

Abstract
Significance: Mitochondria and brain bioenergetics are increasingly thought to play an important role in Alzheimer's disease (AD). Recent Advances: Data that support this view are discussed from the perspective of the amyloid cascade hypothesis, which assumes beta-amyloid perturbs mitochondrial function, and from an opposite perspective that assumes mitochondrial dysfunction promotes brain amyloidosis. A detailed review of cytoplasmic hybrid (cybrid) studies, which argue mitochondrial DNA (mtDNA) contributes to sporadic AD, is provided. Recent AD endophenotype data that further suggest an mtDNA contribution are also summarized. Critical Issues and Future Directions: Biochemical, molecular, cybrid, biomarker, and clinical data pertinent to the mitochondria–bioenergetics–AD nexus are synthesized and the mitochondrial cascade hypothesis, which represents a mitochondria-centric attempt to conceptualize sporadic AD, is discussed. Antioxid. Redox Signal. 16, 1434–1455.
Introduction
Historically, Alzheimer's disease (AD) was defined as a clinical dementia syndrome that occurs in conjunction with brain beta-amyloid (Aβ) plaques and tau tangle deposits (1, 6, 140, 177, 180, 256). More recently, the definition was extended to include a “preclinical” form characterized by Aβ changes and normal cognition (2, 178, 248). The preclinical AD diagnostic criteria designate Aβ an “upstream” marker and all other recognized neuroimaging and biochemical phenomena as “downstream” markers (248).
Aβ's upstream designation is consistent with the amyloid cascade hypothesis (107–109), which postulates the Aβ (99) byproduct of amyloid precursor protein (APP) degradation (135) causes AD. Outside of rare familial autosomal dominant forms, though, it is unclear why Aβ dynamics change in AD. After all, Aβ is constantly produced in brains of young and old people. Extracellular Aβ levels rise during the day and fall during sleep (136). Interstitial Aβ falls after severe closed head injuries, and rising levels signal clinical recovery (27). Clearly, Aβ production is a regulated process and the simple presence of Aβ in the brain does not necessarily initiate AD. What, then, could possibly constitute the “upstream” regulator of brain Aβ? This review argues mitochondria and cell bioenergetics (Fig. 1) regulate Aβ, and that in sporadic AD, changes in mitochondrial function and cell bioenergetics occur upstream to Aβ changes.
FIG. 1.

The mitochondrion and its relationship to bioenergetic fluxes. Under normal conditions, neuron mitochondria may depend heavily on astrocyte-generated lactate as a carbon fuel source, and for this reason the reaction from lactate to pyruvate is explicitly indicated. The conversion of lactate to pyruvate definitely occurs in the cytosol, and some researchers believe this conversion may also occur within the mitochondrion itself. In general, though, carbon from several sources including carbohydrates, fatty acids, and amino acids can feed into the Krebs cycle. Reactions in the Krebs cycle reduce NAD+ to NADH and FAD to FADH2. High-energy electrons from NADH enter the ETC at complex I, and high energy electrons from FADH2 enter the ETC at complex II (not shown). As electrons flow through the ETC from high to low energy states, energy from those electrons is used to pump protons from the matrix to the intermembrane space and create a proton gradient. Due to electrochemical and pH gradients, protons in the intermembrane space are directed to re-access the matrix through complex V (the ATP synthase) and energy captured from this proton flux is used to phosphorylate ADP. Also shown is the mtDNA, which encodes catalytically critical parts of the complex I, III, IV, and V holoenzymes. CoQ, coenzyme; Cyt. C, cytochrome C.
Mitochondria Are Increasingly Implicated in AD and AD Models
Altered oxidative metabolism in AD was reported in the 1960s (89), and abnormal glucose utilization was noted throughout the 1970s and beyond (25, 67, 87, 90, 126, 243, 255). During this time, changes to the main bioenergetics organelle, the mitochondrion, were observed on several levels (263). Mitochondrial ultrastructure was perturbed, and activities of several mitochondria-localized enzymes (including pyruvate dehydrogenase complex and α-ketoglutarate dehydrogenase complex) were reduced (97, 128, 212, 247, 291). Mitochondrial oxygen consumption in AD subject frontal cortex homogenates was shown to differ from control subject homogenates (243). Reduced brain oxygen utilization was demonstrated using oxygen-15 positron tomography (88, 92). Interestingly, bioenergetics and mitochondrial changes were found to extend beyond the brain to nondegenerating tissues such as fibroblasts and lymphocytes (20, 22, 96, 97, 214, 215, 241, 242). Pioneering investigators postulated energy metabolism might constitute an important feature of AD (22, 97, 242), but interest in this line of investigation was largely restricted to the field's periphery.
In 1990, reduced activity of the electron transport chain (ETC) enzyme complex IV (cytochrome oxidase; COX) was demonstrated by Parker et al. (203). The AD COX defect in this study was identified through studies of platelet mitochondria. A similar finding was subsequently demonstrated in independent studies of brain, platelet, and fibroblast mitochondria (24, 31, 40, 62, 144, 165, 176, 189, 204, 205, 240, 280, 281, 284, 292). Some researchers attributed the COX activity reduction to declining COX protein or COX subunit mRNA levels (42, 43, 111, 145). Others reported the enzyme's kinetic properties, and therefore the enzyme structure itself, were altered or that the enzyme was improperly assembled (206, 284). For example, in AD brain mitochondria, COX activity was reduced even when referenced to its own aa3 subunit (206), thus suggesting reduced AD brain COX activity is not simply or solely a consequence of less COX holoenzyme.
Mitochondrial mass, number, and content were shown to be changed in oxidatively stressed hippocampal pyramidal neurons from AD brains (113). These neurons showed increased levels of mtDNA, mtDNA molecules containing a specific 5 kb deletion, and the mtDNA-encoded COX1 protein subunit. This was not due, however, to increased numbers of intact mitochondria or, at least in the case of mtDNA, increased amounts of mtDNA within intact mitochondria. Rather, increases were driven by an expanded pool of autophagocytized mitochondria. While this mitochondrial pool increased, the number and mass of normal mitochondria actually fell. Because the amount of PCR-amplifiable mtDNA also drops in AD brains (28, 55, 65, 113, 220), it is presumed mtDNA within autophagosomes is not extracted or amplified using standard techniques. Additional AD brain morphometric studies further found reduced mitochondrial number, mass, and size; disrupted cristae; and altered intracellular distributions (9). Moreover, these changes were not limited to cells and regions that manifest concomitant Aβ or protein aggregation changes.
Other studies quantifying AD brain mitochondrial mass and mitochondrial COX protein content emphasize that the stage of disease, or at least the stage of disease a given neuron is in, influences what is observed. Using immunochemical and mitochondrial dye approaches, de la Monte et al. noted that, despite an overall reduction in mitochondrial mass, especially in terms of COX content, considerable heterogeneity between individual brains and neurons within brains exist (65). The authors postulated less severely affected neurons mount ultimately unsustainable compensatory increases in COX protein and mitochondrial mass. This interpretation is consistent with the findings of Nagy et al., who found “healthy-appearing” neurons in AD brain hippocampi had increased COX protein levels, while tangle-bearing neurons lacked COX protein (190).
During the 1990s, mitochondrial defects were demonstrated in other neurodegenerative diseases, and it was proposed the brain might be particularly susceptible to mitochondrial dysfunction (259). Oxidative stress was documented in AD brains (138, 160, 170, 172, 199, 245, 246) and it was postulated that mitochondria, a well-recognized source of reactive oxygen species (ROS) production (39), could be responsible (11, 264). Mitochondria were discovered to play a central role in programmed cell death (115, 148, 216, 301), a process felt possibly to mediate neuron loss in AD (57, 65, 66, 151, 194, 222, 231, 244, 251). Age-dependent increases in mtDNA damage, either in the form of oxidative modifications or frank mutations, were documented (54, 179). The age-dependent nature of these changes offered a potential explanation for why AD prevalence increased so much with advancing age. Cytoplasmic hybrid (cybrid) modeling of AD mitochondrial dysfunction further suggested AD-specific mtDNA signatures existed and were physiologically relevant (257). AD cybrid studies are discussed in detail in a following section.
As interest in mitochondria grew, speculation arose over the possible presence of an Aβ-mitochondria nexus. Initial attempts to address this involved exposing cultured cells or isolated mitochondria to Aβ. These studies found Aβ impaired mitochondrial respiration and inhibited COX (30, 35, 61, 207, 210), thereby providing a potential mechanism through which AD brain mitochondrial dysfunction could arise. Subsequently, reduced brain respiration was demonstrated in multiple Aβ-producing transgenic mouse models (74, 76, 112, 298, 299). Related studies from transgenic mice reported expression of genes encoding mitochondrially-located proteins and respiratory chain subunits increased (218), and that brain regions that do not accumulate Aβ may actually show elevated COX activity (249). This suggested Aβ-induced mitochondrial dysfunction triggers a compensatory response. Consistent with this finding, Diana et al. reported that in cultured cells low concentration Aβ exposure decreases mitochondrial mass, fragments mtDNA, and initiates an apparent compensatory increase in mtDNA synthesis (73).
Adding Aβ to cell cultures, though, does not inhibit all bioenergetic pathways. Allaman et al. found exposing cultured astrocytes to Aβ increased glucose uptake as well as glycolysis, pentose phosphate shunt, Krebs cycle, and glycogen synthesis fluxes (4). These flux increases may reflect the fact that from a metabolism perspective the brain is not a homogeneous entity. Instead, astrocytes and neurons likely constitute functionally interactive units (3, 163, 209, 253).
In 2001, Cardoso et al. reported Aβ exposures that were toxic to human teratocarcinoma cells were not toxic to teratocarcinoma cells depleted of endogenous mtDNA (ρ0 cells) (33). Because mtDNA encodes subunits belonging to complexes I, III, IV, and V, these holoenzymes are not fully functional in ρ0 cells. With proper metabolic support, though, ρ0 cells remain viable and expand in culture. In the native teratocarcinoma cells, Aβ decreased MTT reduction, increased extracellular LDH, inhibited ETC enzymes, reduced ATP levels, and depolarized mitochondria. These effects were not observed in the Aβ-exposed ρ0 cells. In the native cells, antioxidant pretreatments minimized some of the Aβ-induced changes, including decreased MTT reduction. While this study did not explicitly demonstrate a physical interaction between Aβ and either mitochondria or the ETC, it showed in cell culture that some aspects of Aβ toxicity are mediated through direct or indirect mitochondrial effects. Following this, a number of studies reported APP and Aβ co-localize with mitochondria in both AD autopsy brains and AD transgenic mice (7, 8, 36, 61, 72, 74, 75, 106, 157, 162, 167, 296, 298). Interestingly, the APP-cleaving gamma secretase complex is also present within mitochondria (105, 271).
Over the last decade, investigators have reported several specific Aβ–mitochondria interactions. In studies involving AD autopsy brains and Aβ-producing transgenic mice, Aβ was found to bind a redox enzyme, Aβ-binding alcohol dehydrogenase (ABAD), within the mitochondrial matrix (162). Aβ-binding to ABAD deformed the enzyme, prevented NAD binding, and interfered with enzyme activity. Blocking the Aβ–ABAD interaction minimized cognitive decline in this model. In a subsequent publication, this team reported Aβ also interacted with cyclophilin D, a constituent of the mitochondrial permeability transition pore, and that binding of Aβ to cyclophilin D activated a mitochondrial permeability transition (75).
Despite the fact and perhaps because the number of intact mitochondria is reduced in degenerating neurons, in AD brains the balance between mitochondrial fission and fusion is shifted in favor of fission (168, 287–289). The proteins that mediate mitochondrial fusion, Opa1, Mfn1, and Mfn2, are reduced. Fis1, which mediates mitochondrial fission, is increased. Drp1, which also facilitates fission, was reduced in one study but increased in another (168, 288). In the study where Drp1 was increased, Drp1 mRNA expression was similarly elevated (168). This study also reported a physical association between Aβ and Drp1. In the studies where Drp1 was reduced, exposing cultured neuronal cells or primary neurons to Aβ fragmented mitochondria and altered mitochondrial fission–fusion protein levels (288). The intracellular mitochondrial distribution shifted from neurites and axons and towards the perinuclear region. Overexpressing Opa1 and Drp1 mitigated Aβ-induced mitochondrial fragmentation and restored a normal mitochondrial distribution pattern.
Overall, multiple cell and animal-based studies show Aβ functionally and structurally alters mitochondria. While the sequence of this relationship is consistent with the amyloid cascade hypothesis, it is important to note the experimental models used for these studies (adding Aβ to cell cultures, and transgenic mice whose mutant APP expression facilitates Aβ production) do not address whether a converse relationship also exists.
This point is critical, as data suggest mitochondrial function affects Aβ, and mitochondrial dysfunction promotes amyloidosis. One study found exposing APP-expressing COS cells to a combination of sodium azide, a COX inhibitor, and 2-deoxyglucose, a glycolysis inhibitor, profoundly affected APP processing (93). This treatment, which presumably reduced ATP levels, shifted cell APP processing to the endoplasmic reticulum (ER) and yielded an “amyloidogenic” derivative. As defined by the authors, this new amyloidogenic derivative consisted of an 11.5 kD APP fragment that contained the entire Aβ amino acid sequence. The sodium azide-2 deoxyglucose treatment therefore appeared to shift APP processing away from its α-secretase cleavage. Similar effects were seen in neuroglioma cells and following treatment with CCCP, a respiratory chain uncoupler.
Other studies have focused on whether bioenergetic manipulation affects APP α-secretase cleavage. One report tested the effect of 2-deoxyglucose and oligomycin, an ATP synthase inhibitor, on differentiated PC12 cell (a rat adrenal pheochromocytoma cell line) APP processing (290). This intervention decreased α-secretase-mediated APP cleavage. The authors speculated reducing α-secretase activity might shift APP processing towards the amyloidogenic direction. Another study performed using COS cells found glucose deprivation, 2-deoxyglucose, and sodium azide independently decreased levels of the soluble α-secretase-derived APP product (94). APP mRNA levels remained unchanged, so diminished amounts of the α-secretase-produced APP product was probably not an artifact of reduced APP. Also, glutathione restored the α-secretase-generated product to normal and even supranormal levels suggesting ROS, conditions that occur in conjunction with ROS, or some other glutathione-sensitive parameter shifts APP processing from the α-secretase cut. In this context, data that show ROS activates the APP β-secretase (BACE) and increases Aβ production are potentially relevant (47, 268, 269).
Fukui et al. evaluated how bioenergetic manipulation affects mouse brain amyloidosis (91). The authors crossed transgenic mice expressing mutated human APP and PSEN1 genes with mice that had a Cre–loxP-mediated knockout of the neuron COX10 gene. COX10 synthesizes a heme component that constitutes part of the COX holoenzyme. The brains of the crossed mice showed reduced numbers of amyloid plaques. Oxidative stress, BACE activity, and Aβ42 levels were also lower. It is unclear why COX10 knockout reduced brain oxidative stress and COX1 levels, since these changes are opposite to what is observed in the AD brain (113). It is perhaps conceivable COX10 knockout lowered levels of an otherwise normal COX holoenzyme, lowered COX holoenzyme levels reduced the overall respiratory flux, a reduced respiratory flux lowered free radical production, lowered free radical production deactivated BACE, and less Aβ was produced. If correct, the Fukui et al. report could join other APP transgenic mouse studies that found manipulating oxidative stress alters brain Aβ dynamics or plaque accumulation. Pertinent studies include those showing paraquat-induced oxidative stress increased brain Aβ levels, increasing periredoxin 3 decreased brain Aβ levels, increasing MnSOD decreased plaque accumulation, and reducing MnSOD increased plaque accumulation (47, 78, 155).
Khan et al. determined Aβ levels in SH-SY5Y cybrid cell lines expressing AD and control subject mtDNA (141). AD cybrid lines had reduced COX activity and increased ROS production. Intracellular and extracellular levels of the 40 and 42 amino acid Aβ species were elevated in AD cybrid cell cultures. These and other data that argue mitochondria play a key role in AD are discussed below.
Could Mitochondria Cause AD?
The first demonstration of reduced AD subject COX activity came from studies of platelet mitochondria (203). Although it is currently recognized that systemic biochemical and clinical changes associate with AD, and therefore AD may not be a brain-limited disease (22, 260), in 1990 this was not a generally acknowledged view. Why, then, did Parker et al. decide to assay ETC activities in platelet mitochondria? In this case, the decision to assay mitochondria from a nondegenerating tissue was based on epidemiologic and mitochondrial genetic principles. Specifically, the authors recognized that most AD cases, and especially late-onset AD cases, typically do not demonstrate recognizable Mendelian inheritance patterns. Parker, who was at that time developing a genetic theory of sporadic disease that was based on mitochondrial DNA (mtDNA) inheritance (201, 202), postulated mtDNA might contribute to the apparently sporadic late-onset form of AD. Immediately prior to his AD platelet study, Parker and colleagues had successfully shown that subjects with Parkinson's disease, another neurodegenerative disorder in which only a minority demonstrate Mendelian inheritance, have reduced platelet and muscle mitochondria complex I activity (17, 202). If his sporadic disease hypothesis was correct, Parker predicted, a systemic ETC defect should also exist in AD.
Parker's sporadic disease hypothesis was based on several mtDNA principles including maternal inheritance, heteroplasmy, threshold, and mitotic segregation (Fig. 2). Because mtDNA is essentially all inherited maternally (98), inherited mtDNA mutations cannot produce Mendelian disease patterns. Maternal inheritance might arise from inherited mtDNA mutations, but only in situations where the mutations are present in adequate amounts in both the germ line and somatic tissues. Due to heteroplasmy, threshold, and mitotic segregation, these conditions might not be met. For example, a heteroplasmic mutation might only be present in a limited number of the mtDNA molecules of an ovum. Because during mitosis mtDNA molecules are stochastically distributed to daughter cells, different tissues may end up with different mtDNA mutation burdens. A relatively high level of mutation, therefore, may end up in the nervous system but not the germ line, in which case a carrier female's chances of developing a neurologic disease would exceed her chance of transmitting a neurologic disease. Alternatively, if a relatively high level of mutation ends up in the germ line but not the nervous system, a carrier female's chances of transmitting a neurologic disease to her offspring would exceed her chances of developing the disease. This latter scenario takes into account the concept of threshold, which assumes that in order to cause disease, the burden of mtDNA mutation in an affected tissue must exceed the threshold at which the mutation or mutations produce biochemical consequences.
FIG. 2.

Maternal inheritance, heteroplasmy, mitotic segregation, and threshold effects let mtDNA play a role in sporadic-appearing diseases. In females, diploid oogonia produce haploid oocytes during embryogenesis. If a heteroplasmic mtDNA mutation is present within an oogonium, oocytes with different mtDNA mutation burdens may result. In general, oocytes with high mutation levels are more likely to produce disease-affected offspring, but due to mitochondrial segregation that occurs after fertilization, different tissues may contain different levels of the mutation. Females with a high mutation burden in the nervous system but not their germ cells will themselves have a high risk of developing the neurologic disease and a low risk of transmission to the next generation. Females with a high mutation burden in their germ cells but not the nervous system have a high risk of transmission to the next generation and a low risk of developing the neurologic disease. Therefore, although the mtDNA mutation is maternally inherited, the disease it associates with appears mostly as a sporadic disorder with perhaps a subtle maternal inheritance bias.
Next, the question of whether mtDNA contributes to low platelet mitochondria COX activity in AD subjects needed to be addressed. COX is a 13-subunit holoenzyme, and three of its subunits are encoded by genes on mtDNA. The transcription and translation of these mtDNA subunits also depends on the integrity of a set of mtDNA rRNA and tRNA genes, as well as a regulatory region that influences mtDNA replication and expression. To evaluate mtDNA's contribution to the AD platelet mitochondria COX defect, a cybrid approach was used.
AD Cybrids
Cybrid cells are generated by mixing the contents of non-nucleated cells, or cytoplasts, with nucleated cells (257) (Fig. 3). This approach was developed during the 1970s to explore the functional consequences of unique mtDNA species (29, 286). In its earliest permutation, investigators created cybrid cell lines with mixed mtDNA populations; the mtDNA in these lines consisted of a nucleated cell's endogenous mtDNA plus the cytoplast's mtDNA. In the 1980s, investigators discovered how to fully deplete cell lines of endogenous mtDNA (70, 71, 181). These lines were called ρ0 cell lines, since prior to its identification as mtDNA cytoplasmic DNA was classified as “ρ” DNA (83). As ρ0 lines became available, investigators began using them to produce cybrid lines whose mtDNA derived entirely from a cytoplast mitochondrial donor (143). In 1994 it was shown that platelets, which contain mitochondria but not nuclei, function effectively as cytoplasts when mixed with ρ0 cells to produce cybrid cells (49).
FIG. 3.

The cybrid technique. mtDNA-depleted (ρ0) cells line and non-nucleated cytoplasts or platelets are mixed in the presence of a plasma membrane-disrupting detergent. After the detergent is diluted, membrane reconstitution generates some cells that contain a nucleus from a ρ0 cell, ρ0 cytosolic contents, and cytoplast cytosolic contents. Only cybrid cells that contain cytoplast mtDNA can go on to produce functional ETCs and survive a subsequent selection process. After the selection process is complete, the resulting cybrid cell line is used for biochemical and molecular analyses. Because cybrid cell lines created from a common ρ0 cell nuclear background have equivalent nuclear genes, and the cell expansion environment is equivalent between cell lines, biochemical or molecular differences between lines are expected to reflect differences between their mtDNA content.
The first studies of cybrid cell lines containing AD subject mtDNA were published in 1997 (64, 233, 264). These cell lines are referred to as “AD cybrid lines” or simply as “AD cybrids”. To produce the AD cybrids, blood samples were taken from subjects diagnosed with AD. Platelets were isolated from each individual blood sample, and the platelet mitochondria from each platelet preparation were incorporated into ρ0 cells derived from either human neuroblastoma SH-SY5Y cells or teratocarcinoma NT2 cells. At the same time that the AD cybrids were generated, platelet mitochondria obtained from non-AD, age-matched control subjects were also used to generate “control cybrids”. In both the neuroblastoma and teratocarcinoma-based studies, mean COX Vmax activities were lower in the AD cybrid group than they were in the corresponding control cybrid group (64, 233, 264). Reduced COX activity was subsequently observed in several independent neuroblastoma and teratocarcinoma AD cybrid series (32, 37, 95, 195–197, 276, 277). Across studies the magnitude of this reduction has ranged from about 15% to 50%. The defect is greatest when assayed in enriched mitochondrial fractions, and least when studied in whole cells. This bioenergetic lesion, either directly or indirectly, is further associated with a reduced cell ATP level; in the NT2 AD cybrid series of Cardoso et al., the ATP level was lowered by 28% (32). This effect is presumed to be COX mediated, since complex I activities between groups, when analyzed, have been comparable (64, 95, 233, 277).
It is important to note that cybrid cell lines are created not through the transfer of isolated mtDNA, but rather through the transfer of whole mitochondria to ρ0 cells. An assumption is made that all transferred cytoplast components that cannot perpetuate independently of the host cell nucleus degrade over time and dilute over the course of repeated cell divisions. Therefore, any transferred cytoplast component that cannot perpetuate itself independent of the cell nucleus should not have a sustainable molecular or biochemical effect. Theoretically, a cytoplast's only self-perpetuating component is its mtDNA. For this reason, the most straightforward explanation of these cybrid studies is that the AD cybrid COX activity reduction is determined by mtDNA.
In AD cybrids, the mtDNA-derived COX defect produces other features that recapitulate phenomena observed in AD subject brains (Fig. 4). Altered mitochondrial morphology, membrane potential, free radical production, calcium handling, movement, and mtDNA synthesis rates are observed. Cell transcription factor activities, calcium homeostasis, intracellular signaling, energy levels, apoptosis markers, and APP processing are perturbed.
FIG. 4.
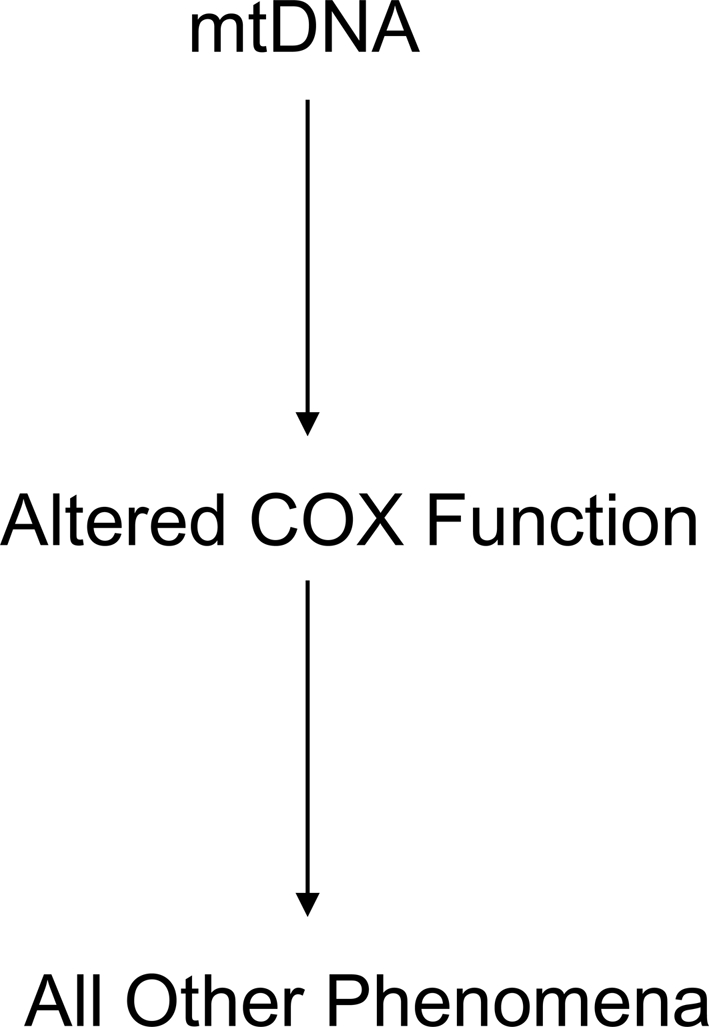
The chain of events in AD cybrids. Altered COX function in AD cybrids presumably arises from and reflects mtDNA differences between the AD and control subjects that provided each cell line's mtDNA. Because mtDNA only encodes components of the respiratory chain, all other unique biochemical and molecular changes that occur in a cybrid line are consequences of its mtDNA-determined respiratory chain function.
Electron microscopy (EM) reveals AD cybrids have increased numbers of enlarged mitochondria with swollen pale matrices; disrupted, reduced cristae; broken outer membranes; and crystal-like inclusions (276, 277, 302). Despite this, relative to control cybrids, the AD cybrid average mitochondrial size is reduced. The overall cell mitochondrial mass is equivalent between AD and control lines, though, because AD cybrid cells appear to contain more mitochondria (276). Preserved mitochondrial mass has been visually demonstrated through EM, and by biochemical assays that find AD cybrids maintain normal citrate synthase and complex I activities (31, 64, 95, 233, 276, 277). Mitochondrial degradation rates in AD cybrids have not been directly characterized, but mitochondrial mtDNA synthesis is increased in AD cybrids and may represent part of a compensatory response to mitochondrial dysfunction (276). Some of these findings reflect changes observed in human AD brains, which show an increased frequency of disrupted mitochondria and generally smaller mitochondria that potentially reflect a shift in the mitochondrial fission–fusion balance towards fission (9, 113, 168, 288). While preservation of mitochondrial mass is not typical of the AD brain, several caveats are worth noting. First, Hirai et al. found that when cell mtDNA is quantified so that intact mitochondria, disrupted mitochondria, and phagosome mtDNA are accounted for, the AD neuron total mtDNA content is actually elevated (113). Second, total cell COX protein is increased in what are apparently healthier AD brain neurons (65, 113, 190). Third, during early AD stages, COX subunit gene expression is increased while complex I subunit gene expression is not (169). Fourth, early in life AD transgenic (tg) 2576 mice show upregulated expression of genes that encode mitochondria-localized proteins (218). Therefore, by increasing their mtDNA synthesis rate and maintaining their mitochondrial mass, AD cybrids are possibly demonstrating a compensatory response that seems to initially occur (but ultimately fail) in AD brain neurons.
Compared to differentiated control cybrids, differentiated SH-SY5Y AD cybrids have a higher percentage of sedentary mitochondria (275). The mitochondria that are moving have a slower mean velocity. Direct studies of AD brain also suggest mitochondrial transport abnormalities occur, and that mitochondria are directed away from axons and neurites (254, 288).
Past fibroblast and lymphocyte studies demonstrate that calcium dynamics are altered in AD (96, 124, 150, 214, 215). The ability of mitochondria to regulate calcium levels is also perturbed in AD cybrids. The total amount of calcium that mitochondria can store, as well as the rate at which they can store it, is reduced (233). This may partly explain why AD cybrids have increased cytosolic calcium levels under basal conditions (233), and why AD cybrid mitochondria, when subjected to cyclosporin A-mediated hyperpolarization, fail to show the typical membrane potential oscillation response (i.e.,“flickering”) that occurs in cyclosporin-treated normal cells (272). Reduced mitochondrial calcium sequestration is also consistent with a relative depolarization of the mitochondrial membrane potential, a phenomenon that has been shown in several AD cybrid studies (37, 141, 276, 277).
Under physiologic conditions, mitochondrial superoxide production is associated with mitochondrial membrane potential hyperpolarization (26). AD cybrid mitochondria, which are relatively depolarized, also generate excess superoxide (16, 32, 68, 195, 233, 264, 302). Mitochondrial ROS overproduction in the setting of a low membrane potential suggests that AD cybrids have a fundamentally defective ETC. Additional evidence of AD cybrid oxidative stress includes increased markers of lipid peroxidation, including thiobarbituric acid reactive substances (TBARS) and 4-hydroxynonenol (4-HNE) protein adducts, and increased protein carbonylation (32, 196). Similar changes are also observed in AD brains (139). AD cybrids also show upregulated activities of several antioxidant enzymes (AOEs), including catalase, copper-zinc SOD, MnSOD, glutathione peroxidase, and glutathione reductase (264). This most likely represents a compensatory, yet ultimately unsuccessful, response to enhanced mitochondrial ROS production. Increased oxidative stress is of course a well-documented feature of the AD brain (113, 193, 213, 245, 250).
In AD cybrids, oxidative stress mediates, at least in part, the observed mitochondrial membrane potential depolarization. Administering trolox, a water-soluble vitamin E analog and antioxidant, to AD cybrids increased their mitochondrial membrane potential (141). Other cybrid data provide potential mechanistic insight into how oxidative stress affects the membrane potential, and also influences other phenomena typical of the AD brain. One particularly interesting finding is that AD cybrids overproduce both Aβ40 and Aβ42. Khan et al. reported a series of SH-SY5Y AD cybrids secreted Aβ40 and Aβ42 at approximately twice the rate of a control cybrid group (141). This study also found AD cybrids contained more intracellular Aβ40 than the control cybrids; intracellular Aβ42 levels trended towards an increase but this difference was not significant. Subsequently, Onyango et al. reported that, compared to SH-SY5Y control cybrids, AD cybrid intracellular and conditioned medium Aβ42 levels were increased (195). Since oxidative stress is believed to activate BACE, the enzyme that processes APP to its Aβ derivative (47, 268), an ROS-mediated increase in AD cybrid Aβ production might be expected. Increased basal Aβ levels may also explain why exposing AD cybrids to exogenous Aβ40 causes an exaggerated decline in their mitochondrial membrane potential (32), and why exposing them to Aβ25–35 exacerbates their already impaired ability to handle an inositol triphosphate (IP3)-mediated ER calcium release (233).
AD cybrids show profound alterations in intracellular stress signaling pathways. Relative to control cybrid lines AKT phosphorylation is increased, as is phosphorylation of the p38, JNK, and ERK1/2 serine-threonine kinases (195, 197, 302). The status of AKT in the AD brain is unclear, as its intracellular distribution and absolute amount seem to change, but available data suggest the AKTser-473 phosphorylated to nonphosphorylated ratio increases (101, 159, 208, 219). ERK, p38, and JNK phosphorylation is also increased in the AD brain (101, 304, 305). AD cybrid studies suggest these activating phosphorylations arise as a consequence of increased oxidative stress, since they are reduced by the isoflavone antioxidant puerarin and other antioxidants (195, 302). Further, in AD cybrids, oxidative stress-mediated p38 and JNK activation may confer harmful consequences, as p38 and JNK inhibition protects these cells from externally-induced oxidative stress (192, 196). AKT pathway and ERK1/2 activation, on the other hand, appear to play a protective role as the specific inhibition of either AKT or ERK1/2 reduces AD cybrid cell viability (196, 197, 228). These viability-reducing and promoting associations are consistent with existing mitogen activated protein kinase (MAPK) data (192, 228). Tyrosine kinase activity, which is also activated by ROS (133), is additionally increased in AD cybrids (195, 197).
AD cybrids manifest evidence of decreased neurotrophin signaling. TrkA and p75NTR receptors are depressed, and the activity of the remaining TrkA receptors is reduced (195). Despite this, exposing AD cybrids to nerve growth factor (NGF) normalizes their elevated cytosolic calcium level and improves their ability to recover from an IP3-mediated ER calcium release. This has led some to argue NGF treatment may benefit AD patients (234). TrkA levels are also decreased in the basal forebrain and cortex of AD brains (23, 60, 226).
The phosphoinositide (PI) hydrolysis-dependent signal transduction system is perturbed in both AD brain (129) and AD cybrids (68). PI signaling occurs when phospholipase C (PLC) cleaves a phosphorylated inositol (an inositol phosphate such as IP3) from a membrane phosphatidylinositol phospholipid; phosphatidylinositol phospholipids are collectively called PIs. De Sarno et al. assessed the integrity of this system in AD and control cybrid lines under basal conditions and following exposure to carbachol, a cholinergic agonist that binds M1 metabotropic receptors and activates PI hydrolysis (68). Although G-protein and PLC levels were equivalent between groups, in AD cybrids basal and carbachol-stimulated PI hydrolysis rates were higher, the G-protein activator sodium fluoride had an overly robust effect, as did the PLC activator ionomycin, and glutathione depletion had a blunted effect. These findings suggest AD cybrids have an overactive PI hydrolysis system. Since acute oxidative stress typically reduces carbachol-induced PI hydrolysis (156), and AD cybrids have elevated PI hydrolysis and chronic oxidative stress, it was postulated PI hydrolysis hyperactivity in these cells represents a direct or indirect adaptive response to chronic oxidative stress.
In addition to producing a phosphorylated inositol molecule, PI hydrolysis by PLC also generates diacylglycerol (DAG). DAG activates protein kinase C (PKC), which promotes AP-1 transcription factor DNA binding; AP-1 regulates aspects of cell differentiation, proliferation, and apoptosis. Although PKC levels are equivalent between groups, less AP-1 DNA binding occurs in AD cybrids treated with carbachol or phorbol 12-myristate 13-acetate (PMA), a DAG substitute that directly activates PKC (68). This may also represent a consequence of oxidative stress, since oxidative stress reduces carbachol-induced AP-1 DNA binding (156). In any case, in AD cybrids, oxidative stress appears to uncouple PKC signaling from PI hydrolysis. It is therefore possible that PI hydrolysis hyperactivity represents an attempt to compensate for this uncoupling. PKC function is reduced and its physical status is altered in the AD brain (51, 175, 236).
In addition to perturbed AP-1 DNA binding, AD cybrids also show altered function of other transcription factors. Activity of the oxidative stress-sensitive NFκB transcription factor (127, 133, 229, 230) is likely increased. This is supported by the finding that phosphorylation of its inhibitor, IκBα, is elevated; IκBα.phosphorylation targets it for proteosomal degradation, which frees NFκB and allows it to promote the expression of generally pro-inflammatory genes (196). NFκB activity is also increased in AD patient brain neurons and astrocytes (132). In AD cybrids, NFκB activation likely plays a protective role, since NFκB inhibition reduces cell viability (195–197). In another study, HSF-1 DNA binding was found to be reduced in AD cybrids (16). It was further shown that in glutathione-depleted cybrid cell lines, hydrogen peroxide exposure induced a less robust HSF-1 activation in AD cybrid lines than it did in control cybrid lines (16). Because acute hydrogen peroxide exposures typically induce HSF-1 DNA binding, these data suggest reduced basal and peroxide-stimulated HSF-1 DNA binding in AD cybrids reflects a compensatory response to chronic oxidative stress.
A final downstream consequence of the AD cybrid COX defect is a reduction in “viability” markers and an increase in apoptosis markers. Relative to control cybrids, AD cybrids show less XTT and MTT reduction, a phenomenon that is partly reversed by antioxidant treatment (195–197, 302). AD cybrid cultures have increased numbers of cells with condensed nuclei, annexin V positive-propidium iodide negative staining, and annexin V positive-propidium iodide positive staining (195–197, 302). AD cybrids show increased PARP cleavage and increased H2A.X phosphorylation; reducing Aβ production through gamma secretase inhibition lowers levels of these apoptosis markers (195–197). The BAX/Bcl2 ratio is increased due to higher BAX and lower Bcl2 levels, while treating AD cybrids with the antioxidant puerarin reduces BAX and increases Bcl2 (302). BAX overexpression in AD neurons has been reported, while Bcl2 levels seem to fluctuate depending on the overall health of the neuron being analyzed (252, 273). AD cybrids have reduced mitochondrial cytochrome c levels, elevated cytosolic cytochrome c levels, and excess caspase 3 activity (32, 141, 302). The greater frequency of swollen mitochondria observed in AD cybrids may also reflect increased apoptosis-related activity, since mitochondrial swelling is associated with mitochondrial permeability transition and permeability transition occurs during apoptosis (114, 276–278, 302).
It is important to note there is one published AD cybrid report in which AD cybrids prepared on a HeLa cervical carcinoma nuclear background were studied (125). In this report, four AD cybrid lines were prepared from platelet mitochondria, three control cybrid lines were prepared from platelet mitochondria, and two control cybrid lines were prepared from fibroblast mitochondria. Only semi-quantitative data in the form of bar graphs are provided, but the bar graphs do indicate COX activity in each cybrid line is comparable to or at least not dramatically different from the COX activity of the native HeLa cell line. Also in this study (125), HeLa ρ0 cells were mixed with synaptosomes prepared from a single AD autopsy brain following a 20 h postmortem interval. After this mixing, three cell colonies that contained mtDNA were later identified and the COX activity from each colony was determined. The data from this experiment, which also are presented only as part of a semi-quantitative bar graph, show the COX activity from these three colonies resembled the COX activity of the native HeLa line. The authors concluded that mtDNA is equivalent between AD and control subjects and that AD and control cybrid lines are functionally equivalent. Because the methods used in this negative AD cybrid study dramatically differ from those of the 17 published positive AD cybrid studies, the degree to which this single study contradicts the positive studies is unclear. The 17 positive studies used SH-SY5Y and NT2 neuronal cell nuclear backgrounds, and in aggregate the positive studies have now assayed over 200 cybrid lines prepared on three continents. To date, the positive studies have compared over 100 AD cybrid lines to over 100 control cybrid lines. Findings from the 18 reported AD cybrid studies are summarized in Table 1.
Table 1.
Findings from 18 Published Alzheimer’s Disease Cybrid Studies
| Parameter | Number of studies concluding “Yes” | Number of studies concluding “No” |
|---|---|---|
| AD cybrids differ from control cybrids | 17 | 1 |
| Reduced cytochrome oxidase activity* | 9 | 1 |
| Complex I activity unchanged | 4 | 0 |
| Increased oxidative stress | 7 | 0 |
| Perturbed calcium handling | 2 | 0 |
| Altered mitochondrial morphology | 3 | 0 |
| Altered mitochondrial synthesis or degradation rate | 1 | 0 |
| Reduced ATP | 1 | 0 |
| Reduced/altered membrane potential | 6 | 0 |
| Mitochondrial mass unchanged | 4 | 0 |
| Increased apoptotic/reduced “viability” markers | 6 | 0 |
| Altered intracellular or stress signaling | 6 | 0 |
| Increased intra- and extracellular Aβ levels | 2 | 0 |
| Altered mitochondrial movement | 1 | 0 |
Three articles by Onyango et al. all report reduced COX activity, but are only counted once since the same cell lines are used in each paper. Similarly, the study of Trimmer and Borland includes a subset of previously reported cell lines, and so is not independently counted. AD, Alzheimer’s disease.
Where Are the mtDNA Mutations?
Cybrid data imply mtDNA can account, at least in part, for low COX activity in AD subjects. They further imply that by influencing ETC function, mtDNA can induce important AD-associated biochemical and molecular phenomena.
While cybrid data do not identify the critical mtDNA feature or features, several inferences are possible. Because complex I activity is not reduced, at least in AD subject platelet mitochondria wholesale mtDNA degradation is unlikely. Similarly, preserved complex I activity as well as preserved mtDNA levels indicate low COX activity in AD cybrids occurs independent of reduced mtDNA levels.
Investigators have argued that somatic acquired mtDNA mutations may contribute to AD (285). An attractive feature of this hypothesis is it provides a mechanism that could account for the striking correlation between AD incidence and advancing age (130). Mitochondrial DNA mutations certainly do accumulate with age, and in animal models somatic mtDNA mutations can drive an aging phenotype (149, 274). Substantial evidence that somatic mtDNA mutations accumulate in AD subjects already exists. Several groups report levels of the 4977 base pair “common” mtDNA deletion, which increases with age in postmitotic tissues such as the brain, is further elevated in the AD brain (54, 104, 113). This particular deletion encompasses mtDNA-encoded COX genes, and so when present in high enough amounts reduced COX activity should result (58, 59). Mitochondrial DNA deletions such as the common deletion, though, are not known to accumulate in blood (18) which suggests it is unlikely to account for findings from AD cybrids.
Lin et al. systematically catalogued low abundance heteroplasmic point mutations in brain mtDNA from young control subjects, aged subjects without dementia, and AD subjects (158). Correlations between subject age, mutation burdens, and brain COX activity were observed. Specifically, the number of low-abundance mutations found in the mtDNA COX1 subunit gene increased with advancing age, and a greater number of mtDNA mutations corresponded with a lower COX Vmax activity. However, between the AD and age-matched control groups, the absolute number of detected COX1 mutations was similar. To date, no study has reliably catalogued low abundance heteroplasmies or compound heteroplasmies in the other two mtDNA-located COX genes (COX2 and COX3).
Other studies, though, have argued that heteroplasmic point mutations are more common in AD. In AD brain but not AD lymphocytes, Chang et al. found a three-fold elevation of a particular mtDNA displacement (D) loop C to T transition (45). The authors felt the nature of the sequence change could represent a consequence of increased AD brain oxidative stress, a view compatible with several studies finding excessive levels of mtDNA oxidative adducts in the AD brain (65, 113, 170, 179). Coskun et al. also reported the frequency of several specific D-loop mutations differed dramatically between AD and control subject brains (55). This group subsequently found that compared to controls, D-loop mutations were also much more frequent in AD subject serum and lymphoblastoid cell line mtDNA (56).
Investigators have also argued inheritance of particular mtDNA sequence variations influences AD risk. One of the earliest deviations to be reported was an A4336G transition in the mtDNA tRNAGln gene (81, 122, 227, 237), although several other studies found no association (48, 80, 221, 294, 306). This transition is characteristic of particular mtDNA haplogroup H subgroups (164, 227). To date, some studies have found haplogroup H is relatively over-represented in AD cohorts (86, 173, 174, 227). As a potentially related finding, haplogroup H may represent a particularly well-coupled haplogroup, which infers that individuals with haplogroup H may produce more ROS than individuals with other haplogroups (171). Other studies, though, have reported associations between AD and less-coupled mtDNA haplogroups (34, 86, 123, 152, 266, 283). Associations between AD and particular rare polymorphisms or polymorphism combinations may also exist (41, 270).
Sequencing of AD subject mtDNA thus far has not revealed any particular high-abundance “smoking gun” mutation that “causes” this disease. This makes sense, since in some demographics AD is an incredibly common disorder. Almost half of those over 85 qualify for a diagnosis of AD or its frequent prodrome, mild cognitive impairment (MCI), as do more than half of those beyond the age of 90 (85, 295). Because AD prevalence is so high, any particular causal variation would probably not qualify as a mutation. This has led some to further consider whether mtDNA and also nuclear COX gene polymorphisms may account for some degree of AD risk.
To assess population variation in COX genes, Lu et al. sequenced 13 COX subunit genes, the three mtDNA COX subunit genes, and ten nuclear COX subunit genes, from 50 nondemented individuals (161). Approximately 20% of individuals carried a nonsynonymous mtDNA COX gene polymorphism. Synonymous mtDNA COX gene polymorphisms were even more frequent. Interestingly, the synonymous polymorphisms were not evenly distributed, but rather were clustered in the less conserved COX3 gene. This suggests that even synonymous polymorphisms could have a functional consequence, a possibility consistent with existing data that indicate synonymous polymorphisms, by changing the rate of protein translation, can alter protein folding and therefore protein function (142, 146, 147). Aside from a common single nucleotide polymorphism (SNP) in the nuclear COX4I1 gene nonsynonymous nuclear changes were rare, but synonymous polymorphisms and especially 5` and 3` untranslated region (UTR) polymorphisms were extremely common. The nonsynonymous COX4I1 SNP and a frequently detected hexanucleotide deletion in the COX7A1 5` UTR were both found to have functional consequences. Expression of the COX4I1 SNP was associated with a lower COX Vmax activity, and in a reporter assay the COX7A1 deletion reduced COX7A1 expression. Underscoring the tremendous degree of genetic variation that was found, when synonymous and UTR polymorphisms were taken into account, no two individuals shared an identical COX holoenzyme genotype.
A considerable degree of inherited and acquired inter-individual COX subunit gene variation, and especially mtDNA COX subunit gene variation, has thus already been demonstrated. Some association studies further report potential differences between AD and non-AD cohorts. In these positive studies, odds ratios are similar to or even exceed those of other genes associated with AD through large nuclear gene genome wide association studies (110, 131, 153, 232). Therefore, instead of asking “where are the mutations”, a more reasonable question is “how much do demonstrable mtDNA sequence variations influence AD risk?”
Data from AD endophenotype studies to some extent indirectly address this question. When a particular trait or biomarker typically found in conjunction with a disease is detected in persons who do not have that disease, the presence of that trait or biomarker is said to constitute an endophenotype. An endophenotype state does not indicate a carrier will develop the full-blown disease, although it infers that compared to persons without the endophenotype, those with the endophenotype carry an increased risk. To date, a number of studies, several of which are neuroimaging-based, have found the adult children of AD-affected mothers are more likely to express AD endophenotypes than the adult children of AD-affected fathers. The first AD endophenotype study, which analyzed fluorodeoxyglucose positron emission tomography (FDG-PET) scans, found the cerebral metabolic rate of glucose (CMRglu) of subjects with AD mothers showed AD-characteristic changes while subjects with AD fathers did not (184). Over time, CMRglu decline rates are greater in subjects with AD mothers (187). Subjects with AD mothers have more atrophy in AD-affected brain regions, as well as faster rates of atrophy progression (15, 118, 119). Pittsburgh compound B (PIB) PET reveals a greater degree of Aβ plaque deposition in those with AD mothers (186, 188). Cerebrospinal fluid (CSF) analysis reveals a lower CSF Aβ42/Aβ40 ratio and CSF isoprostanes, a marker of oxidative stress, are higher (186). On memory tests, APOE4 carriers with AD mothers do not perform as well as APOE4 carriers with AD fathers (69). Platelet mitochondria COX activity is lower in those with AD mothers than it is in those with AD fathers (185).
Cybrid, endophenotype, and positive mtDNA–AD association studies are consistent with epidemiology data that suggest an AD maternal inheritance bias does exist (10, 77, 79, 183). One such study, which took into account that women outlive men and could therefore have a higher lifetime risk of dementia, found greater female longevity did not account for this relationship (79). It therefore seems that although both parents influence an individual's AD risk, mothers have a greater impact.
Synthesizing the Data: The Mitochondrial Cascade Hypothesis
Various explanations for reduced AD subject COX activity have been proposed (Table 2). Some, such as the possibility that reduced AD brain synapse activity induces COX downregulation (293), could account for low brain COX activity but do not explain reduced platelet, fibroblast, or cybrid COX activity.
Table 2.
Potential Causes of Reduced Cytochrome Oxidase Activity in Alzheimer’s Disease
| Cause | Could apply to brain? | Could apply to non-brain tissues? | Could apply to cybrids? |
|---|---|---|---|
| Toxic inhibition by Aβ or another toxin | Yes | Yes | Probably not |
| Toxic downregulation by Aβ or another toxin | Yes | Yes | Probably not |
| Downregulation due to reduced synaptic activity | Yes | No | No |
| Nonspecific consequence of neurodegeneration | Yes | No | No |
| Nuclear gene mutation | Yes | Yes | Not unless it induces a diffuse accumulation of mtDNA mutation |
| mtDNA | Yes | Yes | Yes |
In general, AD cybrid studies suggest mtDNA gives rise to a COX defect, the COX defect causes oxidative stress and reduced ATP levels, and this produces multiple other AD-typical phenomena, including increased Aβ production (Fig. 5). All these features, in turn, influence the overall health of the cell. Because direct correlations between AD cybrid cell line defects and specific mtDNA sequence features are not yet established, this interpretation of the cybrid data assumes that only differences in mtDNA can explain persistent specific differences between individual cybrid cell lines. At this time, no alternative mechanism that causes sustained, inter-cell line biochemical differences has been demonstrated.
FIG. 5.

Consequences of the mtDNA-encoded AD cybrid COX “defect”. Direct consequences of the mtDNA-encoded AD cybrid COX defect include increased ROS production and, perhaps to some extent, reduced ATP. The cell may try to compensate for its relatively tenuous bioenergetic status by increasing mtDNA synthesis. Several cybrid studies indicate ROS in turn activates a series of events, including increased Aβ production, activated stress signaling, and altered gene transcription. ROS, in conjunction with Aβ, also appears to depolarize mitochondria, activate apoptosis, and interfere with calcium homeostasis.
An individual's AD risk is influenced by whether they have an affected parent or parents (137). Although nongenetic factors can and probably do affect risk (103, 198), this strongly implies a genetic contribution. Very rare autosomal dominant forms of AD are recognized (100, 154, 235), but persons with autosomal dominant, familial AD (FAD) tend to present at younger ages than those with sporadic AD; most FAD patients are symptomatic prior to 60 years of age, while most sporadic AD patients develop symptoms after the age of 60 (256). Regarding sporadic AD, which accounts for over 99% of cases, several nuclear DNA nondeterministic polymorphisms are currently believed to influence AD risk. The most strongly associated and extensively studied risk factor gene is APOE on chromosome 19q13.2 (52, 211). AD risk is increased in APOE4, reduced in APOE2, and intermediate in APOE3 carriers. How and why apolipoprotein E, the protein encoded by the APOE gene, modifies AD risk is unknown. One hypothesis is that apolipoprotein E degradation products may directly interfere with mitochondrial function (44, 46, 191). Also, one study reported that between APOE4 carriers and noncarriers, posterior cingulate cortex COX activity was lower in the APOE4 carriers (282).
Interestingly, polymorphic variations in a neighboring gene, TOMM40 (translocase of the outer mitochondrial membrane 40 kDa subunit homolog), were recently shown to track with AD risk (12, 13, 217, 224, 267, 300). Recent studies suggest particular TOMM40 polymorphisms may affect the age of AD onset more stringently than APOE polymorphisms, leading some to speculate this mitochondrial protein is potentially more relevant to AD than apolipoprotein E (223). Several studies have also associated variation in the TFAM gene with AD risk (5, 14, 102, 303). The TFAM gene encodes transcription factor A of the mitochondria, which plays a major role in mtDNA replication and expression (50, 82, 134, 200).
The possibility therefore exists that nuclear genes directly or indirectly related to mitochondrial function mediate a large proportion of a person's nuclear DNA-determined AD risk. As discussed in the previous section, maternal inheritance bias and maternally-defined endophenotypes also suggest mtDNA inheritance modifies risk. If these observations and interpretations are correct, then the inheritance of mitochondrial genes, in conjunction with the inheritance of nuclear genes that specifically influence baseline mitochondrial function and durability, could have a profound impact on whether and when an aging individual develops AD.
Somatic mtDNA mutations have also been shown to accumulate in both the aging and AD brain (53, 54, 158). These mutations appear to influence mitochondrial function (158). The burden of somatic mutation required to affect function is not entirely clear and may depend on multiple factors, such as the type of mutations accumulated, their location on the mitochondrial genome, and the functional baseline upon which they are superimposed (Fig. 6). An important possibility to consider further is that an mtDNA molecule's primary sequence may actually influence the rate at which somatic changes accumulate (117, 279).
FIG. 6.

Bioenergetic failure occurs when mitochondrial function declines below a functional threshold. (A) If mitochochondrial durability and functional decline rates are equivalent, the time required to fall below the disease threshold is determined by the baseline level of mitochondrial function. (B) Given equivalent baseline levels of mitochondrial function, more durable mitochondria with slower rates of age-related decline will remain above the disease threshold longer than less durable mitochondria with accelerated rates of age-related decline.
When a threshold is reached and biochemical consequences manifest, various AD histology and molecular phenomena, as well as clinical signs and symptoms, would result. This rationale constitutes the core assumptions of the mitochondrial cascade hypothesis (Fig. 7), a bioenergetics-centric scheme that places mitochondrial dysfunction at the apex of the AD pathology pyramid. The mitochondrial cascade hypothesis is reviewed in detail elsewhere (258, 260–262). It is important to note other authors have also speculated mitochondrial and bioenergetic dysfunction may represent the primary cause of AD (11, 21, 22, 38, 63, 120, 121, 166, 182, 201, 203, 225, 285).
FIG. 7.

The sporadic Alzheimer's disease mitochondrial cascade hypothesis. The mitochondrial cascade hypothesis postulates inheritance determines an individual's baseline mitochondrial function and durability. Both parents influence these parameters, but because mtDNA is maternally inherited, mothers have a bigger impact. The functional baseline determines the reserve bioenergetic capacity, while durability determines the rate at which an age-related decline in mitochondrial physiology occurs. When a functional threshold is reached, AD-associated histology changes such as Aβ deposition, tangle formation, and synaptic degradation follow. In the mitochondrial cascade hypothesis, Aβ oligomers, which have been shown to interfere with mitochondrial function in AD models, may contribute to the cascade but do not initiate it. This distinguishes the mitochondrial cascade hypothesis from the amyloid cascade hypothesis, which proposes Aβ oligomerization constitutes the most upstream event and initiates a neurodegenerative cascade.
Conclusion
Because the literature supporting a role for mitochondrial and bioenergetic dysfunction in AD has become so extensive, many relevant studies could not be discussed. For instance, mitochondrial uncoupling induces tau-paired helical filament formation (19), in mice ETC inhibition and fasting robustly induce neuron tau phosphorylation (84, 116, 265, 297), and mitochondria may mediate Aβ–tau relationships (238, 239). Apologies are offered to those whose valuable contributions to this field were not cited.
Data from AD cybrids, which effectively model numerous AD phenomena, suggest mtDNA may at least partly account for reduced COX activity and other biochemical changes in nonbrain tissues. An mtDNA contribution is also compatible with the growing number of AD epidemiology, association, endophenotype, and gene analysis studies that implicate a mitochondrial and possibly even mtDNA role in this disease. In an attempt to synthesize these data, and to also acknowledge the central role aging plays in late-onset, sporadic AD, the mitochondrial cascade hypothesis was proposed. The mitochondrial cascade hypothesis postulates mitochondrial dysfunction represents the most upstream pathology in AD.
Abbreviations Used
- Aβ
beta-amyloid
- ABAD
Aβ-binding alcohol dehydrogenase
- AD
Alzheimer's disease
- AOE
antioxidant enzymes
- APOE
apolipoprotein E
- APP
amyloid precursor protein
- BACE
beta secretase
- CMRglu
cerebral metabolic rate of glucose
- Cybrid
cytoplasmic hybrid
- CSF
cerebrospinal fluid
- COX
cytochrome oxidase
- DAG
diacylglycerol
- ETC
electron transport chain
- FAD
familial Alzheimer's disease
- FDG
fluorodeoxyglucose
- HNE
hydroxynonenol
- IP3
inositol triphosphate
- MAPK
mitogen activated protein kinase
- mtDNA
mitochondrial DNA
- NGF
nerve growth factor
- PET
positron emission tomography
- PI
phosphoinositide
- PIB
Pittsburgh compound B
- PKC
protein kinase C
- PLC
phospholipase C
- PMA
phorbol 12-myristate 13-acetate
- ROS
reactive oxygen species
- SNP
single nucleotide polymorphism
- TBARS
thiobarbituric acid reactive substances
- TFAM
transcription factor A of the mitochondria
- TOMM40
translocase of the outer mitochondrial membrane 40 kDa subunit homolog
- UTR
untranslated region
Acknowledgments
The author is supported by the Morgan Family Foundation and NIH P30AG035982.
Author Disclosure Statement
The author reports no conflicts of interest.
References
- 1.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 2.Albert MS. Dekosky ST. Dickson D. Dubois B. Feldman HH. Fox NC. Gamst A. Holtzman DM. Jagust WJ. Petersen RC. Snyder PJ. Carrillo MC. Thies B. Phelps CH. The diagnosis of mild cognitive impairment due to Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allaman I. Belanger M. Magistretti PJ. Astrocyte–neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011;34:76–87. doi: 10.1016/j.tins.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Allaman I. Gavillet M. Belanger M. Laroche T. Viertl D. Lashuel HA. Magistretti PJ. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: Impact on neuronal viability. J Neurosci. 2010;30:3326–3338. doi: 10.1523/JNEUROSCI.5098-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez V. Corao AI. Alonso-Montes C. Sanchez-Ferrero E. De Mena L. Morales B. Garcia-Castro M. Coto E. Mitochondrial transcription factor A (TFAM) gene variation and risk of late-onset Alzheimer's disease. J Alzheimers Dis. 2008;13:275–280. doi: 10.3233/jad-2008-13305. [DOI] [PubMed] [Google Scholar]
- 6.Alzheimer A. Uber eine eigenartige Erkrankung der Hirnrinde. Allg Z Psychiat Psych-Gerichtl Med. 1907;64:146–148. [Google Scholar]
- 7.Anandatheerthavarada HK. Biswas G. Robin MA. Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anandatheerthavarada HK. Devi L. Amyloid precursor protein and mitochondrial dysfunction in Alzheimer's disease. Neuroscientist. 2007;13:626–638. doi: 10.1177/1073858407303536. [DOI] [PubMed] [Google Scholar]
- 9.Baloyannis SJ. Mitochondrial alterations in Alzheimer's disease. J Alzheimers Dis. 2006;9:119–126. doi: 10.3233/jad-2006-9204. [DOI] [PubMed] [Google Scholar]
- 10.Bassett SS. Avramopoulos D. Fallin D. Evidence for parent of origin effect in late-onset Alzheimer disease. Am J Med Genet. 2002;114:679–686. doi: 10.1002/ajmg.10648. [DOI] [PubMed] [Google Scholar]
- 11.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 12.Bekris LM. Galloway NM. Montine TJ. Schellenberg GD. Yu CE. APOE mRNA and protein expression in postmortem brain are modulated by an extended haplotype structure. Am J Med Genet B Neuropsychiatr Genet. 2009;153B:409–417. doi: 10.1002/ajmg.b.30993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bekris LM. Millard SP. Galloway NM. Vuletic S. Albers JJ. Li G. Galasko DR. DeCarli C. Farlow MR. Clark CM. Quinn JF. Kaye JA. Schellenberg GD. Tsuang D. Peskind ER. Yu CE. Multiple SNPs within and surrounding the apolipoprotein E gene influence cerebrospinal fluid apolipoprotein E protein levels. J Alzheimers Dis. 2008;13:255–266. doi: 10.3233/jad-2008-13303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belin AC. Bjork BF. Westerlund M. Galter D. Sydow O. Lind C. Pernold K. Rosvall L. Hakansson A. Winblad B. Nissbrandt H. Graff C. Olson L. Association study of two genetic variants in mitochondrial transcription factor A (TFAM) in Alzheimer's and Parkinson's disease. Neurosci Lett. 2007;420:257–262. doi: 10.1016/j.neulet.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Berti V. Mosconi L. Glodzik L. Li Y. Murray J. De Santi S. Pupi A. Tsui W. De Leon MJ. Structural brain changes in normal individuals with a maternal history of Alzheimer's. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.01.001. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bijur GN. Davis RE. Jope RS. Rapid activation of heat shock factor-1 DNA binding by H2O2 and modulation by glutathione in human neuroblastoma and Alzheimer's disease cybrid cells. Brain Res Mol Brain Res. 1999;71:69–77. doi: 10.1016/s0169-328x(99)00168-0. [DOI] [PubMed] [Google Scholar]
- 17.Bindoff LA. Birch-Machin M. Cartlidge NE. Parker WD., Jr. Turnbull DM. Mitochondrial function in Parkinson's disease. Lancet. 1989;2:49. doi: 10.1016/s0140-6736(89)90291-2. [DOI] [PubMed] [Google Scholar]
- 18.Blanchard BJ. Park T. Fripp WJ. Lerman LS. Ingram VM. A mitochondrial DNA deletion in normally aging and in Alzheimer brain tissue. Neuroreport. 1993;4:799–802. doi: 10.1097/00001756-199306000-00051. [DOI] [PubMed] [Google Scholar]
- 19.Blass JP. Baker AC. Ko L. Black RS. Induction of Alzheimer antigens by an uncoupler of oxidative phosphorylation. Arch Neurol. 1990;47:864–869. doi: 10.1001/archneur.1990.00530080046009. [DOI] [PubMed] [Google Scholar]
- 20.Blass JP. Gibson GE. Nonneural markers in Alzheimer disease. Alzheimer Dis Assoc Disord. 1992;6:205–224. doi: 10.1097/00002093-199206040-00003. [DOI] [PubMed] [Google Scholar]
- 21.Blass JP. Gibson GE. Hoyer S. The role of the metabolic lesion in Alzheimer's disease. J Alzheimers Dis. 2002;4:225–232. doi: 10.3233/jad-2002-4312. [DOI] [PubMed] [Google Scholar]
- 22.Blass JP. Zemcov A. Alzheimer's disease. A metabolic systems degeneration? Neurochem Pathol. 1984;2:103–114. doi: 10.1007/BF02834249. [DOI] [PubMed] [Google Scholar]
- 23.Boissiere F. Lehericy S. Strada O. Agid Y. Hirsch EC. Neurotrophin receptors and selective loss of cholinergic neurons in Alzheimer disease. Mol Chem Neuropathol. 1996;28:219–223. doi: 10.1007/BF02815225. [DOI] [PubMed] [Google Scholar]
- 24.Bosetti F. Brizzi F. Barogi S. Mancuso M. Siciliano G. Tendi EA. Murri L. Rapoport SI. Solaini G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer's disease. Neurobiol Aging. 2002;23:371–376. doi: 10.1016/s0197-4580(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 25.Bowen DM. White P. Spillane JA. Goodhardt MJ. Curzon G. Iwangoff P. Meier-Ruge W. Davison AN. Accelerated ageing or selective neuronal loss as an important cause of dementia? Lancet. 1979;1:11–14. doi: 10.1016/s0140-6736(79)90454-9. [DOI] [PubMed] [Google Scholar]
- 26.Brand MD. Affourtit C. Esteves TC. Green K. Lambert AJ. Miwa S. Pakay JL. Parker N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med. 2004;37:755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 27.Brody DL. Magnoni S. Schwetye KE. Spinner ML. Esparza TJ. Stocchetti N. Zipfel GJ. Holtzman DM. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–1224. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown AM. Sheu RK. Mohs R. Haroutunian V. Blass JP. Correlation of the clinical severity of Alzheimer's disease with an aberration in mitochondrial DNA (mtDNA) J Mol Neurosci. 2001;16:41–48. doi: 10.1385/JMN:16:1:41. [DOI] [PubMed] [Google Scholar]
- 29.Bunn CL. Wallace DC. Eisenstadt JM. Cytoplasmic inheritance of chloramphenicol resistance in mouse tissue culture cells. Proc Natl Acad Sci USA. 1974;71:1681–1685. doi: 10.1073/pnas.71.5.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Canevari L. Clark JB. Bates TE. beta-Amyloid fragment 25-35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999;457:131–134. doi: 10.1016/s0014-5793(99)01028-5. [DOI] [PubMed] [Google Scholar]
- 31.Cardoso SM. Proenca MT. Santos S. Santana I. Oliveira CR. Cytochrome c oxidase is decreased in Alzheimer's disease platelets. Neurobiol Aging. 2004;25:105–110. doi: 10.1016/s0197-4580(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 32.Cardoso SM. Santana I. Swerdlow RH. Oliveira CR. Mitochondria dysfunction of Alzheimer's disease cybrids enhances Abeta toxicity. J Neurochem. 2004;89:1417–1426. doi: 10.1111/j.1471-4159.2004.02438.x. [DOI] [PubMed] [Google Scholar]
- 33.Cardoso SM. Santos S. Swerdlow RH. Oliveira CR. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001;15:1439–1441. doi: 10.1096/fj.00-0561fje. [DOI] [PubMed] [Google Scholar]
- 34.Carrieri G. Bonafe M. De Luca M. Rose G. Varcasia O. Bruni A. Maletta R. Nacmias B. Sorbi S. Corsonello F. Feraco E. Andreev KF. Yashin AI. Franceschi C. De Benedictis G. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer's disease. Hum Genet. 2001;108:194–198. doi: 10.1007/s004390100463. [DOI] [PubMed] [Google Scholar]
- 35.Casley CS. Canevari L. Land JM. Clark JB. Sharpe MA. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- 36.Caspersen C. Wang N. Yao J. Sosunov A. Chen X. Lustbader JW. Xu HW. Stern D. McKhann G. Yan SD. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 37.Cassarino DS. Swerdlow RH. Parks JK. Parker WD., Jr. Bennett JP., Jr Cyclosporin A increases resting mitochondrial membrane potential in SY5Y cells and reverses the depressed mitochondrial membrane potential of Alzheimer's disease cybrids. Biochem Biophys Res Commun. 1998;248:168–173. doi: 10.1006/bbrc.1998.8866. [DOI] [PubMed] [Google Scholar]
- 38.Castellani R. Hirai K. Aliev G. Drew KL. Nunomura A. Takeda A. Cash AD. Obrenovich ME. Perry G. Smith MA. Role of mitochondrial dysfunction in Alzheimer's disease. J Neurosci Res. 2002;70:357–360. doi: 10.1002/jnr.10389. [DOI] [PubMed] [Google Scholar]
- 39.Cecarini V. Gee J. Fioretti E. Amici M. Angeletti M. Eleuteri AM. Keller JN. Protein oxidation and cellular homeostasis: Emphasis on metabolism. Biochim Biophys Acta. 2007;1773:93–104. doi: 10.1016/j.bbamcr.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 40.Chagnon P. Betard C. Robitaille Y. Cholette A. Gauvreau D. Distribution of brain cytochrome oxidase activity in various neurodegenerative diseases. Neuroreport. 1995;6:711–715. doi: 10.1097/00001756-199503270-00002. [DOI] [PubMed] [Google Scholar]
- 41.Chagnon P. Gee M. Filion M. Robitaille Y. Belouchi M. Gauvreau D. Phylogenetic analysis of the mitochondrial genome indicates significant differences between patients with Alzheimer disease and controls in a French-Canadian founder population. Am J Med Genet. 1999;85:20–30. doi: 10.1002/(sici)1096-8628(19990702)85:1<20::aid-ajmg6>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 42.Chandrasekaran K. Giordano T. Brady DR. Stoll J. Martin LJ. Rapoport SI. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Brain Res Mol Brain Res. 1994;24:336–340. doi: 10.1016/0169-328x(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 43.Chandrasekaran K. Hatanpaa K. Brady DR. Rapoport SI. Evidence for physiological down-regulation of brain oxidative phosphorylation in Alzheimer's disease. Exp Neurol. 1996;142:80–88. doi: 10.1006/exnr.1996.0180. [DOI] [PubMed] [Google Scholar]
- 44.Chang S. ran Ma T. Miranda RD. Balestra ME. Mahley RW. Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci USA. 2005;102:18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang SW. Zhang D. Chung HD. Zassenhaus HP. The frequency of point mutations in mitochondrial DNA is elevated in the Alzheimer's brain. Biochem Biophys Res Commun. 2000;273:203–208. doi: 10.1006/bbrc.2000.2885. [DOI] [PubMed] [Google Scholar]
- 46.Chen HK. Ji ZS. Dodson SE. Miranda RD. Rosenblum CI. Reynolds IJ. Freedman SB. Weisgraber KH. Huang Y. Mahley RW. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J Biol Chem. 2011;286:5215–5221. doi: 10.1074/jbc.M110.151084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L. Yoo SE. Na R. Liu Y. Ran Q. Cognitive impairment and increased Abeta levels induced by paraquat exposure are attenuated by enhanced removal of mitochondrial H(2)O(2) Neurobiol Aging. doi: 10.1016/j.neurobiolaging.2011.01.008. [Epub ahead of print, 2011]. [DOI] [PubMed] [Google Scholar]
- 48.Chinnery PF. Taylor GA. Howell N. Andrews RM. Morris CM. Taylor RW. McKeith IG. Perry RH. Edwardson JA. Turnbull DM. Mitochondrial DNA haplogroups and susceptibility to AD and dementia with Lewy bodies. Neurology. 2000;55:302–304. doi: 10.1212/wnl.55.2.302. [DOI] [PubMed] [Google Scholar]
- 49.Chomyn A. Lai ST. Shakeley R. Bresolin N. Scarlato G. Attardi G. Platelet-mediated transformation of mtDNA-less human cells: analysis of phenotypic variability among clones from normal individuals—and complementation behavior of the tRNALys mutation causing myoclonic epilepsy and ragged red fibers. Am J Hum Genet. 1994;54:966–974. [PMC free article] [PubMed] [Google Scholar]
- 50.Clayton DA. Transcription and replication of mitochondrial DNA. Hum Reprod. 2000;15(Suppl 2):11–17. doi: 10.1093/humrep/15.suppl_2.11. [DOI] [PubMed] [Google Scholar]
- 51.Cole G. Dobkins KR. Hansen LA. Terry RD. Saitoh T. Decreased levels of protein kinase C in Alzheimer brain. Brain Res. 1988;452:165–174. doi: 10.1016/0006-8993(88)90021-2. [DOI] [PubMed] [Google Scholar]
- 52.Corder EH. Saunders AM. Strittmatter WJ. Schmechel DE. Gaskell PC. Small GW. Roses AD. Haines JL. Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 53.Corral-Debrinski M. Horton T. Lott MT. Shoffner JM. Beal MF. Wallace DC. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat Genet. 1992;2:324–329. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- 54.Corral-Debrinski M. Horton T. Lott MT. Shoffner JM. McKee AC. Beal MF. Graham BH. Wallace DC. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics. 1994;23:471–476. doi: 10.1006/geno.1994.1525. [DOI] [PubMed] [Google Scholar]
- 55.Coskun PE. Beal MF. Wallace DC. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci USA. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coskun PE. Wyrembak J. Derbereva O. Melkonian G. Doran E. Lott IT. Head E. Cotman CW. Wallace DC. Systemic mitochondrial dysfunction and the etiology of Alzheimer's disease and Down syndrome dementia. J Alzheimers Dis. 2010;20:S293–310. doi: 10.3233/JAD-2010-100351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cotman CW. Apoptosis decision cascades and neuronal degeneration in Alzheimer's disease. Neurobiol Aging. 1998;19:S29–32. doi: 10.1016/s0197-4580(98)00042-6. [DOI] [PubMed] [Google Scholar]
- 58.Cottrell DA. Blakely EL. Johnson MA. Ince PG. Turnbull DM. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology. 2001;57:260–264. doi: 10.1212/wnl.57.2.260. [DOI] [PubMed] [Google Scholar]
- 59.Cottrell DA. Borthwick GM. Johnson MA. Ince PG. Turnbull DM. The role of cytochrome c oxidase deficient hippocampal neurones in Alzheimer's disease. Neuropathol Appl Neurobiol. 2002;28:390–396. doi: 10.1046/j.1365-2990.2002.00414.x. [DOI] [PubMed] [Google Scholar]
- 60.Counts SE. Mufson EJ. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J Neuropathol Exp Neurol. 2005;64:263–272. doi: 10.1093/jnen/64.4.263. [DOI] [PubMed] [Google Scholar]
- 61.Crouch PJ. Blake R. Duce JA. Ciccotosto GD. Li QX. Barnham KJ. Curtain CC. Cherny RA. Cappai R. Dyrks T. Masters CL. Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Curti D. Rognoni F. Gasparini L. Cattaneo A. Paolillo M. Racchi M. Zani L. Bianchetti A. Trabucchi M. Bergamaschi S. Govoni S. Oxidative metabolism in cultured fibroblasts derived from sporadic Alzheimer's disease (AD) patients. Neurosci Lett. 1997;236:13–16. doi: 10.1016/s0304-3940(97)00741-6. [DOI] [PubMed] [Google Scholar]
- 63.Davis JN. Hunnicutt EJ., Jr. Chisholm JC. A mitochondrial bottleneck hypothesis of Alzheimer's disease. Mol Med Today. 1995;1:240–247. doi: 10.1016/s1357-4310(95)91532-x. [DOI] [PubMed] [Google Scholar]
- 64.Davis RE. Miller S. Herrnstadt C. Ghosh SS. Fahy E. Shinobu LA. Galasko D. Thal LJ. Beal MF. Howell N. Parker WD., Jr Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:4526–4531. doi: 10.1073/pnas.94.9.4526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.de la Monte SM. Luong T. Neely TR. Robinson D. Wands JR. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer's disease. Lab Invest. 2000;80:1323–1335. doi: 10.1038/labinvest.3780140. [DOI] [PubMed] [Google Scholar]
- 66.de la Monte SM. Sohn YK. Wands JR. Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer's disease. J Neurol Sci. 1997;152:73–83. doi: 10.1016/s0022-510x(97)00131-7. [DOI] [PubMed] [Google Scholar]
- 67.de Leon MJ. Ferris SH. George AE. Christman DR. Fowler JS. Gentes C. Reisberg B. Gee B. Emmerich M. Yonekura Y. Brodie J. Kricheff II. Wolf AP. Positron emission tomographic studies of aging and Alzheimer disease. AJNR Am J Neuroradiol. 1983;4:568–571. [PMC free article] [PubMed] [Google Scholar]
- 68.De Sarno P. Bijur GN. Lu R. Davis RE. Jope RS. Alterations in muscarinic receptor-coupled phosphoinositide hydrolysis and AP-1 activation in Alzheimer's disease cybrid cells. Neurobiol Aging. 2000;21:31–38. doi: 10.1016/s0197-4580(00)00095-6. [DOI] [PubMed] [Google Scholar]
- 69.Debette S. Wolf PA. Beiser A. Au R. Himali JJ. Pikula A. Auerbach S. Decarli C. Seshadri S. Association of parental dementia with cognitive and brain MRI measures in middle-aged adults. Neurology. 2009;73:2071–2078. doi: 10.1212/WNL.0b013e3181c67833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Desjardins P. de Muys JM. Morais R. An established avian fibroblast cell line without mitochondrial DNA. Somat Cell Mol Genet. 1986;12:133–139. doi: 10.1007/BF01560660. [DOI] [PubMed] [Google Scholar]
- 71.Desjardins P. Frost E. Morais R. Ethidium bromide-induced loss of mitochondrial DNA from primary chicken embryo fibroblasts. Mol Cell Biol. 1985;5:1163–1169. doi: 10.1128/mcb.5.5.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Devi L. Prabhu BM. Galati DF. Avadhani NG. Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Diana A. Simic G. Sinforiani E. Orru N. Pichiri G. Bono G. Mitochondria morphology and DNA content upon sublethal exposure to beta-amyloid(1-42) peptide. Coll Antropol. 2008;32:51–58. [PubMed] [Google Scholar]
- 74.Dragicevic N. Mamcarz M. Zhu Y. Buzzeo R. