

Abstract
Regulator of G protein signaling (RGS) proteins are united into a family by the presence of the homologous RGS domain that binds the α subunits of heterotrimeric G proteins and accelerates their GTPase activity. A member of this family, RGS3 regulates the signaling mediated by Gq and Gi proteins by binding the corresponding Gα subunits. Here we show that RGS3 interacts with the novel partners Smad2, Smad3, and Smad4—the transcription factors that are activated through a transforming growth factor-β (TGF-β) receptor signaling. This interaction is mediated by the region of RGS3 outside of the RGS domain and by Smad’s Mad homology 2 domain. Over-expression of RGS3 results in inhibition of Smad-mediated gene transcription. RGS3 does not affect TGF-β-induced Smad phosphorylation, but it prevents heteromerization of Smad3 with Smad4, which is required for transcriptional activity of Smads. This translates to functional inhibition of TGF-β-induced myofibroblast differentiation by RGS3. In conclusion, this study identifies a novel, noncanonical role of RGS3 in regulation of TGF-β signaling through its interaction with Smads and interfering with Smad heteromerization.
Regulator of G protein signaling RGS3 is a GTPase-activating protein for Gα subunits of heterotrimeric G proteins, which regulates the signaling of Gq- and Gi-coupled receptors for interleukin-8 (Druey et al., 1996; Bowman et al., 1998), endothelin-1 (Dulin et al., 1999; Cho et al., 2003), gonadotropin-releasing hormone (Neill et al., 2001), carbachol (Wang et al., 2002), angiotensin II, and sphingosine-1 phosphate (Cho et al., 2003) in various cellular models. Several isoforms of RGS3 result from an alternative splicing and/or transcription from alternate promoters (Chatterjee et al., 1997; Kehrl et al., 2002). All RGS3 isoforms contain an identical RGS domain that functions as a GTPase-activating protein and an N-terminal region of various lengths, the functions of which are poorly understood. This study focuses on the originally described (519 residues) isoform of RGS3, of which the 380-to-519 region contains the RGS domain (Druey et al., 1996). We and others have previously described the following functions of the non-RGS domain of RGS3: 1) the 1-to-379 region may mediate the calcium-dependent recruitment of RGS3 to the membrane (Dulin et al., 1999), probably through direct calcium binding by the EF hand (the 221–233 region) (Tosetti et al., 2003); 2) the 314-to-379 region may mediate the nuclear localization (Dulin et al., 2000); whereas 3) serine267 provides the interaction of RGS3 with 14-3-3, which controls the regulatory function of RGS3 (Niu et al., 2002; Ward and Milligan, 2005).
Transforming growth factor-β (TGF-β) is a multifunctional cytokine that controls growth, survival, and the phenotype of many cells. The TGF-β signaling is largely mediated by activation of Smads (Moustakas et al., 2001; Feng and Derynck, 2005). This includes phosphorylation of the “receptor-activated” R-Smads (Smad2/3/5/8) by TGF-β receptor family, heteromerization of R-Smads with “common-mediator” CoSmad (Smad4), their accumulation in the nucleus, and activation of specific gene transcription in cooperation with a variety of other coactivators. A high-throughput screening of the components of TGF-β signaling for the interacting partners has identified novel links of the TGF-β pathway to the p21-activated protein kinase, to the polarity complex, and to occludin, a component of tight junctions (Barrios-Rodiles et al., 2005). It is interesting that this screening also predicted the interaction between RGS3 and Smads. Therefore, we sought to examine whether RGS3 interacts with Smads and, if so, the molecular nature and functional significance of this interaction.
Materials and Methods
Cell culture, DNA Transfection and Adenovirus-Mediated Gene Transduction
Chinese hamster ovary (CHO) cells were maintained in Ham’s F12 medium supplemented with 2 mM glutamine, 100 U/ml streptomycin, 100 U/ml penicillin, and 10% fetal bovine serum. The cells were serum-deprived for 24 h in the medium containing 0.1% bovine serum albumin and 2 mM L-glutamine. Primary cultured human pulmonary fibroblasts were isolated from the explanted lungs from patients undergoing lung transplantation for pulmonary fibrosis. Alveolated lung tissue was placed in DMEM and sectioned into ~1-mm3 pieces, washed several times with DMEM, and placed onto 10-cm plates in DMEM with 10% FBS and antibiotics. Expanded populations of fibroblasts were subsequently subcultured after 4 to 5 days, resulting in the development of a homogenous fibroblast population. Transient DNA transfections were performed using LipofectAMINE Plus reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s standard protocol. Adenovirus-mediated gene transduction was performed by incubating cells with desired adenoviruses (100 plaque-forming units per cell) in the medium containing 0.1% bovine serum albumin.
DNA and Reagents
The original cDNA for human RGS3 was provided by Dr. John Kehrl (National Institute of Allergy and Infectious Diseases, Bethesda, MD) (Druey et al., 1996) and was subcloned into either Myc-tag vector (pCMV-tag3B; Stratagene, La Jolla, CA) or Flag-tag vector (pCMV-tag2B; Stratagene) as described previously (Dulin et al., 2000). The cDNAs for Smad proteins were provided by Dr. Liliana Attisano (University of Toronto, Toronto, ON, Canada). The cDNA for type A endothelin receptor was provided by Dr. Masashi Yanagisawa (University of Texas Southwestern Medical Center, Dallas, TX). The plasmid for luciferase reporter driven by four copies of Smad binding elements (SBE4-Luc) was provided by Dr. Bert Vogelstein (The Johns Hopkins University School of Medicine, Baltimore, MD). Recombination-deficient adenovirus encoding green fluorescent protein (AdGFP) was from Vector Biolabs (Philadelphia, PA). Adenovirus encoding RGS3 cDNA was constructed as described previously (Taurin et al., 2007). TGF-β and endothelin-1 (ET1) were from EMD Biosciences (San Diego, CA). Antibodies against Smad4 were from Santa Cruz Biotechnology. Antibodies against Flag or Myc were from Sigma. Antibodies against phosphorylated Smad2 (Ser465/467) and against Smad3 were from Cell Signaling Technology (Danvers, MA). Antibodies against RGS3 were described previously (Dulin et al., 1999).
Immunoprecipitation and Western Blotting
Cells were transfected with cDNAs for desired proteins tagged with either Flag or Myc epitopes. Transfected cells were lysed in an immunoprecipitation (IP) buffer containing 25 mM HEPES, pH 7.5, 150 mM NaCl, 0.5% Nonidet P-40, 1 mM dithiothreitol and protease inhibitors (1 μg/ml leupeptin, 1 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride). The lysates were cleared from insoluble material by centrifugation at 20,000g for 10 min and incubated with agarose-conjugated anti-Flag antibodies for 2 h at 4°C on rotator, followed by three washes with 1 ml of the same buffer. The immune complexes were boiled in Laemmli buffer for 5 min, subjected to electrophoresis, and analyzed by Western blotting with desired primary antibodies, followed by horseradish peroxidase-conjugated secondary antibodies (Calbiochem, San Diego, CA), and developed by enhanced chemiluminescence reaction.
SBE-Luciferase Reporter Assay
CHO cells grown in 24-well plates were cotransfected with 20 ng/well SBE luciferase reporter plasmid, 5 ng/well thymidine kinase promoter (TK)-driven Renilla reniformis luciferase plasmid (Promega) and 50 to 100 ng/well empty vector or cDNA for a desired protein. Cells were serum starved overnight after transfection, stimulated with 2 ng/ml TGF-β for 24 h, washed with phosphate-buffered saline, and lysed in protein extraction reagent. The lysates were assayed for firefly and R. reniformis luciferase activity using the Promega Dual luciferase assay kit (Promega, Madison, WI). To account for differences in transfection efficiency, firefly luciferase activity of each sample was normalized to R. reniformis luciferase activity.
Trans-Luciferase assay for Elk-1 Activation
Endothelin-1 (ET1)-induced activation of Elk-1 was assessed by “PathDetect” trans-reporter system (Stratagene). In brief, cells grown on 24-well plates were transfected with the following plasmids (per): 20 ng/well pFR-Luciferase (reporter plasmid), 1 ng/well pFA2-Elk-1 (fusion trans-activator plasmid), 5 ng/well TK-driven R. reniformis luciferase plasmid (transfection efficiency control), 20 ng/well endothelin receptor cDNA, and 50 to 100 ng/well empty vector or cDNA for a desired protein. Cells were serum-starved overnight after transfection and stimulated with ET-1 for 6 h. The dual luciferase assay was then performed as described above.
Statistical Analysis
All the data represent the results of at least three independent experiments. Quantitative data were analyzed by the Student’s t test, and values of p < 0.05 were considered statistically significant.
Results
Interaction between RGS3 and Smads
To examine the interaction between RGS3 and R-Smad proteins, we first cotransfected CHO cells with Flag-tagged Smad2 or Smad3, together with Myc-tagged RGS3 cDNAs. Immunoprecipitation of Smad2/3 with Flag antibodies followed by Western blotting with Myc antibodies revealed that RGS3 is in the complex with Smad2 or Smad3 (Fig. 1A, left). Immunoprecipitation from cells transfected with empty Flag-vector served as a specificity control. We then performed the similar experiments in a reverse way and examined the interaction of RGS3 with a coSmad, Smad4. Figure 1A, right, shows that Myc-Smad4 readily coimmunoprecipitates with Flag-RGS3. Finally, we assessed the interaction between endogenous RGS3 and Smads, using an EL4 T-thymoma cell line that expresses high levels of endogenous RGS3. Immunoprecipitation of RGS3 from EL4 cell lysates followed by Western blotting with isoform-specific Smad antibodies revealed the interaction between RGS3 and Smad3 or Smad4, respectively (Fig. 1B). Immunoprecipitation using “normal” IgG served as a specificity control. Together, these data demonstrate the interaction between RGS3 and Smads at both overexpressed and endogenous levels of expression.
Fig. 1.

Interaction between RGS3 and Smad proteins. A, CHO cells were transfected with cDNAs for desired Flag-tagged or Myc-tagged proteins, followed by immunoprecipitation (IP) with Flag antibodies and Western blotting (WB) with Myc or Flag antibodies as indicated. B, EL4 cells (50 million cells per condition) were lysed and subjected to immunoprecipitation with “normal” IgG or with antibodies against RGS3. The immune complexes or the original lysates were analyzed by Western blotting with antibodies against RGS3, Smad3 or Smad4 as indicated.
Smad proteins contain two Mad homology (MH) domains (MH1 and MH2) separated by a nonconserved linker region—all serving the designated functions (Feng and Derynck, 2005). To examine which region of Smad proteins mediates the interaction with RGS3, we first generated the MH1+L and L+MH2 fragments of Smad3 tagged with Flag epitope, and examined their interaction with Myc-RGS3. As shown in Fig. 2, left, MH1+L fails to bind RGS3, whereas L+MH2 binding of RGS3 is equal to that of the full-length Smad3. This suggested that MH2 domain mediates the interaction of Smads with RGS3. We then confirmed this by coimmunoprecipitation of Flag-MH2 fragment of Smad3 with Myc-RGS3 (Fig. 2, right). This demonstrates that Smad proteins (Smad3 in our experiments) interact with RGS3 through the MH2 domain.
Fig. 2.

MH2 domain mediates the interaction of Smad3 with RGS3. Cells were transfected with cDNAs for Myc-RGS3 and Flag-tagged full-length Smad3 (FL), the 1- to-225 fragment of Smad3 MH1+L, the 133-to-426 fragment of Smad3 L+MH2, or the 226-to-426 fragment of Smad3 (MH2). Cell lysates were immunoprecipitated (IP) with Flag antibodies followed by Western blotting (WB) with Myc or Flag antibodies as indicated.
RGS3 contains an RGS domain (interacting with G proteins) and a large N-terminal region with no homology to other proteins. To examine which region of RGS3 interacts with Smad proteins, we generated Flag-tagged truncation mutants of RGS3 and examined their ability to bind Myc-tagged Smad3. Figure 3 shows that the full-length RGS3 and the 240–519 deletion mutant of RGS3 bind Smad3 equally well. In contrast, the (379–519) mutant representing the RGS domain of RGS3, which is sufficient for binding G proteins (Dulin et al., 2000), does not interact with Smad3. This suggests that 1) the Smad binding site of RGS3 is located within the 240–379 region but not within the RGS domain of RGS3, and 2) the RGS3-Smad interaction may be functionally unrelated to the regulation of G protein signaling by RGS3.
Fig. 3.

Mapping the Smad-binding region of RGS3. Cells were transfected with cDNAs for Myc-Smad3 and Flag-tagged full-length RGS3 (FL), the 240-to-519 fragment of RGS3, or the 379-to-519 fragment of RGS3. Cell lysates were immunoprecipitated (IP) with Flag antibodies followed by Western blotting (WB) with Myc or Flag antibodies as indicated.
Regulation of Smad-Mediated Gene Transcription by RGS3
We then examined how the interaction between RGS3 and Smads affects the function of RGS3. Consistent with our previous results (Dulin et al., 2000; Niu et al., 2002), overexpression of RGS3 dose-dependently attenuated ET1-induced (G protein-mediated) activation of Elk1-driven luciferase reporter (Fig. 4A), but had little or no effect on the activity of constitutive TK promoter activity (data not shown) or of serum response factor (Taurin et al., 2007). Cotransfection of Smad3 cDNA at concentrations of up to 100-fold higher than that of RGS3 had no significant effect on the ability of RGS3 to regulate the signaling of ET1 (Fig. 4A). This is consistent with our RGS3-Smad binding data (Fig. 3), which show that Smad3 does not interact with the RGS domain of RGS3 that is responsible for regulation of G protein signaling by RGS3 (Dulin et al., 2000).
Fig. 4.

Inhibition of Smad-mediated gene transcription by RGS3. A, Smad3 overexpression had no effect on the regulation of endothelin signaling by RGS3. CHO cells were transfected with Elk1-luciferase reporter plasmids, TK-R. reniformis control plasmid, type A ET1 receptor cDNA (see Materials and Methods for details), and increasing concentrations of RGS3 cDNA (balanced by empty vector), with or without 50 ng of Smad3 cDNA as indicated. Serum-starved cells were stimulated with 100 nM endothelin-1 (ET1) for 6 h and lysed. The luciferase activity of lysates was normalized to the R. reniformis activity and expressed as the mean ± S.D. B and C, inhibition of Smad-mediated gene transcription by RGS3. CHO cells were transfected with SBE-luciferase reporter, TK-R. reniformis control plasmid (see Materials and Methods for details), and 50 ng of empty vector or cDNA for RGS3 or its mutants as indicated. Serum-starved cells were stimulated with 2 ng/ml TGF-β for 24 h and lysed. The luciferase activity of lysates was normalized to the R. reniformis activity and expressed as the mean ± S.D. Shown are the representative results from at least three independent experiments performed in triplicates. D, the N460A mutant of RGS3 does not bind G proteins. CHO cells were transfected with empty vector or with cDNAs for Flag-tagged wild-type (WT) RGS3 or its N460A mutant. Cells were lysed and immunoprecipitated with Flag antibodies in the presence of . The immune complexes or total cell lysates were probed for Gαi3 by Western blotting.
We then examined how RGS3 affects TGF-β-induced (Smad-mediated) gene transcription by using a luciferase reporter driven by four copies of SBEs. As shown in Fig. 3B, RGS3 blocked the activation of SBE by TGF-β. By contrast, the RGS domain of RGS3 (which does not interact with Smads) did not affect the TGF-β-induced SBE activation (Fig. 3B), although it was effective in regulation of G protein signaling (Dulin et al., 2000). Furthermore, the N460A mutant of RGS3 that does not bind G proteins (Fig. 4D) was as effective as the wild-type RGS3 in inhibition of TGF-β-induced SBE activation (Fig. 4C). Together, these data suggest that 1) RGS3 regulates the Smad-dependent gene transcription, and 2) this effect of RGS3 is not related to its known function of regulating the G protein-mediated signaling.
To understand the molecular mechanism by which RGS3 inhibits TGF-β-induced gene transcription, we examined the effect of RGS3 expression on TGF-β signaling. As shown in Fig. 5A, RGS3 expression has no effect on phosphorylation of R-Smads by TGF-β receptor, as assessed by Western blotting with phosphospecific Smad antibodies. In contrast, RGS3 expression nearly abolished the TGF-β -induced binding of Smad3 to Smad4 (Fig. 5B). Given that heteromerization between CoSmads and R-Smad is critical for forming a functional transcriptional complex, the inhibition of Smad3/Smad4 interaction by RGS3 may explain its regulatory effect on TGF-β-induced gene transcription.
Fig. 5.

Effect of RGS3 on TGF-β-induced Smad signaling. A, RGS3 overexpression does not affect TGF-β-induced Smad2 phosphorylation. CHO cells were transfected with Flag-Smad2 cDNA together with Myc-RGS3 cDNA or empty vector. Serum-starved cells were stimulated with or without 2 ng/ml TGF-β for 30 min and lysed. Flag-Smad2 was then immunoprecipitated with Flag antibodies, and the immune complexes were analyzed by Western blotting with desired antibodies as indicated. B, inhibition of Smad3-Smad4 interaction by RGS3. CHO cells were transfected with cDNAs for Flag-Smad3, Myc-Smad4, Myc-RGS3, or with empty vector as indicated. Serum-starved cells were stimulated with 2 ng/ml TGF-β for 30 min and lysed. Flag-Smad3 was then immunoprecipitated with Flag antibodies, and the immune complexes or total cell lysates were analyzed by Western blotting with desired antibodies as indicated.
One established function of TGF-β in fibroblasts is the stimulation of myofibroblast differentiation through the expression of smooth muscle (SM)-specific cytoskeletal proteins, such as SM-α-actin (Gabbiani, 2003). Therefore, we examined whether the regulation of Smad signaling by RGS3 translates to the modulation of SM-α-actin expression by TGF-β in human pulmonary fibroblasts. As shown in Fig. 6A, adenovirus-mediated transduction of RGS3 significantly attenuated TGF-β-induced SM-α-actin expression without affecting the levels Smad3 or Smad4. In contrast, pertussis toxin, which inhibits G protein signaling by ADP-ribosylating Gαi subunits, was without effect. This suggests that RGS3 controls cellular responses to TGF-β by a mechanism unrelated to regulation of Gi signaling.
Fig. 6.
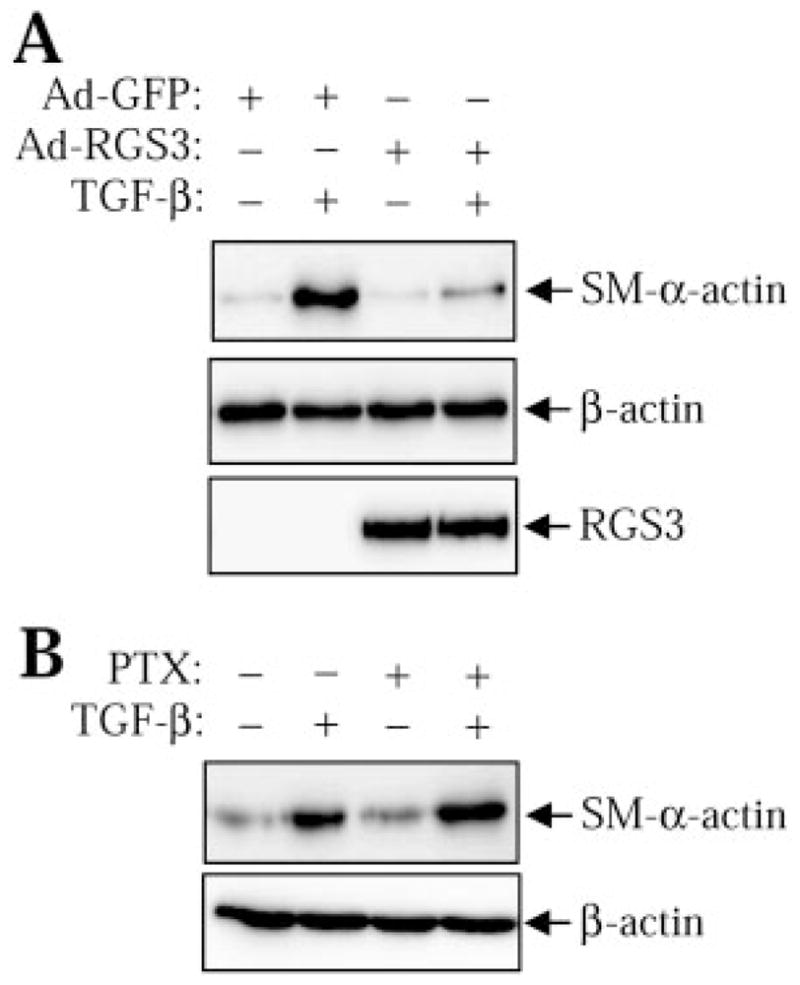
Regulation of TGF-β-induced SM-α-actin expression by RGS3. A, human pulmonary fibroblasts were transduced with RGS3 adenovirus (Ad-RGS3) or with control green fluorescent protein adenovirus (Ad-GFP), followed by stimulation with or without 2 ng/ml TGF-β for 48 h. Cells were then lysed, and the cell lysates were analyzed by Western blotting with desired antibodies as indicated. B, human pulmonary fibro-blasts were pretreated with pertussis toxin (100 ng/ml) overnight, followed by stimulation with or without 2 ng/ml TGF-β for 48 h. Cells were then lysed and the cell lysates were analyzed by Western blotting with desired antibodies as indicated.
