

Abstract
The relative non-toxicity of the diuretic amiloride, coupled with its selective inhibition of the protease urokinase plasminogen activator (uPA), makes this compound class attractive for structure-activity studies. Herein we substituted the C(2)-acylguanidine of C(5)-glycyl-amiloride with amidine and amidoxime groups. The data show the importance of maintaining C(5)-hydrophobicity. The C(5)-benzylglycine analogs containing either C(2)-acylguanidine or amidine inhibited uPA with an IC50 ranging from 3 to 7μM and were cytotoxic to human U87 malignant glioma cells.
Urokinase-type plasminogen activator (uPA, International Union of Biochemistry EC.3.4.21.31) is a serine protease of the trypsin family expressed by a variety of normal migratory cell types and by invasive cancerous cell types. The urokinase plasminogen activation (uPA) pathways are comprised of urokinase-type plasminogen activator (uPA), its plasmalemmal receptor (uPAR), and extracellular plasminogen. uPA is synthesized intracellularly as a single chain, inactive proenzyme (pro-uPA), is secreted, binds to uPAR and is activated more than 30-fold. Activated extracellular uPA cleaves plasminogen to plasmin, which initiates an extracellular protease cascade to degrade the extracellular matrix and facilitate cellular processes such as cellular migration, angiogenesis, tissue remodeling, and wound repair. Highly invasive cancers of breast, brain, prostate and lung notably have increased levels of uPA, its receptor uPAR, or an endogenous inhibitor protein PAI-1, which correlate with the propensity of a cancer cell type to invade and to disseminate.[1] and [2] Numerous small molecule inhibitors of uPA have been developed to inhibit its enzymatic activation of plasminogen, while attempting to minimize off-targeting of the closely related serine protease, tissue plasminogen activator (tPA) promoting clot formation. Small molecule inhibitors of activated uPA bound to its receptor uPAR have been recently reviewed[2] and [3] and include the following classes of small molecules summarized in figure 1.
Figure 1.

Small molecular inhibitors of Urokinase-type plasminogen activator
The parent compound, amiloride (3,5 diamino-6-chloro-N-(diaminomethylene) pyrazinecarboximide) (figure 2) is an FDA-approved anti-hypertensive agent. Amiloride selectively inhibits the protease uPA, but not tPA or other serine protease members of the trypsin superfamily.4 Previously, we demonstrated that amino acids, notably glycine, could be conjugated to the C5 amino group of amiloride to generate a bioactive molecule.5 In figure 2, coupling of the bioactive C5 glycinyl amiloride derivative 1 to the amino terminus of additional peptides generated inactive prodrugs that could be enzymatically activated in a sequence dependent fashion.6 Because of their potential biomedical utility, it was informative to conduct a structure activity relationship study to evaluate the role of substitutions of the more basic amidine 2 or amidoxime moieties for the C2 acylguanidine moiety and to determine alterations in specificity that might be conferred by removal of the C6 chloro group while retaining the aminopyrazine core (figure 2). Substitution of the carbamyl derivatives at the C2 position utilizing aryl cores has been reported to increase chemical stability and potency of uPA inhibition, but with significant retention or increases in the inhibitory potencies of tPA, trypsin, and thrombin[7] and [8] (figure 1). Currently, WXUK1 (figure 1) is a uPA inhibitor with reported nanomolar potency and its oral prodrug is in phase 2 clinical trials as a cytostatic agent that inhibits cancer cell migration.[9] and [10] X-ray crystallographic studies demonstrated that the amiloride C(6) chlorine contributes to selective inhibition of uPA and not of tPA. Amiloride, containing a 6-chloro substitution, and 4-iodobenzo(b)thiophene-2-carboxamidine position their halogen atoms into a unique structural subsite (S1 site) adjacent to the primary binding pocket of uPA comprised of residues Gly218 and Ser146, the Cys191–Cys220 disulfide bridge, the side chain of Lys143 and part of Gln192.[7] and [11] The crystallographic data suggest that halogen substitutions at these sites in amiloride and in analogous C(2)-amidine derivatives could maintain specific uPA inhibition while increasing inhibitory potencies. Amidine substitution can confer chemical stability and maintain necessary protonation of the diamide moiety. However, the more basic C2 substituted amidine can reduce aqueous solubility, as compared with the corresponding acylguanidine derivatives. In this study, we systematically substituted the amide group and generated C6 chloro- and dechloro- derivatives of C5 glycine amiloride derivatives to examine the enzymatic specificities of the resultant amide compounds (figure 2).
Figure 2.

Amiloride, glycine-amiloride conjugate 1 and amidine structural analog 2
Our initial plan sought to install the amidine functionality of 2 via reduction of the corresponding C(2)-amidoxime, a reliable method for synthesis of amidines. Since amidoximes are readily accessible from nitriles, we set out to prepare the C(2)-nitrile as our proximal target. Commercially available amino-dichloropyrazine ester 3 (Scheme 1) was transformed to the corresponding amide by modifying a procedure first reported by Cragoe.21 The literature synthesis of 4 (liq. NH3, room temperature in an autoclave) gives amide 4 as an equimolar mixture with a substitution product arising from ammonia addition to C(5). The desired ester to amide transformation proceeded selectively and in excellent yield when the ammonia reaction was kept cold. Treatment of C(2)-amide 4 with phosphoryl chloride in toluene at reflux effected amide dehydration to afford nitrile 5 in good yield. This direct approach improves on a previously reported two-step procedure wherein the C(3)-amino group of amide 4 was protected as an N-formamidino derivative prior to amide dehyration. Following our protocol for attachment of benzyl-protected glycine to amiloride analogs5, nitrile 5 was converted to glycine-substituted nitrile 6 without incident. Reaction of 6 with hydroxylamine in ethanol at low temperature furnished the desired amidoxime 7 in excellent yield. The nitrile to amidoxime transformation is routinely performed at reflux22, however, we found that reaction of 6 with hydroxylamine at temperatures above 0°C resulted in benzyl ester cleavage.
Scheme 1.

Reagents and conditions: aNH3 (l), −33 °C, 10 h, 100%; bPOCl3 (7.75 eq.), toluene, reflux, 5 h, 88%; cBnO2CCH2NH3•OTs, Et3N, DMF, 60 °C, 16 h, 90%; dH2NOH•HCl (3.7 eq.), Et3N, EtOH, −10°C, 4 h, 98%.
Unfortunately, the reduction of amidoxime 7 using transfer hydrogenolysis conditions failed to deliver target amidine 2 (Scheme 2). Whereas hydrogenolysis of the benzyl ester proceeded smoothly, the amidoxime was resistant to hydrogenation under these conditions, resulting in the formation of amidoxime 8. The formation of 8 was accompanied by further reduction of the C(6)-chloro substituent, which led to formation of 8-H as the major product of the reaction. All subsequent attempts to reduce the amidoxime moiety of 7 to its amidine (e.g., catalytic hydrogenation, SnCl2)23 either failed or gave similar mixtures of 8 and 8-H. Similarly, attempts to reduce the amidoxime group of the dichloropyrazine 9, readily prepared by reaction of nitrile 5 with hydroxylamine, also failed to deliver the corresponding amidine 10. We were gratified to find, however, that reaction of nitrile 5 according to the Garigipati protocol led directly to the C(2)-amidine.24 Treatment of 5 with in situ generated MeAl(Cl)NH2 gave amidine 10 in good yield without complications arising from substitution of the usually reactive C(5)-chloro group. The robust nature of the C(5)-chloro became apparent in the next step as we attempted to substitute it with glycine as in our previous examples. Reaction of amidine 10 with benzyl glycine under the standard conditions gave adduct 11 in 17% yield. Attempts to improve the substitution by subjecting the reactants to more forcing conditions led to lower isolated yields of 11. Hydrogenation of 11 (H2, 10% Pd/C, EtOH) gave a 2.3:1 mixture of the debenzylated products 2.2-H in 81% yield. The amidines 2 and 2-H were readily separated using C18 reverse phase chromatography (CH3CN: 0.1% aq. TFA gradient).
