process. Some questions, however, are beginning to arise for some of the underlying assumptions. With regards to the assumption of neuronal vulnerability based upon a greater number of microglia in the SN, the extensive examination by Lawson (1990) showed the highest number of microglia in the olfactory telencephalon, followed by a higher relative number of microglia with similar phenotype in dentate gyrus of the hippocampus, the substantia nigra, and portions of the basal ganglia (approx. 13%) as compared to a relatively uniform distribution of microglia in the adult rodent brain (cortex - 3%). This observation has been taken to imply that the differential vulnerability of the dopaminergic neurons in the SN is related to the number of microglia present in the structure. Lawson et al (1990) however, also reported that the regional difference in microglia is also evident to how these cells modify their morphology and express cell surface antigens.
In 2006, Sawada and co-workers examined autopsy brains of Parkinson's Disease patients in the attempt to determine whether cytokines produced by activated microglia in the SN and putamen served in a neuroprotective or neurotoxic role. At the early stage of the disease microglia were predominantly associated with tyrosine hydroxylase (TH)-positive neurites in the SN. In the later stages of the disease, activated microglia displaying an amoeboid morphology were associated with dying TH-positive neurons. Interestingly, a microglia response was not limited to the SN but rather was seen in various other regions such as the hippocampus and the cerebral cortex in the absence of neuronal loss. With a Lewy body disease, neuronal loss extended to the hippocampus. Sawada et al. (2006) concluded from these studies that microglia in the SN and putamen may be neuroprotective in the early stages of the disease but become neurotoxic with the degeneration of dopamine neurons. Alternatively, the changes in the microglia response may simply reflect a normal phagocytic activation upon neuronal death. Alternatively, the response of microglia may reflect efforts to regulate the CD8+ and CD4+ T cells known to be present in the SN of PD patients (McGeer et al., 2001; Brochard et al., 2009).
Multiple studies have employed minocycline as a method to study the relationship between microglia activation and neuronal degeneration. Minocycline is a tetracycline derivative antibiotic with a well-described ability to downregulate multiple inflammation-induced secretory products, such as NO, IL-1, cyclooxygenase-2 (COX-2), and prostaglandin E(2) production, as well as expression of inducible nitric oxide synthase, and IL-1beta-converting enzyme. Initial studies reported that blocking minocycline offered neuroprotection against a dose of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) sufficient to induce loss of nigrostriatal dopaminergic neurons (Wu et al., 2002). While this finding could be interpreted as supporting an active role for microglia in neuronal death, there are significant confounders. For example, specificity of an effect of minocycline on microglia is limited as this drug also has direct anti-apoptotic effects, such as inhibition of caspase-1, caspase-3, and cytochrome c release. (Chen et al., 2000; Du et al., 2001; Power et al., 2003; Sanchez Mejia et al., 2001; Tikka and Koistinaho, 2001; Tikka et al., 2002; Yrjanheikki et al., 1998; 1999). In addition, it can modulate ERK1/2 and p38 MAP kinase activity (Corsaro et al., 2009). As, typically, the microglia response is related to the severity of the injury/cell death response, the anti-apoptotic actions of minocycline limit the ability to conclude a causal relationship between microglia activation and neuronal death. The recent finding that minocycline reduced activation of bone marrow-derived cells but did not alter their phagocytic activity (Malm et al., 2008) suggests that the cells maintain the ability to clear neuronal debris. Thus, in any experimental model that induces neuronal death, attempts to draw conclusions regarding a causation of microglia activation on neuronal death are limited if not impossible. The work by Sriram et al. (2006) provides an excellent example for utilizing minocycline to compare the heterogeneity of the microglia response and the impact on specific neuronal populations. In contrast to the study by Wu et al. (2002), in which minocycline protected against MPTP-induced loss of nigrostriatal dopaminergic neurons, Sriram et al (2006) reported that, minocycline did not offer protection against a lower dose of MPTP or methamphetamine (METH) that induces degeneration of striatal dopaminergic nerve terminals. At these lower doses an elevation in TNFα remained with minocycline treatment. The microglia response at the nerve terminals may represent synapse stripping consistent with a phagocytic activity, which is not altered by minocycline (Malm et al., 2008). The results of this study suggested a dual role for TNFα and possibly microglia with promoting degeneration in the striatum and supporting neuronal survival in the hippocampus (Siriram et al., 2006). This is consistent with earlier work from Rousselet et al., (2001) suggesting that TNFα did not participate in the death of dopaminergic neurons following MPTP but that it slightly altered the survival of dopaminergic terminals by a mechanism requiring both TNF receptors. In the Siriram et al (2006) study, the use of minocycline and the low-dose MPTP model of synaptic damage allowed for a clear distinction to be identified between microglia reaction and TNFα production, versus a neuroinflammatory response involving nitric oxide and superoxide.
TMT murine model of hippocampal damage to identify microglia and TNF- related events
Over the years, trimethyltin (TMT) has been used to produce a model of selective hippocampal damage (review, Harry and Lefebvre d'Hellencourt, 2003). Using this neurotoxicant, a significant amount of research has examined the role of microglia in neurotoxicity (Monnet-Tschudi et al., 1995; Maier et al., 1997; Bruccoleri et al., 1998; Viviani et al., 1998; Bruccoleri and Harry, 2000; Haga et al., 2002; Harry et al., 2002; Little et al., 2002; Eskes et al., 2003; Figiel and Dzwonek, 2007; Mao et al., 2007; Koda et al., 2009). These studies have varied from identifying microglia as a sensitive marker for the neurotoxic effect of the chemical to efforts to identify the mechanism of cell death and neuroprotection.
