

Abstract
Significance: Synaptic degeneration, an early pathological feature in Alzheimer's disease (AD), is closely correlated to impaired cognitive function and memory loss. Recent studies suggest that involvement of amyloid-beta peptide (Aβ) in synaptic mitochondrial alteration underlies these synaptic lesions. Thus, to understand the Aβ-associated synaptic mitochondrial perturbations would fortify our understanding of synaptic stress in the pathogenesis of AD. Recent Advances: Increasing evidence suggests that synaptic mitochondrial dysfunction is strongly associated with synaptic failure in many neurodegenerative diseases including AD. Based on recent findings in human AD subjects, AD animal models, and AD cellular models, synaptic mitochondria undergo multiple malfunctions including Aβ accumulation, increased oxidative stress, decreased respiration, and compromised calcium handling capacity, all of which occur earlier than changes seen in nonsynaptic mitochondria before predominant AD pathology. Of note, the impact of Aβ on mitochondrial motility and dynamics exacerbates synaptic mitochondrial alterations. Critical Issues: Synaptic mitochondria demonstrate early deficits in AD; in combination with the role that synaptic mitochondria play in sustaining synaptic functions, deficits in synaptic mitochondria may be a key factor involved in an early synaptic pathology in AD. Future Directions: The importance of synaptic mitochondria in supporting synapses and the high vulnerability of synaptic mitochondria to Aβ make them a promising target of new therapeutic strategy for AD. Antioxid. Redox Signal. 16, 1467–1475.
Introduction
Neurons are distinct from many other eukaryotic cells by the unique architecture of the long processes stemming from the cell body. Synapses are the neuronal contact sites through which neurons receive and send information from/to each other (1, 16). Energy provision and calcium fluctuation in synapses are the prerequisite of inter-neuronal communication (73); to meet the high energy demands and to cope with constant calcium flux, mitochondria are enriched in synapses for on-site energy provision and calcium modulation (20, 55, 81). It follows then that deficits in mitochondrial function and amyloid-beta peptide (Aβ) accumulation in synapses lead to reduced synaptic activity and consequent neuronal perturbations. Such concurrent synaptic alteration and mitochondrial dysfunction have been observed in many neurodegenerative diseases including the Alzheimer's disease (AD).
AD characterized by progressive memory loss and cognitive impairment is the most common type of dementia in aged people. The cognitive impairments of patients with AD are strongly associated with synaptic deficits and synaptic loss (36, 84, 89). Studies of synaptic properties have shown that synaptic damage is an early event in the pathogenesis of AD and worsens with disease progression (44, 89). Although the precise etiology of synaptic failure in AD has not yet been elucidated, Aβ is considered to be an underlying pathogenic factor. Although the list of detrimental impact of Aβ accumulation on synapses/synaptic function is ever expanding, recent studies point to mitochondrial dysfunction as a major player in the synaptic alterations seen in AD (25). Notably, recent efforts to identify the changes in synaptic mitochondria in an Aβ-rich environment are significantly advancing our understanding of the mechanisms of synaptic degeneration in AD, especially in early stages before the presence of Aβ sets in motion the devastating cognitive impairments. In this article, we will focus on the subgroup of synaptic mitochondria and summarize the progress to date in research on synaptic mitochondrial alterations in AD.
Mitochondria at Synapses
Mitochondria are the energy warehouse of eukaryotic cells. In addition to their bioenergetics trait, mitochondria play a crucial role in maintenance of intracellular calcium homeostasis and induction of apoptosis, thus meaning that mitochondria are essential organelles in cell survival. Increasing evidence suggests that mitochondria in different types of cells and even in different subcompartments of one cell differ significantly in their function, morphology, and other properties; accordingly, mitochondria within one cell can be divided into multiple subgroups (43, 83). This recognition of mitochondrial heterogeneity facilitates our understanding of mitochondrial biology and even more so of mitochondrial pathology in many pathological scenarios.
A typical pattern of mitochondrial heterogeneity is seen in neurons. According to their physical position, neuronal mitochondria are categorized into synaptic mitochondria and nonsynaptic mitochondria (19, 25, 82). Although they share the same origin, synaptic mitochondria present in various sizes, trafficking patterns, function, lifespan, and other properties compared with their relatives residing in neuronal soma (4, 12, 46). Synaptic mitochondria are defined as those docked and aggregated in synapses; they play an important role in maintaining normal synaptic function through their ability to meet the high-energy demand of synapses while maintaining synaptic calcium. It is generally accepted that synaptic mitochondria are long lived but are more vulnerable to cumulative damage than nonsynaptic mitochondria (4). Several laboratories including ours have shown that synaptic mitochondria undergo increased oxidation during aging (2, 25, 54). In addition, synaptic mitochondria have higher levels of cyclophilin D (CypD), thus rendering them more susceptible to calcium insult (4, 25, 60). Our recent study demonstrated that synaptic mitochondria had higher levels of Aβ accumulation significantly before such accumulation in nonsynaptic mitochondria in a transgenic AD mouse model overexpressing Aβ (25). Thus, improved understanding of synaptic mitochondrial biology and pathology mechanisms is especially important as we attempt to further elucidate the etiology of synaptic degeneration in pathological scenarios such as aging and AD.
Synaptic mitochondria and synaptic neurotransmission
Neurons form a network to exchange information. The propagation of information is achieved by synaptic transmission, which entails presynaptic neurotransmitter release to synaptic cleft to stimulate postsynaptic neurons. ATP is essential to many steps of synaptic transmission such as the tethering, uncoating, and refilling of synaptic vesicles (50, 59, 81). It has been proposed that ATP per se is a neurotransmitter for the fast synaptic transmission (6). Although the glycolysis process contributes a modest level of ATP, neurons normally derive ATP from aerobic respiration via mitochondrial oxidative phosphorylation (5). However, in many pathological scenarios such as ischemia, deprivation of mitochondrial respiration leads to decreased ATP production and compromised synaptic transmission (18, 68). Further, the administration of glucose to enhance ATP production can efficiently attenuate protonophore-instigated synaptic depression (9, 61). These findings suggest the importance of mitochondrial ATP in supporting normal synaptic transmission.