Scheme 2.

Reagents and conditions: aHCO2−H4N+, Pd/C (10%), EtOH, 86% (Cl:H, 1:4); bH2NOH•HCl, Et3N, EtOH, rt, 12 h, 88%; cAlMe3 (2 eq.), NH4Cl (2 eq.), toluene, 80 °C, 32 h, 95%; dBnO2CCH2NH3+ TsO−, Et3N, DMF, 60 °C, 48h, 17%.
The availability of dichloropyrazine amide 4 led us to prepare the corresponding benzyl glycine conjugate to serve as a neutral C(2)-analog for comparison to the more basic C(2)-amidoxime and C(2)–amidine analogs. In contrast to the reaction of 10, nucleophilic aromatic substitution on 4 (Scheme 3) proceeded smoothly to afford conjugate 12. This ease of this transformation mirrors closely our previously reported synthesis of the benzyl glycine conjugate of amiloride (e.g., 13→ 14).[5] and [6]
Scheme 3.

Additional C(2)-analogs. a12: BnO2CCH2NH3+ TsO−, Et3N, DMF, 60 °C, 18 h, 45%; bH2, cat. 10% Pd/C, EtOH, 48 h, 83% (1.1-H, 1:1.5)
In Table 1, the addition of a glycine moiety to amiloride to form (1) minimally reduced the inhibitory potency of the free acid for uPA (IC50 17μM), as compared with the amiloride (IC50 7μM), while preserving selectivity as a protease inhibitor (Table 1). The absence of the C6 chloro group (1-H) reduced inhibitory potency against uPA (IC50 47μM), but did not alter its selectivity as an uPA inhibitor. Benzylester formation (14) enhanced inhibitory potency (IC50 3μM), while preserving selectivity as a protease inhibitor.
Table 1.
C(2)/C(5)/C(6) amidine analogs and cytotoxicity 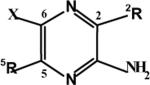
| Compound | 2R | 5R | X | uPA IC50 μM | tPA IC50 μM | Plasmin IC50 μM | Trypsin IC50 μM | Cytoxicity |
|---|---|---|---|---|---|---|---|---|
| amiloride | acylguanidine (H2N)2C=NC(O)– | NH2 | Cl | 7 | 220 | >250 | >250 | 50% cytotoxic at 250μM by 24h |
| 7 amidox-Am-C(5)-Gly-OBn | amidoxime H2NC(NOH)- | -NHCH2CO2Bn | Cl | 74 | 200 | >250 | >250 | Not cytotoxic |
| 8 amidox-Am-C(5)-Gly-OH | amidoxime H2NC(NOH)- | -NHCH2CO2H | Cl | 231 | >250 | >250 | >250 | Not cytotoxic |
| 8-H dechloro-amidox-Am-C(5)-Gly-OH | amidoxime H2NC(NOH)- | -NHCH2CO2H | H | 279 | >250 | 230 | >250 | Not cytotoxic |
| 11 amidine-Am-C(5)-Gly-OBn | amidine H2NC(NH)- | -NHCH2CO2Bn | Cl | 4 | 200 | >250 | >250 | 50% cytotoxic at 250μM by 72h |
| 2 amidine-Am-C(5)-Gly-OH | amidine H2NC(NH)- | -NHCH2CO2H | Cl | 237 | >250 | >250 | >250 | Not cytotoxic |
| 2-H dechloro- amidine-Am-C(5)-Gly-OH | amidine H2NC(NH)- | -NHCH2CO2H | H | 212 | >250 | >250 | >250 | Not cytotoxic |
| 12 amide-Am-C(5)-Gly-OBn | carboxamide H2NC(O)- | -NHCH2CO2Bn | Cl | 69 | >250 | >150 | >250 | Not cytotoxic |
| 14 Am-C(5)-Gly-OBn | acyl guanidine (H2N)2C=NC(O)- | -NHCH2CO2Bn | Cl | 3 | >190 | >250 | >250 | 50% cytotoxic at 100μM by 24h |