In vitro studies demonstrated that TMT activates microglia (Rohl et al., 2009) and induces TNFα production in cultured mixed glia (Maier et al., 1997; Viviani et al., 1998; Harry et al., 2002; Eskes et al., 2003), which has been implicated in the in vivo neurotoxicity (Bruccoleri et al., 1998; Fiedorowicz et al, 2001). Interestingly, this injury model has provided some of the first evidence that the induction of TNFα by microglia may directly contribute to the neuropathology (Harry et al., 2008; Bruccoleri and Harry, 2000; Harry et al., 2002; Figiel and Dzwonek, 2007). However, these studies also suggested a dual role for TNFα similar to what has been reported for MPTP (Siriram et al., 2006) and demonstrated heterogeneity of the microglia response. For example, our previous work characterized the temporal and spatial response of microglia and neurons within the hippocampus following an acute systemic injection of TMT. As early as 6 hrs, microglia cells are found in juxtaposition to dentate granule neurons expressing a low-level of active caspase 3 (Lefebvre d'Hellencourt and Harry 2005; Harry et al., 2008). With the progression of dentate granule cell death occurring over the ensuing 72-hrs post-TMT, an increased staining of both ramified and amoeboid microglia was observed (Bruccoleri et al., 1998; Harry et al., 2008a,b). The amoeboid microglia were in contact with apoptotic dentate granule neurons while the hypertrophied, process-bearing microglia maintained contacts with the healthy dentate neurons. By 72 hrs, a peak level of phagocytic microglia cells occurs in the dentate gyrus coinciding with the actual loss of dentate granule neurons, presumably due to phagocytosis and clearance by microglia (Fig. 1). The morphological response corresponds to the transient elevation of TNFα mRNA (Fig. 2) and localized microglia expression of TNFα as determined by in situ hybridization (Bruccoleri et al., 1998; Bruccoleri and Harry, 2000). In addition, mRNA levels for TNF receptors were elevated selectively in the hippocampus (Harry et al., 2008a,b) and that the level and temporal onset of expression was higher and earlier in the dentate granule cell region as compared to the pyramidal cell layer (Lefebvre d'Hellencourt and Harry, 2005; Harry et al., 2008a).
Figure 1.
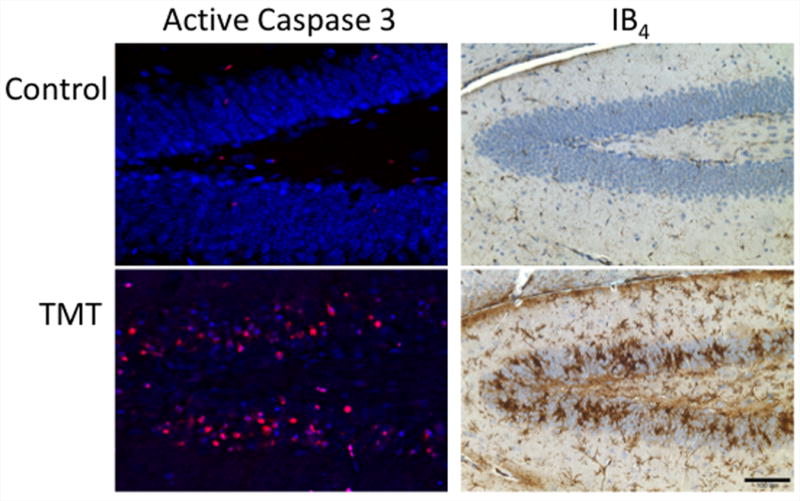
Pathogen free, 23-day-old male mice (CD-1, Charles River; Raleigh, NC) received an intraperitoneal (ip) injection of either 2 mg/kg trimethyltin (TMT) hydroxide (Alfa Products, Danvers MA) or saline (2 ml/kg). Procedures complied with a NIEHS animal care and use committee approved protocol. Rabbit polyclonal anti-active caspase 3 (AC3; 1:1000, 18h, 4°C, #AB3623, Chemicon) was visualized with goat anti-rabbit IgG AF 594 (1:1000, Molecular Probes). Neurons were identified with Neurotrace® blue fluorescent Nissl stain (1:500, 1h, RT; Molecular Probes). Microglia were identified by binding of isolectin B4 (IB4) from Griffonia simplicifolia (Sigma, St. Louis, MO) in 1X Automation Buffer (Biomedia Corp, Foster City, CA) containing 0.1 M CaCl2, MgCl2, MnCl2, and 0.1% Triton X-100 and visualized by 3,3′-diaminobenzidine (DAB) substrate (Harry et al., 2008a,b). AC3 was not evident in the control hippocampus and IB4 staining of microglia in the control brain showed small cells with thin, lightly stained, ramified processes. At 72-hr post-TMT, AC3 staining was prominent within neurons and a severe microglia response was evident characterized by both hypertrophied process bearing microglia and amoeboid microglia within the dentate granule cell layer.
Figure 2.
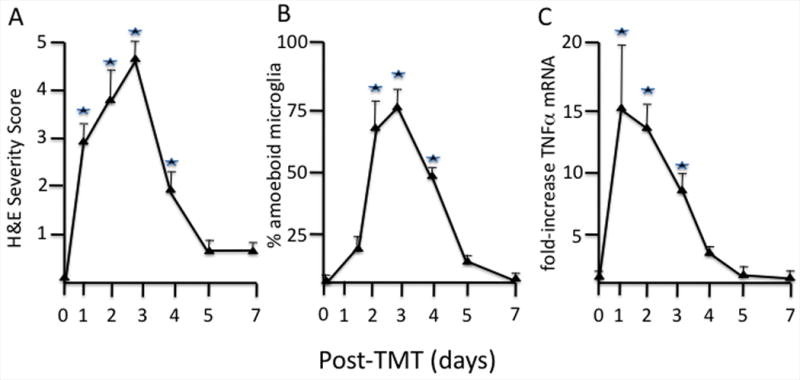
A. The severity of dentate granule cell damage increased over the first 72 hrs post-TMT (2 mg/kg TMT hydroxide ip, PND 23 male CD-1 mice). Scale 1-5 based upon number and location (progressing from inner to outer area of dentate blade) of eosin+ cells (n=10). *p<0.05 Kruskal-Wallis followed by Mann Whitney-U tests for independent group comparisons.