Discussion
RGS proteins have been extensively studied during the past decade as the regulators of signaling mediated by heterotrimeric G proteins. However, emerging data suggest that RGS proteins may serve other functions that are not necessarily related to regulation of G protein signaling. For example, RGS12TS-S elicits transcriptional repressor activity in the nucleus through a unique N-terminal domain, resulting in cell cycle regulation in many cells (Chatterjee and Fisher, 2002). RGS6 promotes neuronal differentiation through the interaction with a neuronal growth-associated protein, SCG10 (Liu et al., 2002). RGS6 can also interact with and inhibit the transcriptional repressor activity of Dnmt1-associated protein, DMAP1 (Liu and Fisher, 2004). RGSZ1 binds protein kinase C interacting protein (PKCI-1) and modulates β-opioid receptor signaling independent of RGSZ1-Gαz interaction (Ajit et al., 2007). RGS2 inhibits adenylyl cyclase activity (Sinnarajah et al., 2001) through its N terminus, also in an RGS domain-independent manner ((Salim et al., 2003; Roy et al., 2006; Gu et al., 2008).
Our study identifies a novel function of RGS3 in regulation of TGF-β-induced gene transcription through the interaction of RGS3 with Smad transcription factors. Within the RGS family, RGS3 seems to be unique in its ability to bind Smads, in that we failed to detect the binding of some other RGS proteins (RGS4, RGS10) to Smad3 (data not shown). In agreement with this notion, we mapped the Smad-binding site to the 240-to-379 region of RGS3 (outside of the RGS domain) that has no significant homology to other RGS proteins. Furthermore, our data suggest that this novel function of RGS3 in controlling TGF-β-induced gene transcription is probably unrelated to regulation of G protein signaling by RGS3, given that 1) the RGS domain of RGS3 (which mediates the interaction of RGS3 with G proteins) does not bind Smad3 (Fig. 3); 2) Smad3 does not affect the regulation of G protein signaling by RGS3 (Fig. 4A); 3) the RGS domain of RGS3 has no effect on Smad signaling (Fig. 4B), whereas it effectively blocks G protein signaling (Dulin et al., 2000); and 4) the N460A mutant of RGS3 that does not bind G proteins (Fig. 4D) is as effective as the wild-type RGS3 in inhibition of Smad-mediated gene transcription (Fig. 4C).
We show here that MH2 domain of Smads mediates the interaction with RGS3. Given that MH2 domain is implicated in heteromerization of Smads that is required for their transcriptional activity, we propose the model wherein RGS3 inhibits Smad-mediated gene transcription through disruption of R-Smad/CoSmad heteromerization (Fig. 5B). It is noteworthy that MH2 domain also mediates the interaction of Smads with many other transcription factors, coactivators, or repressors (Feng and Derynck, 2005). Therefore, it is conceivable that, by interfering with the binding of Smad to some of these proteins, RGS3 may also regulate the specificity of Smad mediated gene transcription.