| 1 Am-C(5)-Gly-OH | acyl guanidine (H2N)2C=NC(O)- | -NHCH2CO2H | Cl | 17 | >250 | >250 | >250 | Not cytotoxic |
| 1-H dechloro-Am-C(5)-Gly-0H | acyl guanidine (H2N)2C=NC(O)- | -NHCH2CO2H | H | 47 | >250 | >250 | >250 | Not cytotoxic |
Amidine substitution for the C(5) benzoylguanidine group of the free acid form of C2-glycinyl amiloride (2) markedly reduced its potency as a uPA inhibitor (IC50 237 μM). The absence of the C6 chloro group (2-H) did not alter the inhibitory potency of the amide glycinyl derivatives, relative to uPA or alter its selectivity against the other proteases. By contrast, formation of the benzyl ester at the C2 substituted glycine moiety (11) restored inhibitory potency (IC50 4 μM) to that of amiloride and of 14 while not altering selectivity as a protease inhibitor (Table 1).
The C2 amidoxime glycinyl amiloride derivatives explored the impact of the more basic amidoxime and amide inhibitors, relative to their benzoylguanidine congeners. The amidoxime analog (8) demonstrated a significant reduction in potency as a uPA inhibitor comparable to that of the amidine derivatives. Dechlorination (8-H) had no impact on altering inhibitory activity. Addition of the benzyl ester formation at the C5-glycine moiety of the amidoxime derivative (7) partially restored uPA inhibitory potency (IC50 74 μM) with very slight inhibition of tPA (IC50 200 μM). The enzymatic assay was done as described.26
Amiloride is cytostatic to several cancer cell types12 and has been reported to produce apoptotic cell death in several others.13 Previously, our group reported that amiloride (~200 μM) causes caspase-independent, non-apoptotic cell death of human malignant glioma cells.[14] and[15] Despite its low potency as a selective anti-cancer agent, amiloride's non-toxicity in humans makes it appealing to explore as a potential candidate for preventing recurrence of highly invasive and metastatic cancers.16 The lactate dehydrogenase (LDH) assay quantifies changes in cytosolic LDH released from dying and dead cells and distinguishes drug-induced anti-glioma cytotoxicity from cytostatic effects, unlike the more commonly reported metabolism-based, tetrazolium live cell assays. Using the LDH assay27, amiloride (250 μM) kills 50% of U87 glioma cells at 24h (Table 1). Benzylglycinyl substitition at the C5 position of amiloride (14) or its amidine homolog (11) confer respective uPA inhibitor potencies of IC50 =3 μM and 4 μM, but 14 is more cytotoxic than 11. The corresponding glycinyl acids at the C5 position (1 and 2) do not demonstrate glioma cytotoxicity, and 2 lost most of its uPA inhibitory activity. Substitution of the amidoxime group at C5 eliminated uPA inhibitory and anti-glioma cytotoxic potencies.