Calcium is another essential player in neurotransmission and is also involved in neuronal plasticity. Synaptic membrane potential depolarization induces activation of voltage-dependent calcium channels, which consequently stimulates calcium entry into presynapses with rapidly increased calcium levels at intra and presynapse sites (38, 40). The rise in calcium levels is the initiating step for synaptic vesicle transport and membrane fusion release as well as for reuptake of neurotransmitters (15, 32, 47, 76); such transient increases in synaptic calcium levels are subsequently quenched to avoid injury due to long-lasting calcium stimulation (17, 20). Prevailing opinion supports the notion that mitochondria play a central role in regulating calcium ions in synapses after calcium influx (3, 66, 67). Although mitochondria and endoplasmic reticulum (ER) are major cellular structures with calcium buffering capacity, it has been shown that synaptic mitochondria have more rapid calcium uptake than ER and that mitochondria more quickly release calcium to sustain prolonged neurotransmitter exocytosis (64, 66, 67). The communication between ER and mitochondria is probably a part of an important mechanism for regulation of intra-synaptic calcium (56, 64). Therefore, synaptic transmission relies largely on mitochondria accumulation in synapses and mitochondrial ability to modulate calcium levels. The density of synaptic mitochondria and their role in energy provision and calcium modulation are key factors working to support synaptic transmissions. Deficits in synaptic mitochondria compromise synaptic activity.
Synaptic mitochondrial generation and trafficking
Mitochondria are membrane-bound mobile organelles that undergo constant movement in neurons and accumulate in synapses to meet synaptic energy demands. Although there are several copies of DNA in mitochondrion, mitochondrial DNA only encodes 13 essential mitochondrial proteins (less than 5% of all mitochondrial proteins). In fact, most essential mitochondrial proteins are encoded by nucleic DNA and then imported into mitochondria. Mitochondrial synthesizing and packaging machineries are located in the perinuclear region of neuronal cytoplasm and are extremely deficient in dendrites and axons (13, 39). Therefore, mitochondria are generated in neuronal soma and transported to synapses via mitochondrial transport systems to achieve their synaptic support function. Based on studies to date, kinesin is responsible for mitochondrial anterograde transport that carries mitochondria from soma to the distal axon tip (35, 63); and dynein is important for mitochondrial retrograde transport that inversely sends mitochondria toward the proximate axon end (63). Adaptor proteins play an important role in binding mitochondria to motor proteins (kinesin and dynein). A variety of adaptor proteins have been identified to connect mitochondria to kinesin, that is, Miro (70), Milton (57) and syntabulin (77). These adaptor proteins play a dual role in mitochondrial trafficking—they tie mitochondria to kinesin, thus enabling mitochondrial transport from soma to synapses; on the other side, these adaptor proteins dissociate from kinesin to enable mitochondrial docking around synapses. For example, Miro, a calcium sensor protein, recognizes a high level of calcium and then discharges mitochondria from kinesin (7, 48). Mitochondrial transport is also regulated by multiple intracellular signaling pathways such as protein kinase A (PKA) (69), glycogen synthase kinase 3β (GSK 3β) (21, 69), and mitogen-activated protein kinase (MAPK) (75) that control mitochondrial undocking from motor proteins. The finely controlled mitochondrial trafficking and docking processes enable mitochondria to be physically adjacent to synapses to modulate synaptic function.
Alterations of Synaptic Mitochondria in AD
Mitochondrial dysfunction is recognized as a predominant AD pathological change that occurs concurrently with synaptic alterations and exacerbates disease progression. Based on the critical role of synaptic mitochondria in sustaining synaptic activity, recent efforts have focused on unraveling the mechanisms driving synaptic mitochondrial dysfunction as a potential player in synaptic failure in AD.
Deficits in synaptic mitochondrial function in AD
Investigations into the pathogenesis of AD in the past century have yielded a large body of evidence showing that Aβ and Aβ-associated cellular changes are important causative factors underlying neuronal perturbation and synaptic distress in AD. Aβ is a small molecule cleaved from amyloid-beta precursor protein (APP) by the proteolysis of β- and γ-secretases. Aβ plaques and intra-cellular, particularly intra-mitochondrial Aβ, are pathological features in AD. Notably, recent studies revealed that Aβ accumulates inside AD brain mitochondria including synaptic mitochondria (Fig. 1) (11, 22–26, 30, 37, 49, 51, 62, 79, 80, 92, 93) and that the levels of mitochondrial Aβ are associated with abnormalities of mitochondrial structure and function. Indeed, mitochondrial malfunction has been documented in the studies on human AD and AD animal models as well as Aβ-overexpressing cell models, thus suggesting that deficits in mitochondrial function occur in an Aβ-rich environment. The most recognized forms of mitochondrial dysfunction in AD include Aβ accumulation in brain mitochondria, decreased mitochondrial ATP provision, elevated mitochondria-associated oxidative stress, increased mitochondrial permeability transition, reduced mitochondrial calcium modulating capacity, impaired respiratory function, and release of pro-apoptogenic factors from mitochondria (10, 24–26, 42, 49, 62, 65, 71, 79, 80, 92). Synaptic mitochondria are an early target of Aβ, thus demonstrating early pathological changes before observed global brain mitochondrial damage.
FIG. 1.
