

Abstract
Rationale
Tissue ischemia/reperfusion (IR) injury underlies several leading causes of death such as heart-attack and stroke. The lack of clinical therapies for IR injury may be partly due to the difficulty of adapting IR injury models to high-throughput screening (HTS).
Objective
To develop a model of IR injury that is both physiologically relevant and amenable to HTS.
Methods and Results
A micro-plate based respirometry apparatus was used. Controlling gas flow in the plate head space, coupled with the instrument’s mechanical systems, yielded a 24 well model of IR injury in which H9c2 cardiomyocytes were transiently trapped in a small volume, rendering them ischemic. Following initial validation with known protective molecules, the model was used to screen a 2000 molecule library, with post IR cell death as an endpoint. pO2 and pH monitoring in each well also afforded metabolic data. Ten protective, detrimental and inert molecules from the screen were subsequently tested in a Langendorff perfused heart model of IR injury, revealing strong correlations between the screening endpoint and both recovery of cardiac function (negative r2=0.66), and infarct size (positive, r2=0.62). Relationships between the effects of added molecules on cellular bioenergetics, and protection against IR injury, were also studied.
Conclusion
This novel cell-based assay can predict either protective or detrimental effects on IR injury in the intact heart. Its application may help identify therapeutic or harmful molecules.
Keywords: Chemical Screen, Library, Myocardial Infarction, Stroke
INTRODUCTION
Diseases of tissue ischemia/reperfusion (IR) injury are the leading causes of death in western societies, with ~1.6 million new incidences of stroke or myocardial infarction (MI) occurring annually in the USA alone 1. The underlying pathologic event in IR injury is the blockade of blood-flow, starving tissue of O2 and substrates and leading to accumulation of metabolites such as lactate. Uncontrolled, tissue reperfusion is also pathogenic, leading to cell death and infarct development.
To date, trial therapies for IR injury have targeted the mechanisms preceding cell death, or the signaling pathways invoked by endogenous protective mechanisms such as ischemic preconditioning (IPC) 2. In the field of cardiac IR, pathologic mechanisms targeted by candidate drugs include Na+ overload 3, Ca2+ overload 4, the mitochondrial permeability transition pore 5, and reactive oxygen species generation 6. Drugs designed to mimic IPC have included adenosine analogs 7, surface and mitochondrial K+ channel openers 8, 9, nitric oxide donors 10, and vasodilators 8, 11. To date however, no drug has been FDA-approved for reduction of myocardial infarct size 12. Similarly, there are no cell-mechanism based therapies for ischemic stroke, with available therapies limited mostly to clot-busters such as tPA.
Unbiased high-throughput screening (HTS) is a mechanism-agnostic approach to drug discovery, but has not been widely applied to the problem of IR injury. The complexity of IR pathology is such that simple cell-based IR models (e.g., chemical ischemia, hypoxia alone, or O2/glucose deprivation) do not reproduce ischemic conditions well. In addition these models often require media or gas exchanges which are incompatible with plate reading devices. In contrast, more physiologically relevant IR models (e.g., the ex-vivo perfused heart 13 or in-vivo murine coronary artery occlusion 14) are expensive, technically challenging and low throughput.
The goal of this study was to overcome the trade-off between physiologic relevance and ease of use, to develop an IR injury model offering both accurate representation of IR conditions and high throughput. To accomplish this, a plate-based respirometry apparatus (Seahorse Bioscience XF-24) was used as a framework 15. The apparatus measures mitochondrial respiration (O2 consumption rate, OCR) and glycolysis (extracellular acidification rate, ECAR) by intact cells on a 24-well plate 15. Atop the cell plate rests a disposable cartridge with 24 plungers that travel in a vertical axis (Fig. 1). Embedded in the plunger tips are fluorescent probes sensitive to pO2 and pH, which are interrogated by fiber-optics. Lowering these plungers traps cells in a transient 7 μl micro-chamber, allowing measurement of changes in pO2 and pH in the extracellular space, and hence the calculation of rates.
Fig 1. Schematic of the Apparatus.

A standard Seahorse XF24 apparatus was used. Adaptation of the disposable cartridge (green) for gas flow was achieved by drilling for Luer fittings (see Online Fig. I). Cartridge is pictured in the upper (A) and lower (B) positions. Lowering the cartridge traps cells in a transient 7 μl micro-chamber.
We hypothesized that upon prolonged lowering of the plungers, cells would consume all available O2 in the micro-chamber, rendering an ischemic-like state. Similarly, raising the plungers would flood cells with bulk media, simulating reperfusion. To gain greater control over O2 levels in the media, the XF apparatus was adapted for argon gas flow in the head space of the cartridge (Online Fig. I). These modifications afforded a 24-well model of IR injury, which was then used to screen a 2000 molecule library for protection against IR-induced cell death. Hits from the screen were validated using a perfused heart model of IR injury. Furthermore, the measurement of cellular bioenergetic function throughout the IR procedure afforded novel insight into the relationship between IR injury and cell metabolism.
METHODS (Full details online)
Reagents and cell culture
The Spectrum Collection™ chemical library was from MS-Discovery Inc. (Gaylordsville, CT), supplied through the University of Rochester HTS core, and stored at −80°C on 96-well plates in 1 mM in DMSO. The cardiomyocyte derived H9c2 cell line was obtained from ATCC (Manassas, VA) at passage 13, and maintained at sub-confluence in DMEM with 25 mM glucose, 1 mM pyruvate, 4 mM glutamine, 10% FBS and pen/strep, at 37°C with 5% CO2. Cells were used between passages 20 and 40, plated on XF-24 V7-PET plates at 15-30,000 cells/well, 24-48 hrs. prior to testing. One hr. prior to assay, media was replaced with 700 μl assay media (DMEM with 25 mM glucose, 1 mM pyruvate, 4 mM glutamine, no serum, no antibiotics, no bicarbonate, pH 7.4 at 37°C).
Adaptation of XF-24 for IR injury
The Seahorse XF-24 measures O2 consumption rate (OCR) and extracellular acidification rate (ECAR) by cells on a 24-well plate 15, using a disposable cartridge of moveable plungers embedded with fluorescent pO2 and pH probes (Fig. 1). Holes were drilled in the cartridge to connect gas lines (Fig. 1 and Online Fig. I). Instrument sensors were disabled to permit removal of the cover, to fit or remove gas lines.
IR injury and end-points
Typical pO2 and pH traces during IR are shown in Fig. 2. Pre-ischemic measurements of OCR and ECAR were obtained both prior to and after injection of test molecules at 1.4 μM. Flow of humidified argon was then initiated at 500 ml/min., followed 35 min. later by lowering plungers for 60 min, modeling ischemia. After 1 hr. of nominal ischemia with the plungers lowered, argon flow to the cartridge was replaced with room air, plungers were raised, and bulk media mixed for 5 min., modeling reperfusion. OCR and ECAR were measured again 1 hr. later. Cell death was then assayed by the luminometric Cytotox-Glo™ assay (Promega, Madison WI) according to the manufacturer’s protocol, on a fluorescent plate reader.
Fig. 2. Representative pO2 (A) and pH (B) traces during an IR experiment.

XF measurements were taken every 11 s. when in read mode. Rates of OCR and ECAR were obtained before and after molecule injection (bold arrow), and at the end of reperfusion. Argon or room air flow, and lowering and raising of plungers are indicated above the traces. The ischemic period, with plungers lowered, was 1 hr. Data are means of 22 wells on a plate, ± SEM, and are representative of the 39 plates screened.
Method work-up and screen validation
Initial studies were undertaken with a variety of molecules known to protect the heart against IR injury : adenosine (50 μM) 16, diazoxide (10μM) 17, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, 50 nM) 18, cyclosporin A (CsA, 0.2 μM) 5, and nitro-linoleic and nitro-oleic acids (1 μM) 19. Online Fig. II shows that five of the 6 molecules resulted in significant reductions in post-IR cell death (p<0.05 by ANOVA), with CsA just failing to achieve significance (p=0.061). Since CsA’s target protein cyclophilin D plays a role in necrotic but not apoptotic cell death 20, the mode of cell death in this model may be weighted toward apoptosis.
First round screen
Test molecules were diluted to 20 μM in assay media on the day of assay, and 70 μl loaded into cartridge injection ports (Fig. 1), for a final injected concentration of 1.43 μM (DMSO 0.57 % vol). A standard multiplexing strategy was used 21, with 4 molecules per well in each of 20 test wells on the XF plate (80 molecules/plate x 25 plates = 2000). Two wells were used for background correction (media alone) and two for vehicle controls (no test molecules). Cell death in each well was expressed relative to the mean for the whole plate, including controls and test wells. This normalization requires that the molecule library is a random distribution of protective/inactive/detrimental molecules, as supported by Online Fig. IIIA.
Second round screen
56 wells (224 molecules) exhibiting the lowest post-IR cell death scores in round 1 were split up, and their components tested at 1-molecule-per-well. Each plate comprised 16 test wells, 2 background correction wells, and 6 vehicle controls (16 molecules/plate x 14 plates = 224). This sub-set of the library was assumed biased toward protective molecules, thus precluding normalization of cell death to the plate average (see first round). Therefore, cell death in the second round was expressed relative to the 6 vehicle controls.
Although well assignments were randomized in both screening rounds, cell culture edge-effects 22 or variations in temperature or gas flow inside the instrument could result in hot spots of cell death on the plate. Thus, the average cell death in each well across all 39 plates was mapped (Online Fig. III), and the map was used to apply a correction factor to cell death values, thus avoiding plate-position bias.
Secondary validation and development of hits
A perfused rat heart model of IR injury was employed, as previously described 13. Male Sprague-Dawley rats (Harlan, Indianapolis IN), 200-250g, were maintained on a 12 hr. light/dark cycle with food and water ad libitum, and all procedures were approved by the AAALAC accredited University Committee on Animal Resources, in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Hearts were perfused with Krebs-Henseleit buffer in constant flow mode (12 ml/min.), with left ventricular pressure monitored by a balloon/transducer and digital data collection. Hearts were subjected to 35 min. global ischemia and 2 hrs. reperfusion, followed by assessment of infarct size by TTC staining 14. Test compounds were administered at 1μM in DMSO (final DMSO <0.2%) for 20 min. prior to ischemia, via a port in the perfusion cannula. Tested compounds are listed in Table 1, and included beneficial, detrimental, and no-effect molecules.
Table 1. Sub-set of molecules tested for cardioprotection in perfused hearts.
The full list of 37 most protective molecules is in Online Table II. Six of those were tested in perfused hearts, as listed above. In addition, one molecule that was without effect (propoxur), and 3 which exhibited a detrimental effect in the screen, were tested. Molecules are listed in the order they were tested.
| Score | Compound | Structure | Mw* | LogP | Bioactivity, notes, references |
|---|---|---|---|---|---|
| 0.546 | Dioxybenzone | 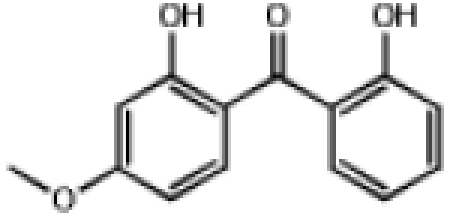 |
244.2 | 2.34 | UVA/UVB Blocker |
| 0.580 | Cloxyquin | 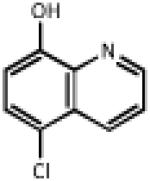 |
179.6 | 2.29 | Antimycobacterial. Similar to clioquinol, a Zn chelator which stimulates autophagy 25 and uncouples mitochondria 51 |
| 0.732 | Methapyrilene | 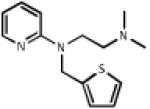 |
261.4 | 3.41 | Antihistamine, anticholinergic, sedative |
| 0.581 | Mevastatin | 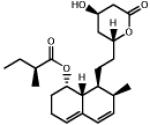 |
390.5 | 3.35 | Cholesterol lowering agent. Protects cardiomyocytes from IR injury via GSK-3β inhibition2 31. |
| 0.687 | Clorsulon | 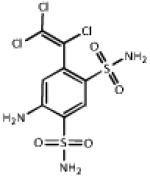 |
380.7 | 0.58 | Antifilarial, used w. ivermectin. Phosphoglycerate kinase inhibitor 36. |
| 0.778 | 7-Hydroxyflavone | 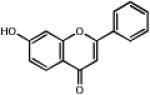 |
238.2 | 2.68 | Vasorelaxant, possibly via KCa channels 39. |
| 1.003 | Propoxur | 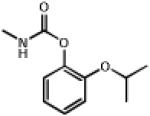 |
209.3 | 1.53 | Insectiside, cholinesterase inhibitor. Elicits pressor response in-vivo 52 |
| 2.040 | Allopregnanolone | 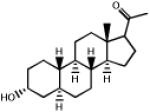 |
304.5 | 3.99 | Neurosteroid. GABAA receptor agonist. Anesthetic (eltanolone 53, minaxolone 54) |
| 2.103 | Apiin |  |
562.5 | −0.98 | Flavonoid glycoside from parsley. |
| 1.850 | 2,4-Dichloro phenoxybutyrate |
 |
249.1 | 2.86 | Herbicide (2,4-DB) metabolized to 2,4-D (Agent Orange) |
Statistics and correlations
All group-wise analyses were conducted with an N of 5-7 per group. For comparison between groups, unless otherwise indicated multiple way ANOVA was used, with significance (p) set at 0.05. For correlations, linear regression curve fit was used.
RESULTS
A Seahorse XF-24 device was adapted for gas flow as shown in Figs. 1 and Online Fig I. The mean pO2 and pH values for all wells on a plate during a typical experiment, are shown in Fig. 2. Following two initial OCR/ECAR measurements, injection of test molecules, and one further OCR/ECAR measurement, argon delivery to the cell plate lowered the bulk media pO2 to <10 mmHg within 35 min. During the subsequent 60 min. period of ischemia, imposed by lowering the plungers, cell respiration brought the pO2 below 0.5 mmHg within the first 10 min., where it remained for the duration. pH in the micro-chamber fell by ~1 unit during the nominal 1 hr. ischemic period. Upon reperfusion (raising plungers and flushing the plate with room air), both pO2 and pH returned to normal levels. A final OCR/ECAR measurement was obtained at the end of the protocol, prior to plate ejection and a cell death assay.
Average OCR prior to test molecule(s) delivery was 172.3 ± 0.01 pmol O2/min, and average ECAR was 14.8 ± 0.01 mpH/min (means ± SEM). Delivery of test molecules caused an average 45.0 ± 0.1 % increase in ECAR, and a 5.9 ± 0.02 % decrease in OCR. Thus, under these cell culture conditions, glycolysis (ECAR) appeared more susceptible to the effects of added molecules than did mitochondrial Ox-Phos (OCR). This observation is pursued further in the discussion and Fig. 6.
Fig. 6. Relationship between XF metabolic parameters and cell death in the second round screen.

Mitochondrial Ox-Phos (OCR) and glycolysis (ECAR) were measured at various time points during the IR protocol, from data of the type shown in Fig. 2. A: Relationship between the effect of added molecules on ECAR vs. effect on OCR. B: Relationship between post-IR cell death and the effect of added molecules on OCR/ECAR ratio. C/D: Data from panel B broken out into the individual relationships of cell death with effects of molecules on either OCR (C) or ECAR (B). E/F: Relationships of cell death with the post-IR recovery of either OCR (E) or ECAR (F). In panels A thru D, each point represents a single well/molecule from the second round (N = 224). In panels E/F, the effect of added molecules is not under investigation, so data from both screening rounds including controls (N=858) is shown. Linear regression fits are shown on all plots (dotted lines) with r2 and p values calculated as detailed in the methods. In each panel, the beneficial or detrimental compounds tested in intact hearts are shown in blue and red symbols, respectively.
The model of IR injury was applied to screen a library of 2000 small molecules. As detailed in the methods, the first round screen was multiplexed, with 4 molecules per well at 1.4 μM each. The mean cell death across all 550 wells (500 test wells, 50 controls) was 17.8 ± 0.02 % (mean ± SEM). Fig. 3A shows normalized cell death scores for the 500 molecules tested, in rank order (Online Fig. IIIA shows results in order of assay, and the full results are also in Online Table I). Scores < 1.0 denote a protective effect, while those >1.0 denote a detrimental effect. Mean cell death was 1.0, and the standard deviation (SD) was ± 0.27, indicated by the gray shading in Fig. 3A. Sixty three wells exhibited scores more than one SD below the mean (i.e., <0.73), and 10 wells exhibited scores more than two SD below the mean (i.e., <0.46, corresponding to z-score >2).
Fig. 3. Overall cell death data.

Normalized cell death score was calculated as described in the methods. A: First round data in rank order. Scores <1 indicate protection from IR injury (less cell death) while scores >1 indicate detriment (more cell death). Each point represents one well containing 4 test molecules. Mean cell death was 1.0. Gray shading = standard deviation (±0.270). B: Second round data in rank order. 56 wells from the first round were broken out to 1 molecule per well (224 molecules total). Each point represents a single well/molecule. Mean cell death was 1.128. Gray shading = one standard deviation (±0.287).
The 56 lowest scoring wells were subjected to a second round screen at 1 molecule per well (224 molecules) to reveal the active constituents. Results from the second round are in Online Table I, and Fig. 3B shows normalized cell death scores in rank order. Mean cell death for the second round was 1.13, and the SD was ± 0.29. A mean cell death value of >1 may seem counterintuitive, since the second round should be biased toward protective molecules (based on selection from the first round). However, if protection in the first round was conferred by only 1 of the 4 molecules in each well, then only ¼ of second round molecules should exhibit protection, with ¾ exhibiting no or a damaging effect. Viewing the second round results in assay order (Online Fig. IIIB) yielded an upward trend over time, consistent with the most protective wells from round 1 being screened first, and less protective wells later.
In the second round screen, 37 molecules exhibited scores more than 1 SD below the mean (i.e., <0.84). These molecules are shown in Online Table II, ranked by score. The table also shows structures and chemical/biological properties. A number of structural motifs were common among the hits, including flavones, chalcones, sterols, terpenoids, and bile-acids. Several pharmacologic classes were also common, including antihelminthics, antibiotics, antihistamines, anticonvulsants, and anti-anginals.
Exclusion criteria were applied to the 37 hits (Online Table II), including whether compounds were commercially available in sufficient quantities, any known cardiovascular pharmacology issues, adherence to rule-of-5 filters for drug like molecules 23, and exclusion of drugs already known to protect against IR injury. This resulted in a refined list of 10 candidates, of which 6 (Table 1) were chosen for further study in a physiologically relevant model of IR injury, the Langendorff perfused rat heart. To further validate the screen, 3 molecules exhibiting the most detrimental effect on cell death, plus one molecule that exhibited no effect in the screen, were also tested in the perfused heart system.
The most protective molecule identified was the UV blocking agent dioxybenzone, but preliminary studies revealed severe negative effects on cardiac contractility during initial infusion, precluding further investigation. As Fig. 4 shows, the remaining 5 protective molecules: cloxyquin, methapyrilene, mevastatin, clorsulon, and 7-hydroxyflavone, at 1 μM yielded either a significant improvement in the post-IR recovery of cardiac function (RPP = heart rate x pressure product), or a significant reduction in myocardial infarct size, or both. Furthermore, Online Table III shows that these molecules also improved post-IR contractility (dP/dtMAX = contraction, dP/dtMIN = relaxation), and lowered the degree of hyper-contracture experienced during ischemia.
Fig. 4. Cardioprotection by hits identified from the screen.

Five molecules from the screen (Table 1) were administered to perfused rat hearts at 1 μM for 20 min., prior to 35 min. ischemia and 2 hr. reperfusion. Contractile function (rate x pressure product, RPP) is expressed as % of the value immediately prior to ischemia. Data are shown for A: cloxyquin (CLX), B: methapyrilene (MPYR), C: mevastatin (MEV), D: clorsulon (CLRS), and E: 7-hydroxyflavone (7OHF). Following reperfusion, infarct size was determined by TTC staining, with representative ventricular cross section images shown in panel F. (white = infarct, red = live tissue). Insets to panels A-E show infarct data for each group. Note that different controls were used for each molecule tested. All data are means ± SEM, N = 5-7, *p<0.05 between treatment group and vehicle controls, by ANOVA.
Further validating the screen, Online Fig. V shows that 3 detrimental molecules (apiin, allopregnanolone, and 2,4-dichlorophenoxybutyrate) all worsened the outcome of IR injury in the perfused heart system, while an ineffective molecule (propoxur) was without effect. Overall, for the 9 molecules tested, strong correlations (r2 >0.62) were observed between the primary end-point of the screening assay (normalized cell-death score), and two outcome measures of IR injury in the intact heart (recovery of rate x pressure product and infarct size). These correlation coefficients (Fig. 5) suggest that the screen has predictive power for ~65% of the effect of a molecule in the intact heart.
Figure 5. Screening score correlates with effect of molecules on intact heart IR injury.

Based on the results of round 2 of the screen, 5 protective molecules (blue), plus 3 detrimental molecules (red), one molecule without effect (propoxur) were tested in an intact perfused heart model of IR injury (see Fig. 4 and Online Fig. V). The outcome of cardiac IR was scored as recovery of rate pressure product (panel A), or infarct size (panel B), relative to untreated controls (filled black symbol in each graph). Line fits are linear regression, with r2 indicated on the graphs.
Finally, an advantage of using the Seahorse XF apparatus in this model of IR injury, was the ability to monitor O2 and pH in every well, and thus to observe metabolic parameters (OCR and ECAR, see Fig. 2) and the effects of test molecules or IR injury on metabolism. Fig. 6 shows relationships between cell death, metabolism and small molecule effects, and these data are be explored in detail in the discussion.
DISCUSSION
The goal of this study was to develop a high throughput screening method for testing the protective efficacy of small molecules in a cell model of IR injury. The model recapitulates several physiologic features of IR, including low pO2 and acidic pH, both of which were independently monitored in each well of the screen. Pre-validation tests indicated that the screen can pick-up known cardioprotective agents (Online Fig. II), and post-validation tests in a perfused heart model of IR indicate that the screen has predictive power (r2 ~0.65) for the beneficial or detrimental effect of molecules in the intact heart.
The protective hits identified by the screen are structurally and pharmacologically diverse (see Table 1), and their bioactivities warrant some discussion. Cloxyquin (Fig. 4A) is an 8-hydroxyquinoline, and its close relative clioquinol is currently under investigation as an Alzheimer’s disease therapeutic 24. Clioquinol stimulates autophagy 25, and it is thought that autophagy plays an important role in cardioprotection triggered by IPC 26. Thus, it is possible that IR protection by cloxyquin and related molecules may proceed via this mechanism.
Methapyrilene (Fig. 4B) is an H1 histamine receptor antagonist, and while histamine is known to confer detrimental effects in IR injury 27, it is unclear whether H1 antagonists in general may be cardioprotective. In a previous study we demonstrated cardioprotection by the H1 antagonist meclizine, but not by pheniramine or pyrilamine 28.
Long term treatment with mevastatin (Fig. 4C) was previously shown to afford protection in a rat model of neuronal IR injury 29. Another statin (simvastatin) is also known to be cardioprotective in-vivo 30. The current investigation administered mevastatin acutely, and in this regard it is known that mevastatin can protect neonatal cardiomyocytes against reoxygenation injury by an Akt/GSK-3β signaling mechanism31. Similarly, simvastatin 32, 33, atorvastatin 34, and rosuvastatin 35 all protected perfused hearts from IR injury when delivered acutely.
Clorsulon (Fig. 4D) is a veterinary antihelminthic. Its proposed mechanism of action is glycolytic inhibition at phosphoglycerate kinase 36, although we observed no effect of clorsulon on ECAR in the XF system. Notably, the antihelminthics clorsulon, albendazole and diethylcarbamazine were all in the top 37 hits, and all are routinely used in combination with ivermectin. Ivermectin enhances P2X4 purinergic receptor channel activity 37, and these channels are known to improve cardiac contractility 38. Thus, the cardioprotective effects of ivermectin in combination with its veterinary cohorts may be worthy of investigation.
7-hydroxyflavone (Fig. 4E) is a vasorelaxant 39, an effect mediated in part by calcium activated potassium (KCa) channels 39. The latter are implicated in the mechanism of IR protection afforded by volatile anesthetic preconditioning 40, and bona-fide KCa channel openers are known to protect the heart and brain against IR injury 41.
The 4 remaining molecules from the refined list of 10 protective hits are also interesting from a cardioprotection standpoint. Phenacemide and Beclamide are anticonvulsants, with good cardiovascular safety records. Phenacemide has been shown to inhibit aldose reductase, and important player in the polyol pathway of oxidative stress 42. While no effects of Beclamide on the heart are known, it is metabolized to 3-chloropropionate, an agonist of the GHB receptor 43 which is expressed on cardiomyocytes and drives a tachycardic response. 3,4′-dimethoxyflavone is an antagonist of the aryl hydrocarbon receptor (AhR) 44, and the known interplay between the AhR and hypoxia-inducible factor (Hif) signaling may have implications for ischemic tolerance. Albendazole inhibits fumarate reductase 45, the invertebrate analog of succinate dehydrogenase. The latter is linked to activation of mitochondrial KATP channels which are important in IPC 46. 3βHydroxy-23,24-bisnorchol-5-enic acid is a bile acid, and many similar molecules have been shown to be protective against oxidative stress.
In addition to the IR protective molecules identified herein, the validity of this screen is further established by the observation that several other known IR protective molecules yielded protective scores in the second round, including: amlodipine, perhexilene, captopril, acadesine, rotenone, amiloride, and digoxin (Online Table I). Although many known protective molecules did not proceed beyond the first round screen (including: meclizine, pravastatin, ranolazine, nitroprusside, adenosine, quinidine, warfarin, cyclosporin, clopidogrel, carvedilol, diazoxide, and resveratrol), this was likely due to their not being present at appropriate concentrations to afford protection, since pre-validation tests (Online Fig. II) confirmed protection by a number of known cardioprotective agents in this model system. Achieving the correct concentration is a common problem in HTS experiments; to re-run the entire screen at several different concentrations would be prohibitive, while choosing a single concentration will inevitably result in some molecules being outside of their effective concentration range.
Alternatively, the protective effects of these molecules in round 1 may have been cancelled out by detrimental effects of other molecules in the same well. Such generation of false-negatives is an acknowledged limitation of pooling approaches in high throughput screens 21. One approach that can mitigate this is an orthogonal pooling strategy, wherein molecules are pooled differently in multiple screening rounds, such that each molecule is tested several times in the presence of different compounds. However, orthoganol pooling is known to generate a higher rate of false positives (e.g. an inactive compound tested alongside positive compounds in all rounds would falsely register as positive), and also requires a larger number of plates or wells, and sophisticated data decoding algorithms 21. Notably, it is important to note that the pooling method used herein does not generate false positives, i.e., any protective scores in round 1 that resulted from drug interactions or synergy would have been eliminated in the round 2 screen at 1 molecule per well.
Among the detrimental and no-effect molecules that were validated in the perfused heart model (the insecticide propoxur, the herbicide 2,4-dichlorophenoxybutyrate, the neurosteroid allopregnanolone, and the flavone glycoside apiin), none to date has been linked with adverse cardiovascular effects. A number of molecules with known detrimental effects in IR injury were seen to have a detrimental effect in round 1 (e.g. the aldehyde dehydrogenase 2 inhibitor disulfiram 47), but due to the nature of the selection process from round 1 to round 2 (i.e., a focus on the protective end of the spectrum), these molecules were not pursued further. Given the current findings, it may be worthwhile to further investigate these and other detrimental molecules from the screen, to determine if they represent cardiac safety concerns.
A major advantage to the use of XF apparatus in this model is the measurement of metabolic parameters throughout the protocol. Fig. 6A shows the relationship between the effect of test molecules on OCR (Ox-Phos) vs. ECAR (glycolysis), revealing two interesting results: First, while an inverse relationship between these parameters was expected (i.e., Ox-Phos inhibition should correlate with glycolytic stimulation and vice-versa), no correlation was observed. Second, glycolysis appeared more plastic than Ox-Phos. This may simply reflect the metabolism of H9c2 cells in this system, but may also have implications for drug targeting, suggesting glycolysis is more amenable to pharmacologic manipulation than Ox-Phos.
Many Ox-Phos inhibitors are known to protect against IR injury 48, and a previous screen for molecules that alter cell metabolism identified an Ox-Phos inhibitor (meclizine) that protected against IR injury in heart and brain 28. However, in the current screen of 224 molecules, a negative correlation was seen between cell death and OCR/ECAR (Fig. 6B), suggesting that molecules which shift metabolism away from Ox-Phos toward glycolysis are detrimental. Breaking this correlation down further into its component phenomena reveals that the bulk of the effect lies within Ox-Phos (Figs. 6C and 6D). Thus, while Ox-Phos is not as amenable to pharmacologic manipulation as glycolysis (see Fig. 6A), altering Ox-Phos appears to have more impact on cell death in IR injury.
The importance of cell metabolism during recovery from IR injury was also examined. Contrary to what might be predicted, no correlation was seen between recovery of Ox-Phos and cell death (Fig. 6E). Conversely however, a significant correlation was seen between recovery of glycolysis and cell death (Fig. 6F). One reason why excessive glycolysis during reperfusion may be detrimental, is the removal of lactic acid via the sodium-hydrogen exchanger, which drives cytosolic Na+ overload 49.
Despite several advantages, there are also limitations to the current model of IR injury – for example it does not reproduce the substrate deprivation seen in ischemia. Based on measured glycolysis rates during ischemia, estimated anaerobic glucose consumption is 18 pmols min.−1 well−1. Thus, the 175 nmols of glucose in the 7 μl micro-chamber are sufficient for 162 hrs of metabolism. A non-physiological glucose level (150 μM) would be required to ensure depletion during 1 hr. of ischemia. Another caveat is the use of the cardiomyocyte-derived H9c2 cell line. While primary cardiomyocytes would be more physiologically relevant, such preparations may not be uniform enough for high-throughput approaches. Finally, the term “high-throughput” is used somewhat casually in reference to the current 24-well system, whereas clearly a 96-well XF instrument would yield a faster screen.
Finally, an important limitation of this screen was the choice to add small molecules prior to ischemia. This effectively limits the clinical applicability of any identified hits, to a prophylactic model. From the perspective of therapeutics for AMI, it would be far more desirable to deliver molecules at reperfusion (e.g. during balloon angioplasty in a percutaneous coronary intervention (PCI) setting). Thus, future iterations of this screen, delivering small molecules at reperfusion, may yield more clinically applicable hits. Furthermore, it has been suggested that the signaling mechanisms of ischemic preconditioning (IPC) and ischemic post-conditioning (IPoC) overlap significantly 50. Therefore, comparison of the cardioprotective efficacy of a large chemical library applied pre vs. post ischemia, could provide novel insight into shared signaling mechanisms between these protective paradigms.
In summary, herein we have adapted a readily available apparatus to afford a 24-well model of IR injury, and screened 2000 molecules for their effects. The screen was validated using a perfused heart model of IR, resulting in identification of 5 new protective molecules. This screening method is applicable to IR in other tissues and cell types, to enhance the discovery of therapeutics for human disease, and may also find use in the identification of molecules with a detrimental effect on cardioprotection.
Supplementary Material
What is known
There is a need for new drugs to treat cardiac ischemia-reperfusion (IR) injury associated with acute myocardial infarction.
Testing for such drugs in current animal and whole-organ models is time-consuming and suboptimal for high throughput analysis
What new information does this article contribute?
We have developed a method to expose cells growing in a 24-well plate to IR injury.
Using this new method, we screened 2000 small drug-like molecules, and identified several new candidates that protect cells from IR injury
By comparing the effects of molecules in our screen, with an established intact heart model of IR injury, we validated that the screen has predictive power for cardioprotection.
Novelty & Significance
The current models of cardiac ischemia-reperfusion (IR) injury are either very complicated and slow (e.g. perfused hearts or open heart surgery on mice), or they are overly simple and do not model the conditions of ischemia well (e.g. hypoxia in cell culture). Therefore, we developed a method to expose cells in a 24-well plate to IR injury. We used this method to screen a library of 2000 small molecules, and identified numerous hits that protected cells from IR. We then validated these molecules in more traditional and physiologically relevant models of IR. The results suggest that this screening method can be used to predict whether a given molecule (or group of molecules) will protect the heart against IR injury. We anticipate that this method will be a useful tool in the search for cardioprotective therapeutics. In addition, the application of this screening method to other types of libraries (e.g. siRNA) may provide new information regarding the involvement of various genes and proteins in the pathology of IR injury.
ACKNOWLEDGEMENTS
We thank Alan Smrcka and the University of Rochester HTS Core for technical assistance and critique of the manuscript.
SOURCES OF FUNDING This work was funded by a grant from the US National Institutes of Health (HL-071158) to PSB.
NON STANDARD ABBREVIATIONS
- CsA
Cyclosporin A
- DMEM
Dubelco’s Modified Eagle Medium
- DMSO
Dimethylsulfoxide
- ECAR
Extracellular Acidifcation Rate
- FCCP
carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- HTS
High Throughput Screening
- IPC
Ischemic Preconditioning
- IR
Ischemia-Reperfusion
- OCR
Oxygen Consumption Rate
- tPA
- TTC
tetrazolium chloride
- XF
Extracellular Flux
Footnotes
DISCLOSURES/CONFLICT OF INTEREST None
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- (1).Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S, de SG, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Greenlund KJ, Hailpern SM, Heit JA, Ho PM, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, McDermott MM, Meigs JB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Rosamond WD, Sorlie PD, Stafford RS, Turan TN, Turner MB, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–36. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- (3).Scholz W, Albus U, Counillon L, Gogelein H, Lang HJ, Linz W, Weichert A, Scholkens BA. Protective effects of HOE642, a selective sodium-hydrogen exchange subtype 1 inhibitor, on cardiac ischaemia and reperfusion. Cardiovasc Res. 1995;29:260–8. [PubMed] [Google Scholar]
- (4).Jain D, Dasgupta P, Hughes LO, Lahiri A, Raftery EB. Ranolazine (RS-43285): a preliminary study of a new anti-anginal agent with selective effect on ischaemic myocardium. Eur J Clin Pharmacol. 1990;38:111–4. doi: 10.1007/BF00265967. [DOI] [PubMed] [Google Scholar]
- (5).Griffiths EJ, Halestrap AP. Protection by Cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993;25:1461–9. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- (6).McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159–63. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- (7).Swain JL, Hines JJ, Sabina RL, Holmes EW. Accelerated repletion of ATP and GTP pools in postischemic canine myocardium using a precursor of purine de novo synthesis. Circ Res. 1982;51:102–5. doi: 10.1161/01.res.51.1.102. [DOI] [PubMed] [Google Scholar]
- (8).Shimshak TM, Preuss KC, Gross GJ, Brooks HL, Warltier DC. Recovery of contractile function in post-ischaemic reperfused myocardium of conscious dogs: influence of nicorandil, a new antianginal agent. Cardiovasc Res. 1986;20:621–6. doi: 10.1093/cvr/20.8.621. [DOI] [PubMed] [Google Scholar]
- (9).Pagel PS, Hettrick DA, Lowe D, Tessmer JP, Warltier DC. Desflurane and isoflurane exert modest beneficial actions on left ventricular diastolic function during myocardial ischemia in dogs. Anesthesiology. 1995;83:1021–35. doi: 10.1097/00000542-199511000-00016. [DOI] [PubMed] [Google Scholar]
- (10).Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, Gladwin MT, Lefer DJ. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–40. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chowdrey HS, Curtis MJ, Hearse DJ. Effects of atrial natriuretic peptide on ischaemia-induced arrhythmias in the rat heart: arrhythmogen or endogenous antiarrhythmic agent? J Cardiovasc Pharmacol. 1989;13:451–4. doi: 10.1097/00005344-198903000-00014. [DOI] [PubMed] [Google Scholar]
- (12).Downey JM, Cohen MV. Why do we still not have cardioprotective drugs? Circ J. 2009;73:1171–7. doi: 10.1253/circj.cj-09-0338. [DOI] [PubMed] [Google Scholar]
- (13).Tompkins AJ, Burwell LS, Digerness SB, Zaragoza C, Holman WL, Brookes PS. Mitochondrial dysfunction in cardiac ischemia-reperfusion injury: ROS from complex I, without inhibition. Biochim Biophys Acta. 2006;1762:223–31. doi: 10.1016/j.bbadis.2005.10.001. [DOI] [PubMed] [Google Scholar]
- (14).Nadtochiy SM, Burwell LS, Ingraham CA, Spencer CM, Friedman AE, Pinkert CA, Brookes PS. In vivo cardioprotection by S-nitroso-2-mercaptopropionyl glycine. J Mol Cell Cardiol. 2009;46:960–8. doi: 10.1016/j.yjmcc.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Gerencser AA, Neilson A, Choi SW, Edman U, Yadava N, Oh RJ, Ferrick DA, Nicholls DG, Brand MD. Quantitative microplate-based respirometry with correction for oxygen diffusion. Anal Chem. 2009;81:6868–78. doi: 10.1021/ac900881z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–6. doi: 10.1161/01.cir.84.1.350. [DOI] [PubMed] [Google Scholar]
- (17).Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, Lodge NJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res. 1997;81:1072–82. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- (18).Brennan JP, Southworth R, Medina RA, Davidson SM, Duchen MR, Shattock MJ. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res. 2006;72:313–21. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- (19).Nadtochiy SM, Zhu Q, Urciuoli W, Rafikov R, Black SM, Brookes PS. Nitroalkenes Confer Acute Cardioprotection via Adenine Nucleotide Translocase 1. J Biol Chem. 2012;287:3573–80. doi: 10.1074/jbc.M111.298406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- (21).Kainkaryam RM, Woolf PJ. Pooling in high-throughput drug screening. Curr Opin Drug Discov Devel. 2009;12:339–50. [PMC free article] [PubMed] [Google Scholar]
- (22).Lundholt BK, Scudder KM, Pagliaro L. A simple technique for reducing edge effect in cell-based assays. J Biomol Screen. 2003;8:566–70. doi: 10.1177/1087057103256465. [DOI] [PubMed] [Google Scholar]
- (23).Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- (24).Gouras GK, Beal MF. Metal chelator decreases Alzheimer beta-amyloid plaques. Neuron. 2001;30:641–2. doi: 10.1016/s0896-6273(01)00330-0. [DOI] [PubMed] [Google Scholar]
- (25).Park MH, Lee SJ, Byun HR, Kim Y, Oh YJ, Koh JY, Hwang JJ. Clioquinol induces autophagy in cultured astrocytes and neurons by acting as a zinc ionophore. Neurobiol Dis. 2011;42:242–51. doi: 10.1016/j.nbd.2011.01.009. [DOI] [PubMed] [Google Scholar]
- (26).Gottlieb RA, Finley KD, Mentzer RM., Jr Cardioprotection requires taking out the trash. Basic Res Cardiol. 2009;104:169–80. doi: 10.1007/s00395-009-0011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Valen G, Kaszaki J, Szabo I, Nagy S, Vaage J. Histamine release and its effects in ischaemia-reperfusion injury of the isolated rat heart. Acta Physiol Scand. 1994;150:413–24. doi: 10.1111/j.1748-1716.1994.tb09706.x. [DOI] [PubMed] [Google Scholar]
- (28).Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249–55. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Amin-Hanjani S, Stagliano NE, Yamada M, Huang PL, Liao JK, Moskowitz MA. Mevastatin, an HMG-CoA reductase inhibitor, reduces stroke damage and upregulates endothelial nitric oxide synthase in mice. Stroke. 2001;32:980–6. doi: 10.1161/01.str.32.4.980. [DOI] [PubMed] [Google Scholar]
- (30).Scalia R, Gooszen ME, Jones SP, Hoffmeyer M, Rimmer DM, III, Trocha SD, Huang PL, Smith MB, Lefer AM, Lefer DJ. Simvastatin exerts both anti-inflammatory and cardioprotective effects in apolipoprotein E-deficient mice. Circulation. 2001;103:2598–603. doi: 10.1161/01.cir.103.21.2598. [DOI] [PubMed] [Google Scholar]
- (31).Bergmann MW, Rechner C, Freund C, Baurand A, El JA, Dietz R. Statins inhibit reoxygenation-induced cardiomyocyte apoptosis: role for glycogen synthase kinase 3beta and transcription factor beta-catenin. J Mol Cell Cardiol. 2004;37:681–90. doi: 10.1016/j.yjmcc.2004.05.025. [DOI] [PubMed] [Google Scholar]
- (32).Szarszoi O, Maly J, Ostadal P, Netuka I, Besik J, Kolar F, Ostadal B. Effect of acute and chronic simvastatin treatment on post-ischemic contractile dysfunction in isolated rat heart. Physiol Res. 2008;57:793–6. doi: 10.33549/physiolres.931559. [DOI] [PubMed] [Google Scholar]
- (33).Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation. 1999;100:178–84. doi: 10.1161/01.cir.100.2.178. [DOI] [PubMed] [Google Scholar]
- (34).Efthymiou CA, Mocanu MM, Yellon DM. Atorvastatin and myocardial reperfusion injury: new pleiotropic effect implicating multiple prosurvival signaling. J Cardiovasc Pharmacol. 2005;45:247–52. doi: 10.1097/01.fjc.0000154376.82445.06. [DOI] [PubMed] [Google Scholar]
- (35).D’Annunzio V, Donato M, Erni L, Miksztowicz V, Buchholz B, Carrion CL, Schreier L, Wikinski R, Gelpi RJ, Berg G, Basso N. Rosuvastatin given during reperfusion decreases infarct size and inhibits matrix metalloproteinase-2 activity in normocholesterolemic and hypercholesterolemic rabbits. J Cardiovasc Pharmacol. 2009;53:137–44. doi: 10.1097/FJC.0b013e318197c5e9. [DOI] [PubMed] [Google Scholar]
- (36).Schulman MD, Ostlind DA, Valentino D. Mechanism of action of MK-401 against Fasciola hepatica: inhibition of phosphoglycerate kinase. Mol Biochem Parasitol. 1982;5:133–45. doi: 10.1016/0166-6851(82)90016-0. [DOI] [PubMed] [Google Scholar]
- (37).Priel A, Silberberg SD. Mechanism of ivermectin facilitation of human P2X4 receptor channels. J Gen Physiol. 2004;123:281–93. doi: 10.1085/jgp.200308986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hu B, Mei QB, Yao XJ, Smith E, Barry WH, Liang BT. A novel contractile phenotype with cardiac transgenic expression of the human P2X4 receptor. FASEB J. 2001;15:2739–41. doi: 10.1096/fj.01-0445fje. [DOI] [PubMed] [Google Scholar]
- (39).Calderone V, Chericoni S, Martinelli C, Testai L, Nardi A, Morelli I, Breschi MC, Martinotti E. Vasorelaxing effects of flavonoids: investigation on the possible involvement of potassium channels. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:290–8. doi: 10.1007/s00210-004-0964-z. [DOI] [PubMed] [Google Scholar]
- (40).Redel A, Lange M, Jazbutyte V, Lotz C, Smul TM, Roewer N, Kehl F. Activation of mitochondrial large-conductance calcium-activated K+ channels via protein kinase A mediates desflurane-induced preconditioning. Anesth Analg. 2008;106:384–91. doi: 10.1213/ane.0b013e318160650f. table. [DOI] [PubMed] [Google Scholar]
- (41).Cao CM, Xia Q, Gao Q, Chen M, Wong TM. Calcium-activated potassium channel triggers cardioprotection of ischemic preconditioning. J Pharmacol Exp Ther. 2005;312:644–50. doi: 10.1124/jpet.104.074476. [DOI] [PubMed] [Google Scholar]
- (42).Whittle SR, Turner AJ. Anti-convulsants and brain aldehyde metabolism: inhibitory characteristics of ox brain aldehyde reductase. Biochem Pharmacol. 1981;30:1191–6. doi: 10.1016/0006-2952(81)90296-3. [DOI] [PubMed] [Google Scholar]
- (43).Macias AT, Hernandez RJ, Mehta AK, MacKerell AD, Jr., Ticku MK, Coop A. 3-chloropropanoic acid (UMB66): a ligand for the gamma-hydroxybutyric acid receptor lacking a 4-hydroxyl group. Bioorg Med Chem. 2004;12:1643–7. doi: 10.1016/j.bmc.2004.01.025. [DOI] [PubMed] [Google Scholar]
- (44).Lee JE, Safe S. 3′,4′-dimethoxyflavone as an aryl hydrocarbon receptor antagonist in human breast cancer cells. Toxicol Sci. 2000;58:235–42. doi: 10.1093/toxsci/58.2.235. [DOI] [PubMed] [Google Scholar]
- (45).Barrowman MM, Marriner SE, Bogan JA. The fumarate reductase system as a site of anthelmintic attack in Ascaris suum. Biosci Rep. 1984;4:879–83. doi: 10.1007/BF01138170. [DOI] [PubMed] [Google Scholar]
- (46).Wojtovich AP, Brookes PS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol. 2009;104:121–9. doi: 10.1007/s00395-009-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–5. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Burwell LS, Nadtochiy SM, Brookes PS. Cardioprotection by metabolic shut-down and gradual wake-up. J Mol Cell Cardiol. 2009;46:804–10. doi: 10.1016/j.yjmcc.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Karmazyn M. The role of the myocardial sodium-hydrogen exchanger in mediating ischemic and reperfusion injury. From amiloride to cariporide. Ann N Y Acad Sci. 1999;874:326–34. doi: 10.1111/j.1749-6632.1999.tb09248.x. [DOI] [PubMed] [Google Scholar]
- (50).Hausenloy DJ, Tsang A, Yellon DM. The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med. 2005;15:69–75. doi: 10.1016/j.tcm.2005.03.001. [DOI] [PubMed] [Google Scholar]
- (51).Yamanaka N, Imanari T, Tamura Z, Yagi K. Uncoupling of oxidative phosphorylation of rat liver mitochondria by chinoform. J Biochem. 1973;73:993–8. doi: 10.1093/oxfordjournals.jbchem.a130183. [DOI] [PubMed] [Google Scholar]
- (52).Futagawa H, Kakinuma Y, Takahashi H. The role of cholinergic and noncholinergic mechanisms in the cardiorespiratory failure produced by N-methylcarbamate cholinesterase inhibitors in rabbits. Toxicol Appl Pharmacol. 2000;165:27–36. doi: 10.1006/taap.2000.8924. [DOI] [PubMed] [Google Scholar]
- (53).Riou B, Ruel P, Hanouz JL, Langeron O, Lecarpentier Y, Viars P. In vitro effects of eltanolone on rat myocardium. Anesthesiology. 1995;83:792–8. doi: 10.1097/00000542-199510000-00019. [DOI] [PubMed] [Google Scholar]
- (54).Sear JW, Prys-Roberts C, Dye J. Cardiovascular studies during induction with minaxolone. Acta Anaesthesiol Belg. 1979;30(Suppl):161–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.