B. The response of IB4 + microglia changed over time with a transient increase in the percent of microglia displaying an amoeboid morphology between 24 and 72 hr (Fig. 2B). These cells were characterized by an increase in cell body size to greater than 5 microns, rounding-up of the cell, and the lack of ramified processes representative of a phagocytic phenotype. *p<0.05 Kruskal-Wallis followed by Mann Whitney-U tests for independent group comparisons
C. mRNA levels for TNFα demonstrated a peak elevation at 1 day preceding the peak response of amoeboid microglia (B). This was followed by a decline at 3 days corresponding to the peak level of amoeboid microglia, and a return to within control levels by 5 days consistent with the downregulation of the morphological response of microglia. Procedures were as previously reported (Harry et al., 2008a,b). From amplification plots, threshold cycle values were determined; fold changes were calculated for TNFp75R transcript level induced by TMT over saline-treated controls using the comparative CT method. GAPDH levels were determined as an internal control.
*p<0.05 ANOVA followed by a Dunnett's t-test
Work examining the osteopetrotic mouse deficient in colony stimulating factor and thus severely deficient in macrophages and microglia, demonstrated that the physical presence of microglia was not the deciding factor in TMT-induced toxicity but that rather the significant elevation in TNFα mRNA by microglia was a major contributing factor (Bruccoleri and Harry, 2000). Further reports demonstrating that neutralizing antibodies to TNFα provide neuroprotection against TMT (Harry et al., 2002; 2003; 2008a) suggest a direct causative effect in neuronal death. Additional work supported the conclusion that the microglia response represented resident brain cells rather than infiltrating cells from the periphery as an inhibition of systemic TNFα by pentoxifylline (Shohami et al., 1996) altered mRNAs associated with the vasculature system but did not provide neuroprotection (Harry et al., 2003). In addition, the systemic administration of ebselen also did not provide neuroprotection (Harry et al., 2003). Ebselen is known to inhibit neutrophil recruitment and activation through an inhibitory effect on TNF, IL-1, and ICAM-1 expression (Haddad et al., 2002). In models of brain injury such as ischemia, ebselen has shown promise as a protective agent due to anti-inflammatory and antioxidant properties (Parnham and Sies, 2000; Imai et al., 2001; Namura et al., 2001). Thus, its ineffectiveness in the TMT acute injury may be because of a lack of infiltration of systemic immune cells, as well as, the absence of a significant free radical activation, as suggested by the absence of elevation in iNOS mRNA levels either in vivo (Bruccoleri et al., 1998; Harry et al., 2003) or isolated microglia (Maier et al., 1997). When neurotoxicity was examined in TNF receptor deficient mice a shift in the dose response was observed (Harry et al. 2008a). Mice deficient for one of the receptors demonstrated a more severe neuropathology while the absence of both receptors provided neuroprotection (Harry et al. 2008a). When the cellular distribution of the receptors was examined by immunohistochemistry, it was found that early in the process of cell death, dentate granule neurons show a transient expression and internalization of TNFp55R followed closely by expression and internalization of TNFp75R. In mice deficient for TNFp55R, active caspase 3 positive dentate granule neurons showed an early internalization of TNFp75R. These data suggested both an interaction between the receptors in apoptosis and the ability of TNFp75R to act in an apoptotic fashion in the absence of TNFp55R. This would be consistent with other data on TNF receptor expression as mentioned earlier but the first time the sequence of events and the relationship to neuronal survival versus death has been described.
Within this model of delayed-neuronal death (2-3 days) of dentate granule neurons, the CA pyramidal neurons are spared. Given the circuitry of the hippocampus, including the trisynaptic circuit of the dentate gyrus, CA3 pyramidal cells, and the CA1 pyramidal cells (Amaral and Witter, 1989), this model allows us to compare microglia responses between the two regions (Fig. 3). In the CA1 region, a pronounced microglia response occurs characterized by hypertrophied process bearing ramified microglia in contact with pyramidal neurons (Bruccoleri et al., 1998; Harry et al., 2008a,b). By in situ hybridization, TNFα transcript was detected in these ramified microglia (Bruccoleri et al., 1998; Bruccoleri and Harry, 2000). Using laser capture microscopy to extract specific hippocampal regions, RNA levels for TNFα and TNF receptors were elevated in the CA region; however, this was lower or delayed as compared to the dentate granule cell region (Lefebvre d'Hellencourt and Harry, 2005; Harry et al., 2008a). Immunohistochemistry for receptor distribution showed that, while expression was elevated, neither receptor showed evidence of internalization as occurred in the dentate granule cells. Diverging consequences of TNFα/TNF receptor-activation can be dependent on subtle differences in stimulus intensity, duration, or the extra/intracellular environment (Shohami et al., 1999). Thus, the effects of any elevation in TNFα vary and may depend on the actual level of ligand produced. The differential response observed between the DG and the CA1 following TMT suggests the possibility of a threshold requirement for TNFα levels to initiate receptor internalization and activation of the cell death pathway. Alternatively, at the lower level of TNFα expression the differential response of microglia cells in the CA layer that may represent trophic factor support for neurons such as insulin like growth factor (Wine et al., 2009).
Figure 3.
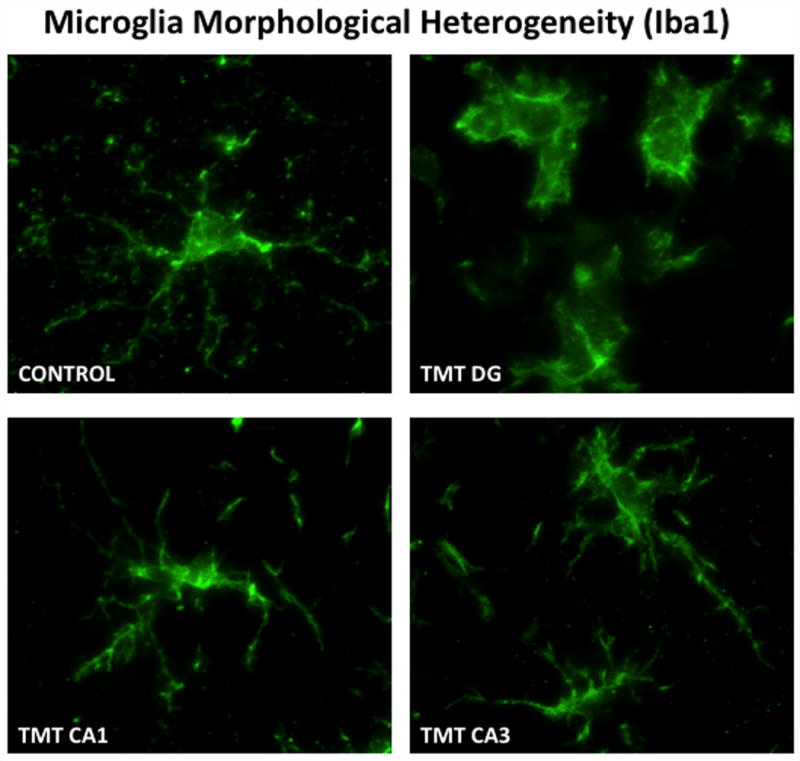
Heterogeneity of microglia responses within various hippocampal regions were detected at 72 hrs post-TMT (2mg/kg) injection. In the hippocampus of normal mice, Iba-1 staining of microglia showed small cells with thin, lightly stained, ramified processes. Within the dentate granule cell layer, microglia displayed a more rounded morphology with retraction of processes consistent with an amoeboid phagocytic phenotype. Within the CA1 and the CA3 pyramidal cell layers, microglia showed hypertrophy of processes with indication of retraction. Microglia were detected by rabbit polyclonal antibody to ionized calcium-binding adaptor molecule 1 (Iba1; 1:500; 1h, RT; Wako Chemicals, Richmond, VA) detected with IgG Alexafluor 488 (1:1000, Molecular Probes). Digital images were acquired using a SpotRT™ cooled, charged-couple device camera (Diagnostic Instruments, Sterling Heights, MI) on a Leica DMRBE microscope (Wetzlar, Germany) equipped with epifluorescence and Z-control and Metamorph™ (Universal Imaging Co., Downingtown, PA). 100x image stacks were acquired, deconvolved and 3D reconstruction by maximum projection shown at 20 degree angle.
Conclusions
From the expanding number of studies on microglia functions, we are beginning to appreciate the diverse nature of these cells. Overall the data suggest that distinct and individual microglia responses depend upon the types of changes in the microenvironment. The heterogeneity of normal microglia across brain regions and in response to injury may preclude any assumption of a common mechanism of action for brain microglia/macrophages; whether neurotoxic or neuroprotective. One of the major current issues with regards to neuroinflammation is its contribution to chronic neurodegenerative diseases. As with any disease, it is very difficult to model a human neurological disease. One can model different features of the disease process such as loss of dopaminergic neurons for this rarely is considered to reflect all aspects of the disease condition. In any long-term exposure or degenerative process, it becomes difficult to identify a causal effect of microglia activation on neuronal death. Many of the acute/short term injury studies are currently providing critical information with regards to the different types of responses of microglia and the temporal sequence of events. One of the interesting comparisons is between the various models that do not involve the infiltration of blood-borne macrophages and thus, the associated high levels of various stimulatory and damaging factors. For example, the low dose MPTP and methamphetamine models used by Siriram et al. (2006) and the trimethyltin model that we have examined in our laboratory demonstrate similar findings of a microglia and TNFα involvement in neuronal injury in the absence of other “inflammatory factors” such as iNOS, SOD, IFNγ. Characterizing the heterogeneity and temporal pattern of morphological and functional changes becomes important in interpreting data on the nature of the microglia response. Temporal and spatial relationships defined in acute or short-term studies will provide a significant database from which to determine critical events and outcomes in more chronic exposures or conditions. In addition, determining neurotoxicity associated with a microglia response and elevations in TNFα in the absence of other classic inflammatory markers such as nitric oxide or infiltrating blood borne cells will significantly contribute, not only to our understanding of the heterogeneity of microglia, but also to identification of critical signaling events leading to neuronal death or survival.
Acknowledgments
This research was funded by the Division of Intramural Research, National Institute of Environmental Health Sciences, National Institutes of Health.
References
- Ajami Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Akassoglou K, Douni E, Bauer J, Lassmann H, Kollias G, Probert L. Exclusive tumor necrosis factor (TNF) signaling by the p75TNF receptor triggers inflammatory ischemia in the CNS of transgenic mice. Proc Natl Acad Sci USA. 2003;100:709–714. doi: 10.1073/pnas.0236046100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DG, Witter MP. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neurosci. 1989;31:571–591. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- Andersson PB, Perry VH, Gordon S. Intracebral injection of proinflammatory cytokines or leucocyte chemotaxins induces minimal myelomonocytic cell recruitment to the parenchyma of the CNS. J Exper Med. 1992;176:255–259. doi: 10.1084/jem.176.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badie B, Schartner J, Vorpahl J, Preston K. Interferon-gamma induces apoptosis and augments the expression of Fas and Fas ligand by microglia in vitro. Exp Neurol. 2000;162:290–296. doi: 10.1006/exnr.1999.7345. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Jones F, Kubota ES, Pocock JM. Stimulation of microglia metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor α-induced neurotoxicity in concert with microglial-derived fas ligand. J Neurosci. 2005;25:2945–2964. doi: 10.1523/JNEUROSCI.4456-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–337. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- Bigda J, Beletsky I, Brakebusch C, Varfolomeev Y, Engelmann H, Bigda J, Holtmann H, Wallach D. Dual role of the p75 tumor necrosis factor (TNF) receptor in TNF cytotoxicity. J Exp Med. 1994;180:445–460. doi: 10.1084/jem.180.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson's disease. Neurosci Lett. 1994;172:151–154. doi: 10.1016/0304-3940(94)90684-x. [DOI] [PubMed] [Google Scholar]
- Botchkina GI, Geimonen E, Bilof ML, Villarreal O, Tracey KJ. Loss of NF-kappaB activity during cerebral ischemia and TNF cytotoxicity. Mol Med. 1999;5:372–381. [PMC free article] [PubMed] [Google Scholar]
- Botchkina GI, Meistrell ME, 3rd, Botchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med. 1997;3:765–781. [PMC free article] [PubMed] [Google Scholar]
- Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119:182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruccoleri A, Harry GJ. Chemical-induced hippocampal neurodegeneration and elevations in TNFα, TNFβ, IL-1β, IP-10, and MCP-1 mRNA in osteopetrotic (op/op) mice. J Neurosci Res. 2000;62:146–155. doi: 10.1002/1097-4547(20001001)62:1<146::AID-JNR15>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Bruccoleri A, Brown HW, Harry GJ. Cellular localization and temporal elevation of tumor necrosis factor- α, interleukin-1α, and transforming growth factor-β1 mRNA in hippocampal injury response induced by trimethyltin. J Neurochem. 1998;71:1577–1587. doi: 10.1046/j.1471-4159.1998.71041577.x. [DOI] [PubMed] [Google Scholar]
- Carson MJ. Microglia as liaisons between the immune and central nervous systems, functional implications for multiple sclerosis. Glia. 2002;40:218–231. doi: 10.1002/glia.10145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Bilousova TV, Puntambekar SS, Melchior B, Doose JM, Ethell IM. A rose by any other name? The potential consequences of microglial heterogeneity during CNS health and disease. Neurotherapeutics. 2007;4:571–579. doi: 10.1016/j.nurt.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Lenardo MJ. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur J Immunol. 2000;30:652–660. doi: 10.1002/1521-4141(200002)30:2<652::AID-IMMU652>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Corsaro A, Thellung S, Chivitti K, Villa V, Simi A, Raggi F, Paludi D, Russo C, Aceto A, Florio T. Dural modulation of ERK1/2 and p38 MAP kinase activities induced by minocycline reverses the neurotoxic effects of the prion protein fragment 90-231. Neurotoxicity Res. 2009;15:102–141. doi: 10.1007/s12640-009-9015-3. [DOI] [PubMed] [Google Scholar]
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6:797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- Cunningham O, Sampion S, Perry VH, Murray C, Sidenius N, Docagne F, Cunningham C. Microglia and the urokinase plasminogen activator receptor/uPA system in innate brain inflammation. Glia. 2009 doi: 10.1002/glia.20892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol. 2003;62:127–136. doi: 10.1093/jnen/62.2.127. [DOI] [PubMed] [Google Scholar]
- Davalos D, Gruntzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Declerecq W, Denecker G, Fiers W, Vandenabeele P. Cooperation of both TNF receptors in inducing apoptosis: Involvement of the TNF receptor-associated factor binding domain of the TNF receptor 75. J Immunol. 1998;161:390–399. [PubMed] [Google Scholar]
- Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997;75:104–112. doi: 10.1016/s0165-5728(97)00009-x. [DOI] [PubMed] [Google Scholar]
- Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc Natl Acad Sci USA. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskes C, Juillerat-Jeanneret L, Leuba G, Honegger P, Monnet-Tschudi F. Involvement of microglia-neuron interactions in the tumor necrosis factor-alpha release, microglial activation, and neurodegeneration induced by trimethyltin. J Neurosci Res. 2003;71:583–590. doi: 10.1002/jnr.10508. [DOI] [PubMed] [Google Scholar]
- Fiedorowicz A, Figiel I, Kaminska B, Zaremba M, Wilk S, Oderfeld-Nowak B. Dentate granule neuron apoptosis and glia activation in murine hippocampus induced by trimethyltin exposure. Brain Res. 2001;912:116–127. doi: 10.1016/s0006-8993(01)02675-0. [DOI] [PubMed] [Google Scholar]
- Fiers W, Beyaert R, Boone E, Cornelis S, Declercq W, Decoster E, Denecker G, Depuydt B, De Valck D, De Wilde G, Goossens V, Grooten J, Haegeman G, Heyninck K, Penning L, Plaisance S, Vancompernolle K, Van Criekinge W, Vandenabeele P, Vanden Berghe W, Van de Craen M, Vandevoorde V, Vercammen D. TNF-induced intracellular signaling leading to gene induction or to cytotoxicity by necrosis or by apoptosis. J Inflamm. 1995-1996;47:67–75. [PubMed] [Google Scholar]
- Figiel I, Dzwonek K. TNFalpha and TNF receptor 1 expression in the mixed neuronal-glial cultures of hippocampal dentate gyrus exposed to glutamate or trimethyltin. Brain Res. 2007;1131:17–28. doi: 10.1016/j.brainres.2006.10.095. [DOI] [PubMed] [Google Scholar]
- Flaris NA, Densmore TL, Molleston mC, Hickey WF. Characterization of microglia and macrophages in the central nervous system of rats: Definition of the differential expression of molecules using standard and novel monoclonal antibodies in normal CNS and in four models of parenchymal reaction. Glia. 1993;7:34–40. doi: 10.1002/glia.440070108. [DOI] [PubMed] [Google Scholar]
- Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, Scheurich P, Schmid JA, Wajant H. Apoptotic crosstalk of TNF receptors: TNF-R2 induces depletion of TRAF2 and IAP proteins and accelerated TNF-R1-dependent activation of caspase 8. J Cell Sci. 2002;115:2757–2770. doi: 10.1242/jcs.115.13.2757. [DOI] [PubMed] [Google Scholar]
- Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Gebicke-Haerter PJ, Van Calker D, Nörenberg W, Illes P. Molecular mechanisms of microglial activation. A. Implications for regeneration and neurodegenerative diseases. Neurochem International. 1996;29:1–12. [PubMed] [Google Scholar]
- Gehrmann J, Mies G, Bonnekoh P, Banati R, Iijima T, Kreutzberg GW, Hossmann KA. Microglial reaction in the rat cerebral cortex induced by cortical spreading depression. Brain Pathol. 1993;3:11–18. doi: 10.1111/j.1750-3639.1993.tb00720.x. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia, intrinsic immuneffector cell of the brain. Brain Res Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Folks DG. Innate and acquired immunity in neurodegenerative disorders. J Leukoc Biol. 1999;65:407–408. doi: 10.1002/jlb.65.4.407. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- Gregersen R, Lambertsen K, Finsen B. Microglia and macrophages are the major source of tumor necrosis factor in permanent middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2000;20:53–65. doi: 10.1097/00004647-200001000-00009. [DOI] [PubMed] [Google Scholar]
- Grell M, Scheurich P, Meager A, Pfizenmaier K. TR60 and TR80 tumor necrosis factor (TNF)-receptors can independently mediate cytolysis. Lymphokine Cytokine Res. 1993;12:143–148. [PubMed] [Google Scholar]
- Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- Grell M, Zimmermann G, Gottfried E, Chen CM, Grünwald U, Huang DC, Wu Lee YH, Dürkop H, Engelmann H, Scheurich P, Wajant H, Strasser A. Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J. 1999;18:3034–3043. doi: 10.1093/emboj/18.11.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci USA. 1998;95:570–575. doi: 10.1073/pnas.95.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad ElB, Mccluskie K, Birrell MA, Dabrowski D, Pecoraro M, Underwood S, Chen B, DeSanctis GT, Webber SE, Foster ML, Belvisi MG. Differential effects of ebselen on neutrophil recruitment, chemokine, and inflammatory mediator expression in a rat model of lipopolysaccharide-induced pulmonary inflammation. J Immunol. 2002;169:974–982. doi: 10.4049/jimmunol.169.2.974. [DOI] [PubMed] [Google Scholar]
- Haga S, Haga C, Aizawa T, Ikeda K. Neuronal degeneration and glial cell-responses following trimethyltin intoxication in the rat. Acta Neuropathol. 2002;103:575–582. doi: 10.1007/s00401-001-0505-5. [DOI] [PubMed] [Google Scholar]
- Hanisch UW. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Lefebvre d'Hellencourt C. Dentate Gyrus: Alterations that occur with hippocampal injury. Neurotoxicology. 2003;24:343–356. doi: 10.1016/S0161-813X(03)00039-1. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Bruccoleri A, Lefebvre d'Hellencourt C. Differential modulation of hippocampal chemical-induced injury response by ebselen, pentoxifylline, and TNFalpha-, IL-1alpha-, and IL-6-neutralizing antibodies. J Neurosci Res. 2003;73:526–536. doi: 10.1002/jnr.10653. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Lefebvre d'Hellencourt C, McPherson CA, Funk JA, Aoyama M, Wine RN. Tumor necrosis factor p55 and p75 receptors are involved in chemical-induced apoptosis of dentate granule neurons. J Neurochem. 2008a;106:281–298. doi: 10.1111/j.1471-4159.2008.05382.x. [DOI] [PubMed] [Google Scholar]
- Harry GJ, Funk JA, Lefebvre d'Hellencourt C, McPherson CA, Aoyama M. The type 1 interleukin 1 receptor is not required for the death of murine hippocampal dentate granule cells and microglia activation. Brain Res. 2008b;1194:8–20. doi: 10.1016/j.brainres.2007.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry GJ, Tyler K, Lefebvre d'Hellencourt C, Tilson HA, Maier WE. Morphological alterations and elevations in tumor necrosis factor-alpha, interleukin (IL)-1alpha, and IL-6 in mixed glia cultures following exposure to trimethyltin, modulation by proinflammatory cytokine recombinant proteins and neutralizing antibodies. Toxicol Appl Pharmacol. 2002;180:205–218. doi: 10.1006/taap.2002.9390. [DOI] [PubMed] [Google Scholar]
- Hanisch UW. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Simon RP. Epilepsy and apoptosis pathways. J Cereb Blood Flow Metab. 2005;25:1557–1572. doi: 10.1038/sj.jcbfm.9600149. [DOI] [PubMed] [Google Scholar]
- Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- Imai H, Masayasu H, Dewar D, Graham DI, Macrae IM. Ebselen protects both gray and white matter in a rodent model of focal cerebral ischemia. Stroke. 2001;32:2149–2154. doi: 10.1161/hs0901.095725. [DOI] [PubMed] [Google Scholar]
- Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- Kimoto H, Eto R, Abe M, Kato H, Araki T. Alterations of glial cells in the mouse hippocampus during postnatal development. Cell Mol Neurobiol. 2009 doi: 10.1007/s10571-009-9412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinouchi K, Brown G, Pasternak G, Donner DB. Identification and characterization of receptors for tumor necrosis factor-alpha in the brain. Biochem Biophys Res Commun. 1991;181:1532–1538. doi: 10.1016/0006-291x(91)92113-x. [DOI] [PubMed] [Google Scholar]
- Koda T, Kuroda Y, Imai H. Rutin Supplementation in the Diet has Protective Effects Against Toxicant-Induced Hippocampal Injury by Suppression of Microglial Activation and Pro-Inflammatory Cytokines: Protective Effect of Rutin Against Toxicant-Induced Hippocampal Injury. Cell Mol Neurobiol. 2009 doi: 10.1007/s10571-008-9344-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer PH. CD95's deadly mission in the immune system. Nature. 2000;407:789–795. doi: 10.1038/35037728. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia, a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–328. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, Haugaard LS, Wirenfeldt M, Nielsen M, Dagnaes-Hansen F, Bluethmann H, Faergeman NJ, Meldgaard M, Deierborg T, Finsen B. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci. 2009;29:1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Clausen BH, Fenger C, Wulf H, Owens T, Dagnaes-Hansen F, Meldgaard M, Finsen B. Microglia and macrophages express tumor necrosis factor receptor p75 following middle cerebral artery occlusion in mice. Neurosci. 2007;144:934–949. doi: 10.1016/j.neuroscience.2006.10.046. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Schmied M, Vass K, Hickey WF. Bone marrow derived elements and resident microglia in brain inflammation. Glia. 1993;7:19–24. doi: 10.1002/glia.440070106. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neurosci. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Lefebvre d'Hellencourt C, Harry GJ. Molecular profiles of mRNA levels in laser capture microdissected murine hippocampal regions differentially responsive to TMT-induced cell death. J Neurochem. 2005;93:206–220. doi: 10.1111/j.1471-4159.2004.03017.x. [DOI] [PubMed] [Google Scholar]
- Little AR, Benkovic SA, Miller DB, O'Callaghan JP. Chemically induced neuronal damage and gliosis, enhanced expression of the proinflammatory chemokine, monocyte chemoattractant protein (MCP)-1, without a corresponding increase in proinflammatory cytokines. Neurosci. 2002;115:307–320. doi: 10.1016/s0306-4522(02)00359-7. [DOI] [PubMed] [Google Scholar]
- Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- Lu MO, Duan RS, Quezada HC, Chen ZG, Mix E, Jin T, Yang X, Ljunggren HG, Zhu J. Aggravation of experimental autoimmune neuritis in TNF-a receptor 1 deficient mice. J Neuroimmunol. 2007;186:19–26. doi: 10.1016/j.jneuroim.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Maier WE, Bartenbach MJ, Brown HA, Tilson HA, Harry GJ. Induction of tumor necrosis factor alpha in cultured glial cells by trimethyltin. Neurochem Int'l. 1997;30:385–392. doi: 10.1016/s0197-0186(96)00073-3. [DOI] [PubMed] [Google Scholar]
- Malm TM, Magga J, Kuh GF, Vatanen T, Koistinaho M, Koistinaho J. Minocycline reduces engraftment and activation of bone marrow-derived cells but sustains their phagocytic activity in a mouse model of Alzheimer's disease. Glia. 2008;56:1767–1779. doi: 10.1002/glia.20726. [DOI] [PubMed] [Google Scholar]
- Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, Krammer PH. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Diff. 2001;8:679–686. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- Mao H, Fang X, Floyd KM, Polcz JE, Zhang P, Liu B. Differential activation of microglia and astrocytes following trimethyl tin-induced neurodegeneration. Brain Res. 2007;1186:267–274. [Google Scholar]
- Matsumoto Y, Fujiwara M. Absence of donor-type major histocompatibility complex class I antigen bearing microglia in the rat central nervous system of radiation bone marrow chimeras. J Neuroimmunol. 1987;17:71–82. doi: 10.1016/0165-5728(87)90032-4. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Yasojima K, McGeer EG. Inflammation in Parkinson's disease. Adv Neurol. 2001;86:83–89. [PubMed] [Google Scholar]
- Medvedev AE, Espevik T, Ranges G, Sundan A. Distinct roles of the two tumor necrosis factor (TNF) receptors in modulating TNF and lymphotoxin alpha effects. J Biol Chem. 1996a;271:9778–9784. doi: 10.1074/jbc.271.16.9778. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Laegreid A, Sundan A, Espevik T. A non-competitive P55 TNF receptor antibody enhances the specific activity of lymphotoxin-alpha. Scand J Immunol. 1996b;43:439–448. doi: 10.1046/j.1365-3083.1996.d01-58.x. [DOI] [PubMed] [Google Scholar]
- Medvedev AE, Sundan A, Espevik T. Involvement of the tumor necrosis factor receptor p75 in mediating cytotoxicity and gene regulating activities. Eur J Immunol. 1994;24:2842–2849. doi: 10.1002/eji.1830241139. [DOI] [PubMed] [Google Scholar]
- Monnet-Tschudi F, Zurich MG, Pithon E, van Melle G, Honegger P. Microglial responsiveness as a sensitive marker for trimethyltin (TMT) neurotoxicity. Brain Res. 1995;690:8–14. doi: 10.1016/0006-8993(95)00509-o. [DOI] [PubMed] [Google Scholar]
- Micheau O, Tschopp J. Induction of TNF receptor 1-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- Namura S, Nagata I, Takami S, Masayasu H, Kikuchi H. Ebselen reduces cytochrome c release from mitochondria and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Stroke. 2001;32:1906–1911. doi: 10.1161/01.str.32.8.1906. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Parnham M, Sies H. Ebeselen: prospective therapy for cerebral ischaemia. Expert Opin Investig Drugs. 2000;9:607–619. doi: 10.1517/13543784.9.3.607. [DOI] [PubMed] [Google Scholar]
- Perry VH, Andersson PB, Gordon S. Macrophages and inflammation in the central nervous system. Trends Neurosci. 1993;16:268–273. doi: 10.1016/0166-2236(93)90180-t. [DOI] [PubMed] [Google Scholar]
- Power C, Henry S, Del Bigio MR, Larsen PH, Corbett D, Imai Y, Yong VW, Peeling J. Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Ann Neurol. 2003;53:731–742. doi: 10.1002/ana.10553. [DOI] [PubMed] [Google Scholar]
- Orr CF, Rowe DB, Halliday GM. An inflammatory review of Parkinson's Disease. Prog Neurobiol. 2002;68:325–340. doi: 10.1016/s0301-0082(02)00127-2. [DOI] [PubMed] [Google Scholar]
- Quintana A, Giralt M, Rojas S, Penkowa M, Campbell IL, Hidalgo J, Molinero A. Differential role of tumor necrosis factor receptors in mouse brain inflammatory responses in cryolesion brain injury. J Neurosci Res. 2005;82:701–716. doi: 10.1002/jnr.20680. [DOI] [PubMed] [Google Scholar]
- Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005;28:571–573. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Raivich G, Liu ZQ, Kloss CU, Labow M, Bluethmann H, Bohatschek M. Cytotoxic potential of proinflammatory cytokines: combined deletion of TNF receptors TNFR1 and TNFR2 prevents motoneuron cell death after facial axotomy in adult mouse. Exp Neurol. 2002;178:186–193. doi: 10.1006/exnr.2002.8024. [DOI] [PubMed] [Google Scholar]
- Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- Rohl C, Grell M, Maser E. The organotin compounds trimethyltin (TMT) and triethyltin (TET) but not tributyltin (TBT) induce activation of microglia co-cultivated with astrocytes. Toxicol In Vitro. 2009 doi: 10.1016/j.tiv.2009.04.013. [DOI] [PubMed] [Google Scholar]
- Rousselet E, Callebert J, Parain K, Joubert C, Hunot S, Hartmann A, Jacque C, Perez-Diaz F, Cohen-Salmon C, Launay JM, Hirsch EC. Role of TNF-alpha receptors in mice intoxicated with the parkinsonian toxin MPTP. Exp Neurol. 2002;177:183–192. doi: 10.1006/exnr.2002.7960. [DOI] [PubMed] [Google Scholar]
- Sairanen TR, Lindsberg PJ, Brenner M, Carpén O, Sirén A. Differential cellular expression of tumor necrosis factor-alpha and Type I tumor necrosis factor receptor after transient global forebrain ischemia. J Neurol Sci. 2006;186:87–99. doi: 10.1016/s0022-510x(01)00508-1. [DOI] [PubMed] [Google Scholar]
- Saito K, Suyama K, Nishida K, Sei Y, Basile AS. Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci Lett. 1996;206:149–152. doi: 10.1016/s0304-3940(96)12460-5. [DOI] [PubMed] [Google Scholar]
- Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, Nakano H. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48:1393–1399. doi: 10.1097/00006123-200106000-00051. [DOI] [PubMed] [Google Scholar]
- Sawada M, Imamura K, Nagatsu T. Role of cytokines in inflammatory process in Parkinson's disease. J Neural Transm Suppl. 2006;70:373–381. doi: 10.1007/978-3-211-45295-0_57. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Butovsky O, Brück W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. 2006;29:68–74. doi: 10.1016/j.tins.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, Mentlein R, Kabelitz D, Schutze S. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity. 2004;21:415–428. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Li R, Shiosaki K. Inhibition of p75 tumor necrosis factor receptor by antisense oligonucleotides increases hypoxic injury and beta-amyloid toxicity in human neuronal cell line. J Biol Chem. 1997;272:3550–3553. [PubMed] [Google Scholar]
- Shohami E, Ginis I, Hallenbeck JM. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 1999;10:119–130. doi: 10.1016/s1359-6101(99)00008-8. [DOI] [PubMed] [Google Scholar]
- Shohami E, Bass R, Wallach D, Yamin A, Gallily R. Inhibition of tumor necrosis factor (TNF) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab. 1996;16:378–384. doi: 10.1097/00004647-199605000-00004. [DOI] [PubMed] [Google Scholar]
- Sriram K, Miller DB, O'Callaghan JP. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J Neurochem. 2006;96:706–718. doi: 10.1111/j.1471-4159.2005.03566.x. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglial senescence: does the brain's immune system have an expiration date? Trends Neurosci. 2006;29:506–510. doi: 10.1016/j.tins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Kreutzberg GW. Response of endogenous glial cells to motor neurodegeneration induced by toxic ricin. J Comp Neurol. 1988;268:248–263. doi: 10.1002/cne.902680209. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflam. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PG, Bruce-Keller AJ, Rabchevsky AG, Christakos S, Clair DK, Mattson MP, Scheff SW. Exacerbation of damage and altered NF-kappaB activation in mice lacking tumor necrosis factor receptors after traumatic brain injury. J Neurosci. 1999;19:6248–6256. doi: 10.1523/JNEUROSCI.19-15-06248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvannavejh GC, Lee HO, Padilla J, Dal Canto MC, Barrett TA, Miller SD. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35-55)-induced experimental autoimmune encephalomyelitis. Cell Immunol. 2000;205:24–33. doi: 10.1006/cimm.2000.1706. [DOI] [PubMed] [Google Scholar]
- Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13:151–153. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Jones F, Kubota ES, Pocock JM. Stimulation of microglia metabotropicglutamate receptor mGlu2 triggers tumor necrosis factor α-induced neurotoxicity in concert with microglial-derived fas ligand. J Neurosci. 2005;25:2945–2964. doi: 10.1523/JNEUROSCI.4456-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchelingerian JL, Le Saux F, Jacque C. Identification and topography of neuronal cell populations expressing TNFα and IL-1α in response to hippocampal lesion. J Neurosci Res. 1996;43:99–106. doi: 10.1002/jnr.490430113. [DOI] [PubMed] [Google Scholar]
- Thomas WE. Brain microphages: evaluation of microglia and their functions. Brain Res Brain Res Rev. 1992;17:61–74. doi: 10.1016/0165-0173(92)90007-9. [DOI] [PubMed] [Google Scholar]
- Thorburn A. Death receptor-induced cell killing. Cell Signal. 2004;16:139–144. doi: 10.1016/j.cellsig.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166:7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- Tikka TM, Vartiainen NE, Goldsteins G, Oja SS, Andersen PM, Marklund SL, Koistinaho J. Minocycline prevents neurotoxicity induced by cerebrospinal fluid from patients with motor neurone disease. Brain. 2002;125(Part 4):722–731. doi: 10.1093/brain/awf068. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 1995;5:392–399. doi: 10.1016/s0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- Viviani B, Corsini E, Galli CL, Marinovich M. Glia increase degeneration of hippocampal neurons through release of tumor necrosis factor-alpha. Toxicol Appl Pharmacol. 1998;150:271–276. doi: 10.1006/taap.1998.8406. [DOI] [PubMed] [Google Scholar]
- Wajant H, Grell M, Scheurich P. TNF receptor associated factors in cytokine signaling. Cytokine Growth Factor Rev. 1999;10:15–26. doi: 10.1016/s1359-6101(98)00023-9. [DOI] [PubMed] [Google Scholar]
- Wajant H, Henkler F, Scheurich P. The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. 2001;13:389–400. doi: 10.1016/s0898-6568(01)00160-7. [DOI] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- Weishaupt A, Gold R, Hartung T, Gaupp S, Wendel A, Brück W, Toyka KV. Role of TNF-alpha in high-dose antigen therapy in experimental autoimmune neuritis: inhibition of TNF-alpha by neutralizing antibodies reduces T-cell apoptosis and prevents liver necrosis. J Neuropathol Exp Neurol. 2000;59:368–376. doi: 10.1093/jnen/59.5.368. [DOI] [PubMed] [Google Scholar]
- Weiss T, Grell M, Hessabi B, Bourteele S, Müller G, Scheurich P, Wajant H. Enhancement of the TNF receptor p60-mediated cytotoxicity by TNF receptor p80. Requirement of the TNF receptor-associated factor-2 binding site. J Immunol. 1997;158:2398–2404. [PubMed] [Google Scholar]
- Whitton PS. Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol. 2007;150:963–976. doi: 10.1038/sj.bjp.0707167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine RN, McPherson CA, Harry GJ. IGF-1 and pAKT signaling promote hippocampal CA1 neuronal survival following injury to dentate granule cells. Neurotox Res. 2009 doi: 10.1007/s12640-009-9060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22:3025–3032. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, Liao W, Chen Z, Liu Z, Su B. The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. 2001 Jul;2:620–624. doi: 10.1038/89769. [DOI] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Tikka T, Keinanen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci USA. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]