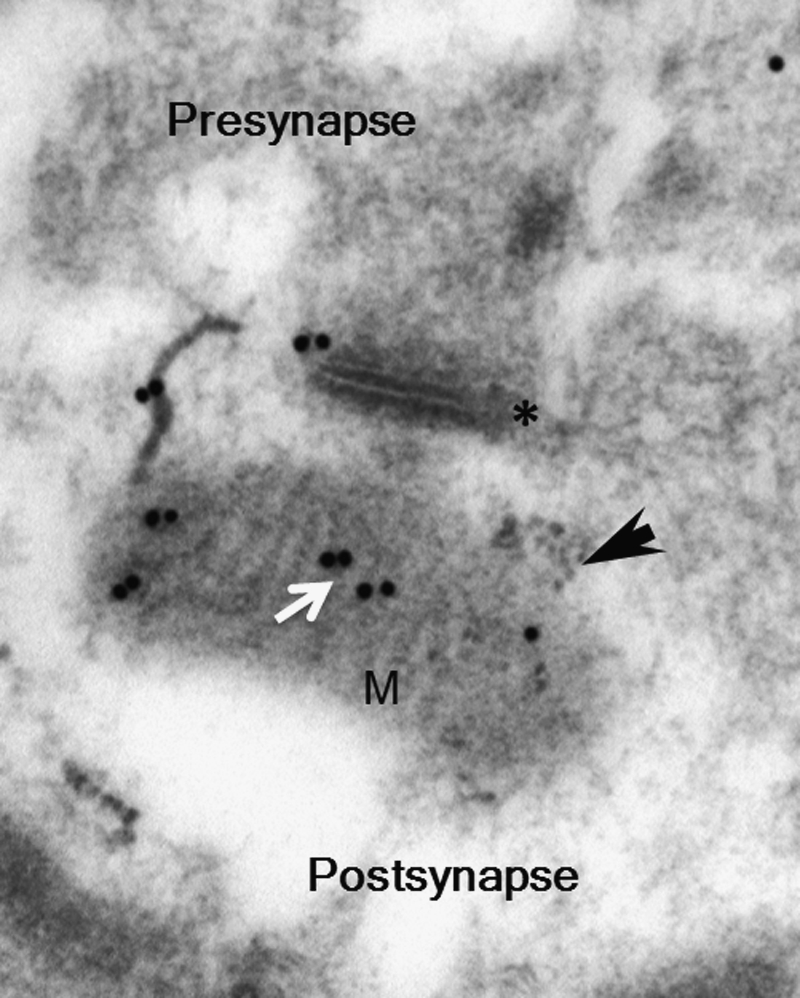
Accumulation of Aβ in synaptic mitochondria. Immunogold electron microscopy images with a specific Aβ1–42 antibody followed by gold-conjugated antibody (18 nm) to show the presence of intra-mitochondrial Aβ accumulation (white arrow) in 12-month-old Tg mAPP. The black arrow denotes mitochondria. The asterisk (*) denotes a synapse. M indicated mitochondria. Aβ, amyloid-beta peptide; APP, amyloid-beta precursor protein.
Synaptosomes are membrane-sealed neuronal terminals containing synaptic mitochondria and other synapse-related structures such as synaptic vesicles and lysosomes (29, 72). In a Mungarro-Menchaca et al. study, incubation of synaptosomes from Wistar rats with Aβ25–35 (58) altered ultrastructure of synaptosomes with swollen synaptic mitochondria and significantly reduced the number of synaptic vesicles. The combination of Aβ with Ryanodine, an agonist of Ryanodine receptor, exacerbated the aforementioned ultrastructural changes of synaptic mitochondria and synaptic vesicles, thus suggesting that Aβ toxicity is more predominant in calcium-stressed synaptosomes (58). Additionally, an independent in vitro experiment using Aβ1–40 to treat synaptosomes isolated from C57B mice demonstrated remarkable synaptic mitochondrial changes, including membrane potential collapse, mitochondrial calcium accumulation, and increased free radical production (41); these studies raise the possibility of the potential impact of Aβ on synaptic activity and synaptic mitochondrial properties. Lastly, an in vivo study in human patients with AD and an AD mouse model (Tg2576 mice) provided direct evidence of the existence of Aβ and Tau pathology in synaptosomes (33, 34).
Since synaptic mitochondria are more vulnerable to cumulative damage induced by deleterious factors such as Aβ, it is possible that synaptic mitochondria undergo earlier changes than detectable alterations in global brain mitochondria. An AD mouse model (Tg mAPP mice) demonstrates age-dependent brain Aβ accumulation starting in 4–5-month-old mice and exacerbating with age, thus mimicking the process of Aβ pathology in human AD (25). Tg mAPP synaptic mitochondria from young mice in which no brain Aβ plaques were found showed significant accumulation of Aβ; in contrast, the Aβ levels in the same preparations of Tg mAPP nonsynaptic mitochondria were significantly lower (Fig. 2). Both Tg mAPP mouse synaptic and nonsynaptic mitochondria demonstrated elevated Aβ levels with age, but Tg mAPP synaptic mitochondria had a more prominent increase in Aβ levels than nonsynaptic mitochondria in Tg mAPP mice at the same age. These results provide evidence that synaptic mitochondria are more vulnerable to Aβ accumulation than nonsynaptic mitochondria and that synaptic mitochondrial Aβ aggregation is an early mitochondrial pathological process relevant to the Aβ pathology.
FIG. 2.

Synaptic mitochondria are more vulnerable to Aβ accumulation. Synaptic mitochondria and nonsynaptic mitochondria were prepared from Tg mAPP mice at age of 4 months and subjected to measure the levels of Aβ1–40 and Aβ1–42. The data showed that levels of both Aβ species were significantly increased in synaptic mitochondria.
As a result, Aβ-insulted synaptic mitochondria from young Tg mAPP mice undergo significant decline in respiratory function and cytochrome c oxidase activity, increased oxidative stress, and enhanced probability of mitochondrial permeability transition pore processes (25). Further, Aβ-insulted synaptic mitochondria also showed increased expression of CypD and Aβ-binding alcohol dehydrogenase (ABAD). Both mitochondrial proteins (CypD and ABAD) have been shown to interact with Aβ, thereby accelerating and exacerbating mitochondrial stress in an Aβ-rich environment (24, 26, 27, 49, 60, 80, 91). In contrast, nonsynaptic mitochondria isolated from young Tg mAPP mice demonstrated preserved function comparable to those in age-matched wild type mice. However, both synaptic and nonsynaptic mitochondria from aged Tg mAPP mice presented compromised function, but changes in synaptic mitochondria were more profound. These findings suggest that synaptic mitochondria are an early victim of Aβ and undergo pathological changes before nonsynaptic mitochondria. Consistent with our observation, Gillardon's study reported detectable Aβ accumulation in synaptosomal fractions from young Tg2576 mice (another AD mouse model) before detectable brain Aβ plaques; accordingly, synaptic mitochondria were functionally compromised in these young Tg2576 mice (34). Taken together, these data indicate that synaptic mitochondrial functional disturbances are early deficits in the progress of AD and synaptic mitochondrial dysfunction is a key player in Aβ-mediated mitochondrial and neuronal toxicity.
Synaptic mitochondrial transport and dynamics change in AD
Mitochondria are mobile and dynamic organelles. The accumulation of mitochondria in synapses depends on mitochondrial transport to neuronal terminals. The constant mitochondrial fusion and fission regulates mitochondrial density and morphology and is closely related to mitochondrial function. Indeed, concomitant mitochondrial dysfunction and motility change has been observed in patients with AD and Aβ overexpression cell lines as well as in AD animal models. As a major causative factor of AD, Aβ disrupts mitochondrial motility and dynamics in neurites, thus resulting in disorganized synaptic mitochondrial distribution. Rui et al. showed that acute treatment with 20 μM Aβ25–35 on cultured cortical neurons caused a significant decrease in neuronal mitochondrial movement (69). Several other laboratories have obtained similar results regarding the impact of Aβ on neuritic mitochondrial movement including axonal mitochondrial transport (8, 21, 25, 85). Two recent studies using chronic treatment of low concentration, for example, 200 nM, oligomeric Aβ on neurons, which mimics the Aβ toxicity in AD brains, reported significant alterations in axonal mitochondrial transport (21, 25). Impairment on anterograde movement of Aβ–superimposed axonal mitochondria is more pronounced than on retrograde movement (8, 25) as shown by decreased percentage of anterograde mitochondria and reduced mobility of anterograde speed. In contrast, the retrograde speed is not significantly decreased after treatment with 200 nM oligomeric Aβ for 24 h (Fig. 3) (25). The precise mechanism of this discrepancy in impairment on mitochondrial anterograde and retrograde movement remains unknown. Given that PKA and GSK 3β signaling cascades are implicated in Aβ-induced mitochondrial trafficking impairments (21, 69) and that activation of PKA or suppression of GSK 3β rescues Aβ-injured mitochondrial motility (21, 69), interference with the mitochondria-dependent signal transduction pathway might be a key factor involved in synaptic mitochondrial transport and trafficking such as Aβ-mediated injury.
FIG. 3.

Aβ affects axonal mitochondrial movement. Representative kymograph images of the vehicle and Aβ1–42-treated axonal mitochondrial movement showing injured axonal mitochondrial movement. The arrows denote the traces of mitochondrial movement.
In addition to changes in mitochondrial movement, Aβ also is an instigating factor in abnormal mitochondrial dynamics, thus leading to defects in mitochondrial fusion and fission. A series of studies from the Zhu's laboratory demonstrated the altered expression levels of mitochondrial fusion- and fission-related proteins that are thought to be associated with increased mitochondrial fission and lowered mitochondrial fusion in an Aβ-rich environment (86–88). This imbalance in mitochondrial dynamics resulted in increased mitochondrial fragmentation, decreased neuritic mitochondrial density, and altered synaptic mitochondrial distribution. Similar changes have been further observed in patients with AD, APP overexpressing cell lines, and Aβ-treated primary cultured neurons in several studies (8, 25, 52, 87, 88) including ours (Fig. 4). Among many changes in mitochondrial fusion and fission proteins in AD, dynamin-like protein 1 (Dlp1) has been intensively studied—reports showed changes in Dlp1 expression levels (87), increased Drp1 S-nitrosylation (14), and Drp1 interaction with Aβ (52), thus implicating the role of Dlp1 in neuronal mitochondrial dynamics changes seen in AD.
FIG. 4.

Aβ alters axonal mitochondrial distribution. The axonal mitochondrial density is decreased after Aβ1–42 (200 nM) treatment for 24 h on cultured hippocampal neurons (Lower panel). *p<0.05 vs. cells treated with vehicle or reversed Aβ42-1. The upper panel shows representative images for vehicle- or Aβ-treated axonal mitochondrial distribution. Double immunostaining with Mitotracker (red, mitochondrial marker) and Tau (green, axonal marker) was performed. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
Changes in Aβ-related neuronal/synaptic mitochondrial motility and dynamics are closely related to synaptic mitochondrial malfunction as well as consequent impaired synaptic function. Therefore, although the studies on mitochondrial motility and dynamics change in AD are still at an early stage, elucidation of the detailed mechanisms will be likely to fully depict the role of synaptic mitochondrial morphological and transport changes in mitochondrial and neuronal injury in AD.
Summary and Perspectives
Increasing evidence suggests the close correlation of brain mitochondrial dysfunction with synaptic degeneration, both of which are early lesions in AD. Here, we focused on changes in synaptic mitochondria in AD, particularly on the exposure to Aβ and summarized recent progress in the study of Aβ-related synaptic mitochondrial pathology. Due to their physical proximity to synapses, synaptic mitochondria demonstrate a critical role in directly supporting synaptic activity; in addition, synaptic mitochondria undergo constant activation to meet energy demands and control calcium modulation in synapses. Correspondingly, in comparison to somatic mitochondria, synaptic mitochondria are more vulnerable to cumulative damages due to Aβ insult (4, 25, 60). Synaptic mitochondria demonstrate early Aβ accumulation, impaired respiration, lowered calcium handling capacity, and enhanced oxidative stress (25). Importantly, synaptic mitochondria undergo pathological changes before nonsynaptic mitochondria in presymptomatic AD animal models even in conditions of low or undetectable levels of Aβ in nonsynaptic mitochondria. In scenarios with substantial Aβ pathology, synaptic mitochondrial injury is still more predominant than nonsynaptic mitochondrial damage. Moreover, the negative effect of Aβ on synaptic mitochondrial motility and dynamics significantly and adversely impacts normal synaptic distribution, density, and morphology, which, in turn, substantially compromises effects of synaptic mitochondria on synaptic function (Figs. 5 and 6). Thus, although alterations of synaptic mitochondria in AD are a nascent target of AD pathogenesis, the resultant early deficits and high vulnerability of synaptic mitochondria in response to Aβ toxicity have attracted intensive research efforts. A more complete understanding of synaptic mitochondrial dysfunction in AD will also broaden our knowledge regarding the pathogenesis of this neurodegenerative disease. Current study in this field is just beginning, and more detailed mechanisms of synaptic mitochondrial pathology in AD should be elucidated to address the following questions: (i) what are the reasons/mechanisms of early Aβ deposition in synaptic mitochondria; (ii) what are the precise mechanisms controlling synaptic mitochondrial motility and dynamics in “normal” and Aβ-related scenarios; and (iii) what is the impact of early synaptic mitochondrial dysfunction on synapses. We believe that the answers to these questions will provide new insights into synaptic mitochondrial structure/properties relevant to AD pathogenesis and, in particular, Aβ-mediated synaptic pathology.
FIG. 5.

Schematic figure to show the role of synaptic mitochondria in supporting synaptic activity. Synaptic mitochondria are generated in neuronal soma and further transported to synapses. Normal synaptic mitochondrial movement, docking, and dynamics are crucial features for mitochondria to exert their function on ATP production, calcium modulation, and regulation of cell signaling cascades, consequently maintaining synaptic plasticity and transmission. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
FIG. 6.

Working hypothesis. In the presence of Aβ, mitochondrial transport and dynamics are injured with compromised synaptic mitochondrial structure and function, thus leading to decreased energy metabolism, dysregulated calcium homeostasis, and perturbed cell signaling cascades, eventually leading to synaptic injury and cognitive dysfunction.
Early changes in synaptic mitochondria before devastating Aβ pathology in AD and the importance of synaptic mitochondria for normal synaptic activity make synaptic mitochondria a preferential target for the treatment of AD. Previous studies have provided evidence for the protection of mitochondrial function in ameliorating synaptic changes and the consequent cognitive impairments in AD animal models. A representative example of the many mitochondria-protecting approaches is the application of antioxidants to human AD, AD animal and cell models, which demonstrates significant amelioration of mitochondrial/neuronal dysfunction against Aβ toxicity (28, 31, 74, 90). More recent studies suggested the striking efficacy of mitochondria-targeting antioxidants including MitoQ, SS peptides, and MitoE in strengthening mitochondria in their respiration, membrane potential, and calcium-handling capacity from toxic insults including the Aβ (45, 53, 78). It is noted that MitoQ and SS31 significantly reversed Aβ-induced CypD elevation, mitochondrial fusion/fission proteins imbalance, and neurite growth in AD cell models, thus suggesting the close relationship between neuronal mitochondrial dysfunction and neuronal/synaptic perturbation and the value of eliminating neuronal mitochondrial oxidative stress in the treatment of neuronal/synaptic alterations in AD (53). Thereby, interventions affecting mitochondrial activity are likely promising therapeutic strategies for halting and treating AD. Rescuing and protecting synaptic mitochondria in presymptomatic subjects or in patients suffering from AD with mild cognitive impairments may be appropriate targets for amelioration of synaptic alterations at the early stage of AD.
Abbreviations Used
- Aβ
amyloid-beta peptide
- ABAD
amyloid beta-peptide binding alcohol dehydrogenase
- AD
Alzheimer's disease
- APP
amyloid-beta precursor protein
- CypD
cyclophilin D
- Dlp1
dynamin-like protein 1
- ER
endoplasmic reticulum
- GSK 3β
glycogen synthase kinase 3β
- MAPK
mitogen-activated protein kinase
- PKA
protein kinase A
Acknowledgments
This work was supported by the National Institute on Aging (NIH/NIA PO1AG017490, R37AG037319, R21AG040011, and K99AG037716) and the Alzheimer's Association.
References
- 1.Alonso-Nanclares L. Gonzalez-Soriano J. Rodriguez JR. DeFelipe J. Gender differences in human cortical synaptic density. Proc Natl Acad Sci U S A. 2008;105:14615–14619. doi: 10.1073/pnas.0803652105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banaclocha MM. Hernandez AI. Martinez N. Ferrandiz ML. N-acetylcysteine protects against age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res. 1997;762:256–258. doi: 10.1016/s0006-8993(97)00493-9. [DOI] [PubMed] [Google Scholar]
- 3.Billups B. Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown MR. Sullivan PG. Geddes JW. Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. J Biol Chem. 2006;281:11658–11668. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- 5.Buchanan ME. Davis RL. A distinct set of Drosophila brain neurons required for neurofibromatosis type 1-dependent learning and memory. J Neurosci. 2010;30:10135–10143. doi: 10.1523/JNEUROSCI.0283-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnstock G. Historical review: ATP as a neurotransmitter. Trends Pharmacol Sci. 2006;27:166–176. doi: 10.1016/j.tips.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Cai Q. Sheng ZH. Moving or stopping mitochondria: Miro as a traffic cop by sensing calcium. Neuron. 2009;61:493–496. doi: 10.1016/j.neuron.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calkins MJ. Reddy PH. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer's disease neurons. Biochim Biophys Acta. 2011;1812:507–513. doi: 10.1016/j.bbadis.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calupca MA. Prior C. Merriam LA. Hendricks GM. Parsons RL. Presynaptic function is altered in snake K+-depolarized motor nerve terminals containing compromised mitochondria. J Physiol. 2001;532:217–227. doi: 10.1111/j.1469-7793.2001.0217g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cardoso SM. Santos S. Swerdlow RH. Oliveira CR. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001;15:1439–1441. doi: 10.1096/fj.00-0561fje. [DOI] [PubMed] [Google Scholar]
- 11.Caspersen C. Wang N. Yao J. Sosunov A. Chen X. Lustbader JW. Xu HW. Stern D. McKhann G. Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 12.Chang DT. Honick AS. Reynolds IJ. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci. 2006;26:7035–7045. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang DT. Reynolds IJ. Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience. 2006;141:727–736. doi: 10.1016/j.neuroscience.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 14.Cho DH. Nakamura T. Fang J. Cieplak P. Godzik A. Gu Z. Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collin T. Marty A. Llano I. Presynaptic calcium stores and synaptic transmission. Curr Opin Neurobiol. 2005;15:275–281. doi: 10.1016/j.conb.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Connors BW. Long MA. Electrical synapses in the mammalian brain. Annu Rev Neurosci. 2004;27:393–418. doi: 10.1146/annurev.neuro.26.041002.131128. [DOI] [PubMed] [Google Scholar]
- 17.Csordas G. Thomas AP. Hajnoczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999;18:96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dale N. Frenguelli BG. Release of adenosine and ATP during ischemia and epilepsy. Curr Neuropharmacol. 2009;7:160–179. doi: 10.2174/157015909789152146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dave KR. DeFazio RA. Raval AP. Torraco A. Saul I. Barrientos A. Perez-Pinzon MA. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase C epsilon. J Neurosci. 2008;28:4172–4182. doi: 10.1523/JNEUROSCI.5471-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.David G. Barrett EF. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol. 2003;548:425–438. doi: 10.1113/jphysiol.2002.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Decker H. Lo KY. Unger SM. Ferreira ST. Silverman MA. Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci. 2010;30:9166–9171. doi: 10.1523/JNEUROSCI.1074-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devi L. Prabhu BM. Galati DF. Avadhani NG. Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dragicevic N. Mamcarz M. Zhu Y. Buzzeo R. Tan J. Arendash GW. Bradshaw PC. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer's transgenic mice. J Alzheimers Dis. 2010;20(Suppl 2):S535–S550. doi: 10.3233/JAD-2010-100342. [DOI] [PubMed] [Google Scholar]
- 24.Du H. Guo L. Fang F. Chen D. Sosunov AA. McKhann GM. Yan Y. Wang C. Zhang H. Molkentin JD. Gunn-Moore FJ. Vonsattel JP. Arancio O. Chen JX. Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du H. Guo L. Yan S. Sosunov AA. McKhann GM. Yan SS. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du H. Guo L. Zhang W. Rydzewska M. Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2011;32:398–406. doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du H. Yan SS. Mitochondrial permeability transition pore in Alzheimer's disease: cyclophilin D and amyloid beta. Biochim Biophys Acta. 2010;1802:198–204. doi: 10.1016/j.bbadis.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dumont M. Lin MT. Beal MF. Mitochondria and antioxidant targeted therapeutic strategies for Alzheimer's disease. J Alzheimers Dis. 2010;20(Suppl 2):S633–S643. doi: 10.3233/JAD-2010-100507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dunkley PR. Jarvie PE. Robinson PJ. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc. 2008;3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- 30.Eckert A. Hauptmann S. Scherping I. Rhein V. Muller-Spahn F. Gotz J. Muller WE. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener Dis. 2008;5:157–159. doi: 10.1159/000113689. [DOI] [PubMed] [Google Scholar]
- 31.Escribano L. Simon AM. Perez-Mediavilla A. Salazar-Colocho P. Del Rio J. Frechilla D. Rosiglitazone reverses memory decline and hippocampal glucocorticoid receptor down-regulation in an Alzheimer's disease mouse model. Biochem Biophys Res Commun. 2009;379:406–410. doi: 10.1016/j.bbrc.2008.12.071. [DOI] [PubMed] [Google Scholar]
- 32.Faas GC. Adwanikar H. Gereau RWt. Saggau P. Modulation of presynaptic calcium transients by metabotropic glutamate receptor activation: a differential role in acute depression of synaptic transmission and long-term depression. J Neurosci. 2002;22:6885–6890. doi: 10.1523/JNEUROSCI.22-16-06885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fein JA. Sokolow S. Miller CA. Vinters HV. Yang F. Cole GM. Gylys KH. Co-localization of amyloid beta and tau pathology in Alzheimer's disease synaptosomes. Am J Pathol. 2008;172:1683–1692. doi: 10.2353/ajpath.2008.070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillardon F. Rist W. Kussmaul L. Vogel J. Berg M. Danzer K. Kraut N. Hengerer B. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics. 2007;7:605–616. doi: 10.1002/pmic.200600728. [DOI] [PubMed] [Google Scholar]
- 35.Glater EE. Megeath LJ. Stowers RS. Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gouras GK. Tampellini D. Takahashi RH. Capetillo-Zarate E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 2010;119:523–541. doi: 10.1007/s00401-010-0679-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansson Petersen CA. Alikhani N. Behbahani H. Wiehager B. Pavlov PF. Alafuzoff I. Leinonen V. Ito A. Winblad B. Glaser E. Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105:13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hendricson AW. Thomas MP. Lippmann MJ. Morrisett RA. Suppression of L-type voltage-gated calcium channel-dependent synaptic plasticity by ethanol: analysis of miniature synaptic currents and dendritic calcium transients. J Pharmacol Exp Ther. 2003;307:550–558. doi: 10.1124/jpet.103.055137. [DOI] [PubMed] [Google Scholar]
- 39.Hollenbeck PJ. The pattern and mechanism of mitochondrial transport in axons. Front Biosci. 1996;1:d91–d102. doi: 10.2741/a118. [DOI] [PubMed] [Google Scholar]
- 40.Huang X. Senatore A. Dawson TF. Quan Q. Spafford JD. G-proteins modulate invertebrate synaptic calcium channel (LCa(v)2) differently from the classical voltage-dependent regulation of mammalian Ca(v)2.1 and Ca(v)2.2 channels. J Exp Biol. 2010;213:2094–2103. doi: 10.1242/jeb.042242. [DOI] [PubMed] [Google Scholar]
- 41.Keller JN. Lauderback CM. Butterfield DA. Kindy MS. Yu J. Markesbery WR. Amyloid beta-peptide effects on synaptosomes from apolipoprotein E-deficient mice. J Neurochem. 2000;74:1579–1586. doi: 10.1046/j.1471-4159.2000.0741579.x. [DOI] [PubMed] [Google Scholar]
- 42.Kim HS. Lee JH. Lee JP. Kim EM. Chang KA. Park CH. Jeong SJ. Wittendorp MC. Seo JH. Choi SH. Suh YH. Amyloid beta peptide induces cytochrome C release from isolated mitochondria. Neuroreport. 2002;13:1989–1993. doi: 10.1097/00001756-200210280-00032. [DOI] [PubMed] [Google Scholar]
- 43.Kuznetsov AV. Margreiter R. Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int J Mol Sci. 2009;10:1911–1929. doi: 10.3390/ijms10041911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lacor PN. Buniel MC. Furlow PW. Clemente AS. Velasco PT. Wood M. Viola KL. Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leo S. Szabadkai G. Rizzuto R. The mitochondrial antioxidants MitoE(2) and MitoQ(10) increase mitochondrial Ca(2+) load upon cell stimulation by inhibiting Ca(2+) efflux from the organelle. Ann N Y Acad Sci. 2008;1147:264–274. doi: 10.1196/annals.1427.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Z. Okamoto K. Hayashi Y. Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 47.Liu SQ. Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- 48.Liu X. Hajnoczky G. Ca2+ -dependent regulation of mitochondrial dynamics by the Miro-Milton complex. Int J Biochem Cell Biol. 2009;41:1972–1976. doi: 10.1016/j.biocel.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lustbader JW. Cirilli M. Lin C. Xu HW. Takuma K. Wang N. Caspersen C. Chen X. Pollak S. Chaney M. Trinchese F. Liu S. Gunn-Moore F. Lue LF. Walker DG. Kuppusamy P. Zewier ZL. Arancio O. Stern D. Yan SS. Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 50.Ly CV. Verstreken P. Mitochondria at the synapse. Neuroscientist. 2006;12:291–299. doi: 10.1177/1073858406287661. [DOI] [PubMed] [Google Scholar]
- 51.Manczak M. Anekonda TS. Henson E. Park BS. Quinn J. Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 52.Manczak M. Calkins MJ. Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet. 2011;13:2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Manczak M. Mao P. Calkins MJ. Cornea A. Reddy AP. Murphy MP. Szeto HH. Park B. Reddy PH. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J Alzheimers Dis. 2010;20(Suppl 2):S609–S631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez M. Hernandez AI. Martinez N. Ferrandiz ML. Age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res. 1996;731:246–248. doi: 10.1016/0006-8993(96)00708-1. [DOI] [PubMed] [Google Scholar]
- 55.Miller KE. Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- 56.Mironov SL. Symonchuk N. ER vesicles and mitochondria move and communicate at synapses. J Cell Sci. 2006;119:4926–4934. doi: 10.1242/jcs.03254. [DOI] [PubMed] [Google Scholar]
- 57.Misko A. Jiang S. Wegorzewska I. Milbrandt J. Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30:4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mungarro-Menchaca X. Ferrera P. Moran J. Arias C. beta-Amyloid peptide induces ultrastructural changes in synaptosomes and potentiates mitochondrial dysfunction in the presence of ryanodine. J Neurosci Res. 2002;68:89–96. doi: 10.1002/jnr.10193. [DOI] [PubMed] [Google Scholar]
- 59.Murthy VN. De Camilli P. Cell biology of the presynaptic terminal. Annu Rev Neurosci. 2003;26:701–728. doi: 10.1146/annurev.neuro.26.041002.131445. [DOI] [PubMed] [Google Scholar]
- 60.Naga KK. Sullivan PG. Geddes JW. High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J Neurosci. 2007;27:7469–7475. doi: 10.1523/JNEUROSCI.0646-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nicholls DG. Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- 62.Pagani L. Eckert A. Amyloid-Beta interaction with mitochondria. Int J Alzheimers Dis. 2011;2011:925050. doi: 10.4061/2011/925050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pilling AD. Horiuchi D. Lively CM. Saxton WM. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell. 2006;17:2057–2068. doi: 10.1091/mbc.E05-06-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pivovarova NB. Pozzo-Miller LD. Hongpaisan J. Andrews SB. Correlated calcium uptake and release by mitochondria and endoplasmic reticulum of CA3 hippocampal dendrites after afferent synaptic stimulation. J Neurosci. 2002;22:10653–10661. doi: 10.1523/JNEUROSCI.22-24-10653.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reddy PH. Manczak M. Mao P. Calkins MJ. Reddy AP. Shirendeb U. Amyloid-beta and mitochondria in aging and Alzheimer's disease: implications for synaptic damage and cognitive decline. J Alzheimers Dis. 2010;20(Suppl 2):S499–S512. doi: 10.3233/JAD-2010-100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rizzuto R. Calcium mobilization from mitochondria in synaptic transmitter release. J Cell Biol. 2003;163:441–443. doi: 10.1083/jcb.200309111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rizzuto R. Bernardi P. Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol. 2000;529(Pt 1):37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robin E. Simerabet M. Hassoun SM. Adamczyk S. Tavernier B. Vallet B. Bordet R. Lebuffe G. Postconditioning in focal cerebral ischemia: role of the mitochondrial ATP-dependent potassium channel. Brain Res. 2011;1375:137–146. doi: 10.1016/j.brainres.2010.12.054. [DOI] [PubMed] [Google Scholar]