Amiloride, an acylguanidine, is one of the most selective inhibitors of uPA, relative to other proteases. Our data is in agreement with studies showing that amiloride has an IC50 reported to be between 7 and 50 μM for uPA with IC50 >1000 for tPA or plasmin.17 Compared with other trypsin-like serine proteases, the S2 and S3/S4 pockets of uPA are reduced in size because of the 99-insertion loop.18 Nienaber and colleagues utilized the crystalline structure of re-engineered urokinase to demonstrate that amiloride utilized its C(6) chloride to occupy an auxillary structural subsite adjacent to the primary binding pocket and which conferred the inhibitory specificity observed with amiloride.11 The unique binding mode of this class of uPA inhibitors utilizes interactions at the S1'/S2' subsites, which enhance their inhibitory selectivity and potency.[19] and [20] Our selection of amiloride as the initial intracellular uPA inhibitor is based upon its high selectivity for uPA inhibition and its minimal toxicity profile in humans.
C2-amidine derivatives of amiloride would appear to be structurally analogous to the acylguanidine inhibitors of uPA (figure 1). A significant increase in chemical stability and potency as a uPA inhibitor has been reported by incorporating amidine moities, but with a proportional gain in inhibitory potencies of tPA, trypsin, and thrombin.7 However, it has been proposed that selectivity of C2-amidine derivatives for uPA should be maintained by the introduction of a halogen at the C6 position.4 While maintaining the pyrazine core of amiloride we demonstrated that de-chlorination significantly reduced uPA inhibitory potencies of the C2-amidine and C2- amidoxime derivatives. Furthermore, no gain in inhibitory potencies was noted with other related proteases. Inclusion of the benzyl ester on the 2R glycinyl group significantly improved inhibitory potency of uPA while maintaining selectivity towards other proteases. This suggests that hydrophobic stabilization by the 2R group contributes towards uPA inhibitory activity of these pyrazine-based derivatives and agrees with independent data obtained using amidine aryl cores.[3] and [8]
Amiloride has been reported to kill human glioma cells and other cancer cells, including malignant breast cancer cell lines, in micromolar concentrations.14 Here we demonstrate that compounds 14 and 11 are cytotoxic to human U87 glioma cells in a fashion comparable to amiloride, but not their respective free acid derivatives 1 and 1-H, 2 and 2-H. Inhibition of uPA by 14 and 11 is comparable to that of amiloride and corresponds with the cytotoxicity demonstrated by all three compounds. It would appear from 14 and 11 and their derivatives that substitution of the more basic amidine group for the acylguanidine moiety is associated with a loss of uPA activity and anti-glioma cytotoxicity and that derivatization of the C(5) glycine with benzyl helps the molecule to regain the necessary hydrophobicity required for enzymatic and biological activities.
Our conception to replace the acyl guanidine of 14 with the chemically more stable amidine did not deliver a more potent inhibitor, but the study did reveal for this class of inhibitors the importance of the addition of benzyl groups to the carboxyl terminus. Compounds 14 and 11 demonstrated comparable selective inhibition of uPA and anti-glioma cytotoxicity, as compared with their free acid forms. Also, this study demonstrated improvements in accessing C2 amidines of the widely studied amiloride core and surprisingly demonstrated that the enzymatic selectivity conferred by the C6 chloride in amiloride cannot be uniformly applied to these amidine and amidoxime derivatives.
Supplementary Material
Acknowledgement
This research funded by the National Institutes of Health (NS040489, NS060880 F.G.).
Glossary
- uPA
urokinase-type plasminogen activator
- uPAR
urokinase-type plasminogen activator receptor
- tPA
tissue plasminogen activator
- PAI-1
plasminogen activator inhibitor protein -1
- LDH
lactate dehydrogenase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The experimental procedures and corresponding analytical data for 2-amidino analogs of 5-glycinylamiloride conjugates is provided in the Supporting Information section.
References and notes
- (1).Annecke K, Schmitt M, Euler U, Zerm M, Paepke D, Paepke S, von Minckwitz G, Thomssen C, Harbeck N. Adv. Clin. Chem. 2008;45:31. doi: 10.1016/s0065-2423(07)00002-9. [DOI] [PubMed] [Google Scholar]
- (2).Ulisse S, Baldini E, Sorrenti S, D'Armiento M. Curr. Cancer Drug Targets. 2009;9:32. doi: 10.2174/156800909787314002. [DOI] [PubMed] [Google Scholar]
- (3).Rockaway TW. Expert Opin. Ther. Patents. 2003;13:773. [Google Scholar]
- (4).Nienaber V, Wang J, Davidson D, Henkin J. J. Biol. Chem. 2000;275:7239. doi: 10.1074/jbc.275.10.7239. [DOI] [PubMed] [Google Scholar]
- (5).Palandoken H, By K, Hegde M, Harley WR, Gorin FA, Nantz MH. J. Pharmacol. Exp. The. 2005;312:961. doi: 10.1124/jpet.104.076984. [DOI] [PubMed] [Google Scholar]
- (6).Gorin FA, Nantz MH. U.S. patent 7863415. 2011 Jan 4; [Google Scholar]
- (7).Wendt MD, Rockway TW, Geyer A, McClellan W, Weitzberg M, Zhao X, Mantei R, Nienaber VL, Stewart K, Klinghofer V, Giranda V. J. Med. Chem. 2004;47:303. doi: 10.1021/jm0300072. [DOI] [PubMed] [Google Scholar]
- (8).Schweinitz A, Steinmetzer T, Banke IJ, Arlt MJ, Sturzebecher A, Schuster O, Geissler A, Giersiefen H, Zeslawska E, Jacob U, Kruger A, Stürzebecher J. J. Biol. Chem. 2004;279:33613. doi: 10.1074/jbc.M314151200. [DOI] [PubMed] [Google Scholar]
- (9).Muehlenweg B, Sperl S, Magdolen V, Schmitt M, Harbeck N. Expert Opin. Biol. Ther. 2001;1:683. doi: 10.1517/14712598.1.4.683. [DOI] [PubMed] [Google Scholar]
- (10).Sperl S, Buergle M, Schmalix W, Wosikowski K, Clement B. U.S. patent 2/105,975 filed. 2008 Apr 18; [Google Scholar]
- (11).Nienaber VL, Davidson D, Edalji R, Giranda VL, Klinghofer V, Henkin J, Magdalinos P, Mantei R, Merrick S, Severin JM, Smith RA, Stewart K, Walter K, Wang J, Wendt M, Weitzberg M, Zhao X, Rockway T. Struct. 2000;8:553. doi: 10.1016/s0969-2126(00)00136-2. [DOI] [PubMed] [Google Scholar]
- (12).Wong P, Kleemann HW, Tannock IF. Br. J. Cancer. 2002;87:238. doi: 10.1038/sj.bjc.6600424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Berdiev BK, Xia J, McLean LA, Markert JM, Gillespie GY, Mapstone TB, Naren AP, Jovov B, Bubien JK, Ji HL, Fuller CM, Kirk KL, Benos DJ. J. Biol. Chem. 2003;278:15023. doi: 10.1074/jbc.M300991200. [DOI] [PubMed] [Google Scholar]
- (14).Hegde M, Roscoe J, Cala P, Gorin F. J. Pharmacol. Exp. Ther. 2004;310:67–74. doi: 10.1124/jpet.103.065029. [DOI] [PubMed] [Google Scholar]
- (15).Harley W, Floyd C, Dunn T, Zhang XD, Chen TY, Hegde M, Palandoken H, Nantz MH, Leon L, Carraway KL, III, Lyeth B, Gorin FA. Brain Res. 2010;1363:159–169. doi: 10.1016/j.brainres.2010.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Helenius MA, Savinainen KJ, Bova GS, Visakorpi T. BJU Int. 2006;97:404–4098. doi: 10.1111/j.1464-410X.2005.05912.x. [DOI] [PubMed] [Google Scholar]
- (17).Bridges AJ, Lee A, Schwartz CE, Towle MJ, Littlefield BA. Bioorg. Med. Chem. 1993;1:403. doi: 10.1016/s0968-0896(00)82150-1. [DOI] [PubMed] [Google Scholar]
- (18).Sperl S, Jacob U, Arroyo de Prada N, Stürzebecher J, Wilhelm OG, Bode W, Magdolen V, Huber R, Moroder L. Proc. Natl. Acad. Sci. USA. 2000;97:5113. doi: 10.1073/pnas.97.10.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Katz BA, Mackman R, Luong C, Radika K, Martelli A, Sprengeler PA, Wang J, Chan H, Wong L. Chem. Biol. 2000;7:299. doi: 10.1016/s1074-5521(00)00104-6. [DOI] [PubMed] [Google Scholar]
- (20).Mackman RL, Katz BA, Breitenbucher JG, Hui HC, Verner E, Luong C, Liu L, Sprengeler PA. J. Med. Chem. 2001;44:3856. doi: 10.1021/jm010244+. [DOI] [PubMed] [Google Scholar]
- (21).Cragoe EJ, Jr., Wolterdorsf O, Bicking J, Kwong S, Jones JH. J. Med., Chem. 1967;10:66. doi: 10.1021/jm00313a014. [DOI] [PubMed] [Google Scholar]
- (22).Jendralla H, Seuring B, Herchen J, Kulitzscher B, Wunner J, Stueber W, Koschinsky R. Tetrahedron. 1995;51:12047. [Google Scholar]
- (23).Cesar J, Nadrah K, Dolenc MS. Tetrahedron Lett. 2004;45:7445. [Google Scholar]
- (24).Garigipati RS. Tetrahedron Lett. 1990;31:1969–1972. [Google Scholar]
- (25).Enzymes and substrates were purchased as follows: high molecular weight (HMW) uPA, tPA (Sigma), trypsin (Worthington), thrombin (Calbiochem), plasmin (Enzyme Research Labs), and factor Xa (Haematological Technologies, Inc.). The substrates for trypsin, plasmin, and thrombin (tosyl-Gly-Pro-Lys-pNA (pNA = p-nitroanilide) and for factor Xa (CH3OCO-D-CHA-Gly-Arg-pNA-AcOH,) were purchased from Centerchem, Inc. The substrate for HMW uPA (Glt-Gly-Arg-AMC) was fluorogenic urokinase substrate III (Calbiochem), and the substrate for tPA (CH3SO2-D-HHT-Gly-Arg-pNA, “Spectrozyme tPA”) was from American Diagnostica.
- (26).Enzymatic Assays: uPA was assayed spectrofluorometrically, and the other proteases were assayed spectrophotometrically in 96 well microtiter plates using a SpectraMax M2 with SoftmaxPro software. All enzymes were assayed at 22°C temperature in 50 mM Tris, 150 mM NaCl, 0.05% (v/v) Tween-20, 10% (v/v) DMSO, 0.002% antifoam, and EDTA (1 mM) at pH 7.4. The thrombin and factor Xa assays also contained 5.0 mM CaCl2. Substrate concentrations of for each enzyme were selected based upon their respective Km determinations. For uPA activity, the rate of change in relative fluorescence units (RFUs) emitted at 460 nm (excitation at 355 nm) was measured immediately after addition of enzyme. For all other enzymatic activities, the rate of change in absorbance at 405 nm was measured immediately following enzyme addition. Apparent inhibition constants, Ki', were calculated from the velocity data generated at the various inhibitor concentrations using 4-parameter logistic function equation using Sigma Plot software (Systat Software, Inc., Chicago IL.).
- (27).Cell death assay: Lactate dehydrogenase (LDH) assay was used to measure the release of cytosolic LDH from dying glioma cells. Ten thousand cells per well were plated in 96 well microtiter plate and treated with the drugs for 24–48 h at 37°C, 5% CO2. LDH assay was performed as per the manufacturer's protocol (Sigma Diagnostics, St Louis, MO). Absorbance was read at 492 nm using a SpectraMax M2 with SoftmaxPro software.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.