Finally, we show that RGS3-Smad interaction translates functionally to inhibition of TGF-β-induced myofibroblast differentiation (SM-α-actin expression) in pulmonary fibroblasts (Fig. 6A). Given the multiple, cell-specific roles of TGF-β in the control of cell growth, survival, and phenotype, the regulation of Smad signaling by RGS3 may have multiple functional outcomes in various cell types, which is the subject of our future studies.
Acknowledgments
This study was supported by National Institutes of Health grants HL071755 (to N.O.D.), HL07605 (to D.M.Y), and American Heart Association (to S.T. and N.S.).
We thank Drs. John Kehrl, Liliana Attisano, Masashi Yanagisawa, and Bert Vogelstein for providing the DNA reagents.
ABBREVIATIONS
- RGS
regulator of G protein signaling
- TGF-β
transforming growth factor-β
- CHO
Chinese hamster ovary
- DMEM
Dulbecco’s modified Eagle’s medium
- SBE
Smad binding element
- TK
thymidine kinase
- ET1
endothelin-1
- SM
smooth muscle
- L
linker
- MH
mad homology
References
- Ajit SK, Ramineni S, Edris W, Hunt RA, Hum W-T, Hepler JR, Young KH. RGSZ1 interacts with protein kinase C interacting protein PKCI-1 and modulates mu opioid receptor signaling. Cellular Signalling. 2007;19:723–730. doi: 10.1016/j.cellsig.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Barrios-Rodiles M, Brown KR, Ozdamar B, Bose R, Liu Z, Donovan RS, Shinjo F, Liu Y, Dembowy J, Taylor IW, et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621–1625. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]
- Bowman EP, Campbell JJ, Druey KM, Scheschonka A, Kehrl JH, Butcher EC. Regulation of chemotactic and proadhesive responses to chemoattractant receptors by RGS (regulator of G-protein signaling) family members. J Biol Chem. 1998;273:28040–28048. doi: 10.1074/jbc.273.43.28040. [DOI] [PubMed] [Google Scholar]
- Chatterjee TK, Eapen AK, Fisher RA. A truncated form of RGS3 negatively regulates G protein-coupled receptor stimulation of adenylyl cyclase and phosphoinositide phospholipase C. J Biol Chem. 1997;272:15481–15487. doi: 10.1074/jbc.272.24.15481. [DOI] [PubMed] [Google Scholar]
- Chatterjee TK, Fisher RA. RGS12TS-S localizes at nuclear matrix-associated subnuclear structures and represses transcription: structural requirements for subnuclear targeting and transcriptional repression. Mol Cell Biol. 2002;22:4334–4345. doi: 10.1128/MCB.22.12.4334-4345.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Harrison K, Schwartz O, Kehrl JH. The aorta and heart differentially express RGS (regulators of G-protein signalling) proteins that selectively regulate sphingosine 1-phosphate, angiotensin II and endothelin-1 signalling. Biochem J. 2003;371:973–980. doi: 10.1042/BJ20021769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druey KM, Blumer KJ, Kang VH, Kehrl JH. Inhibition of G-protein-mediated MAP kinase activation by a new mammalian gene family. Nature. 1996;379:742–746. doi: 10.1038/379742a0. [DOI] [PubMed] [Google Scholar]
- Dulin NO, Pratt P, Tiruppathi C, Niu J, Voyno-Yasenetskaya T, Dunn MJ. Regulator of G protein signaling RGS3T is localized to the nucleus and induces apoptosis. J Biol Chem. 2000;275:21317–21323. doi: 10.1074/jbc.M910079199. [DOI] [PubMed] [Google Scholar]
- Dulin NO, Sorokin A, Reed E, Elliott S, Kehrl JH, Dunn MJ. RGS3 inhibits G protein-mediated signaling via translocation to the membrane and binding to Gα11. Mol Cell Biol. 1999;19:714–723. doi: 10.1128/mcb.19.1.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Derynck R. Specificity and versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–503. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- Gu S, Anton A, Salim S, Blumer KJ, Dessauer CW, Heximer SP. Alternative translation initiation of human RGS2 yields a set of functionally distinct proteins. Mol Pharmacol. 2008;73:1–11. doi: 10.1124/mol.107.036285. [DOI] [PubMed] [Google Scholar]
- Kehrl JH, Srikumar D, Harrison K, Wilson GL, Shi CS. Additional 5′ exons in the RGS3 locus generate multiple mRNA transcripts, one of which accounts for the origin of human PDZ-RGS3. Genomics. 2002;79:860–868. doi: 10.1006/geno.2002.6773. [DOI] [PubMed] [Google Scholar]
- Liu Z, Chatterjee TK, Fisher RA. RGS6 interacts with SCG10 and promotes neuronal differentiation. Role of the Gγ subunit-like (GGL) domain of RGS6. J Biol Chem. 2002;277:37832–37839. doi: 10.1074/jbc.M205908200. [DOI] [PubMed] [Google Scholar]
- Liu Z, Fisher RA. RGS6 interacts with DMAP1 and DNMT1 and inhibits DMAP1 transcriptional repressor activity. J Biol Chem. 2004;279:14120–14128. doi: 10.1074/jbc.M309547200. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-{beta} signal transduction. J Cell Sci. 2001;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- Neill JD, Duck LW, Sellers JC, Musgrove LC, Kehrl JH. A regulator of G protein signaling, RGS3, inhibits gonadotropin-releasing hormone (GnRH)-stimulated luteinizing hormone (LH) secretion. BMC Cell Biol. 2001;2:21. doi: 10.1186/1471-2121-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Scheschonka A, Druey KM, Davis A, Reed E, Kolenko V, Bodnar R, Voyno-Yasenetskaya T, Du X, Kehrl J, et al. RGS3 interacts with 14-3-3 via the N-terminal region distinct from the RGS (regulator of G-protein signalling) domain. Biochem J. 2002;365:677–684. doi: 10.1042/BJ20020390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy AA, Baragli A, Bernstein LS, Hepler JR, Hebert TE, Chidiac P. RGS2 interacts with Gs and adenylyl cyclase in living cells. Cellular Signalling. 2006;18:336–348. doi: 10.1016/j.cellsig.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Salim S, Sinnarajah S, Kehrl JH, Dessauer CW. Identification of RGS2 and type V adenylyl cyclase interaction sites. J Biol Chem. 2003;278:15842–15849. doi: 10.1074/jbc.M210663200. [DOI] [PubMed] [Google Scholar]
- Sinnarajah S, Dessauer CW, Srikumar D, Chen J, Yuen J, Yilma S, Dennis JC, Morrison EE, Vodyanoy V, Kehrl JH. RGS2 regulates signal transduction in olfactory neurons by attenuating activation of adenylyl cyclase III. Nature. 2001;409:1051–1055. doi: 10.1038/35059104. [DOI] [PubMed] [Google Scholar]
- Taurin S, Hogarth K, Sandbo N, Yau DM, Dulin NO. Gβγ-mediated prostacyclin production and cAMP-dependent protein kinase activation by endothelin-1 promotes vascular smooth muscle cell hypertrophy through inhibition of glycogen synthase kinase-3. J Biol Chem. 2007;282:19518–19525. doi: 10.1074/jbc.M702655200. [DOI] [PubMed] [Google Scholar]
- Tosetti P, Pathak N, Jacob MH, Dunlap K. RGS3 mediates a calcium-dependent termination of G protein signaling in sensory neurons. Proc Natl Acad Sci U S A. 2003;100:7337–7342. doi: 10.1073/pnas.1231837100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liu M, Mullah B, Siderovski DP, Neubig RR. Receptor-selective effects of endogenous Rgs3 and Rgs5 to regulate mitogen-activated protein kinase activation in rat vascular smooth muscle cells. J Biol Chem. 2002;277:24949–24958. doi: 10.1074/jbc.M203802200. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Milligan G. A key serine for the GTPase-activating protein function of regulator of G protein signaling proteins is not a general target for 14-3-3 interactions. Mol Pharmacol. 2005;68:1821–1830. doi: 10.1124/mol.105.015073. [DOI] [PubMed] [Google Scholar]