- 69.Rui Y. Tiwari P. Xie Z. Zheng JQ. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26:10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saotome M. Safiulina D. Szabadkai G. Das S. Fransson A. Aspenstrom P. Rizzuto R. Hajnoczky G. Bidirectional Ca2+ -dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saraiva LM. Seixas da Silva GS. Galina A. da-Silva WS. Klein WL. Ferreira ST. De Felice FG. Amyloid-beta triggers the release of neuronal hexokinase 1 from mitochondria. PloS One. 2010;5:e15230. doi: 10.1371/journal.pone.0015230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sato Y. Yamanaka H. Toda T. Shinohara Y. Endo T. Comparison of hippocampal synaptosome proteins in young-adult and aged rats. Neurosci Lett. 2005;382:22–26. doi: 10.1016/j.neulet.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 73.Shepherd GM. Harris KM. Three-dimensional structure and composition of CA3—>CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci. 1998;18:8300–8310. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siedlak SL. Casadesus G. Webber KM. Pappolla MA. Atwood CS. Smith MA. Perry G. Chronic antioxidant therapy reduces oxidative stress in a mouse model of Alzheimer's disease. Free Radic Res. 2009;43:156–164. doi: 10.1080/10715760802644694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stagi M. Gorlovoy P. Larionov S. Takahashi K. Neumann H. Unloading kinesin transported cargoes from the tubulin track via the inflammatory c-Jun N-terminal kinase pathway. FASEB J. 2006;20:2573–2575. doi: 10.1096/fj.06-6679fje. [DOI] [PubMed] [Google Scholar]
- 76.Stevens CF. Sullivan JM. The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron. 2003;39:299–308. doi: 10.1016/s0896-6273(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 77.Su Q. Cai Q. Gerwin C. Smith CL. Sheng ZH. Syntabulin is a microtubule-associated protein implicated in syntaxin transport in neurons. Nat Cell Biol. 2004;6:941–953. doi: 10.1038/ncb1169. [DOI] [PubMed] [Google Scholar]
- 78.Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006;8:E521–E531. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Takuma K. Fang F. Zhang W. Yan S. Fukuzaki E. Du H. Sosunov A. McKhann G. Funatsu Y. Nakamichi N. Nagai T. Mizoguchi H. Ibi D. Hori O. Ogawa S. Stern DM. Yamada K. Yan SS. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc Natl Acad Sci U S A. 2009;106:20021–20026. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takuma K. Yao J. Huang J. Xu H. Chen X. Luddy J. Trillat AC. Stern DM. Arancio O. Yan SS. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 2005;19:597–598. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- 81.Verstreken P. Ly CV. Venken KJ. Koh TW. Zhou Y. Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 82.Vos M. Lauwers E. Verstreken P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front Synaptic Neurosci. 2010;2:139. doi: 10.3389/fnsyn.2010.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Waagepetersen HS. Hansen GH. Fenger K. Lindsay JG. Gibson G. Schousboe A. Cellular mitochondrial heterogeneity in cultured astrocytes as demonstrated by immunogold labeling of alpha-ketoglutarate dehydrogenase. Glia. 2006;53:225–231. doi: 10.1002/glia.20276. [DOI] [PubMed] [Google Scholar]
- 84.Wakabayashi K. Honer WG. Masliah E. Synapse alterations in the hippocampal-entorhinal formation in Alzheimer's disease with and without Lewy body disease. Brain Res. 1994;667:24–32. doi: 10.1016/0006-8993(94)91709-4. [DOI] [PubMed] [Google Scholar]
- 85.Wang X. Perry G. Smith MA. Zhu X. Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener Dis. 2010;7:56–59. doi: 10.1159/000283484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang X. Su B. Fujioka H. Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol. 2008;173:470–482. doi: 10.2353/ajpath.2008.071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang X. Su B. Lee HG. Li X. Perry G. Smith MA. Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang X. Su B. Siedlak SL. Moreira PI. Fujioka H. Wang Y. Casadesus G. Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wilcox KC. Lacor PN. Pitt J. Klein WL. Abeta oligomer-induced synapse degeneration in Alzheimer's disease. Cell Mol Neurobiol. 2011 doi: 10.1007/s10571-011-9691-4. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu H. Wang H. Zhuang L. Yan B. Yu Y. Wei Z. Zhang Y. Dyck LE. Richardson SJ. He J. Li X. Kong J. Li XM. Demonstration of an anti-oxidative stress mechanism of quetiapine: implications for the treatment of Alzheimer's disease. FEBS J. 2008;275:3718–3728. doi: 10.1111/j.1742-4658.2008.06519.x. [DOI] [PubMed] [Google Scholar]
- 91.Yan SD. Stern DM. Mitochondrial dysfunction and Alzheimer's disease: role of amyloid-beta peptide alcohol dehydrogenase (ABAD) Int J Exp Pathol. 2005;86:161–171. doi: 10.1111/j.0959-9673.2005.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yao J. Du H. Yan S. Fang F. Wang C. Lue LF. Guo L. Chen D. Stern DM. Gunn Moore FJ. Xi Chen J. Arancio O. Yan SS. Inhibition of amyloid-{beta} (A{beta}) peptide-binding alcohol dehydrogenase-A{beta} interaction reduces A{beta} accumulation and improves mitochondrial function in a mouse model of Alzheimer's disease. J Neurosci. 2011;31:2313–2320. doi: 10.1523/JNEUROSCI.4717-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yao J. Irwin RW. Zhao L. Nilsen J. Hamilton RT. Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]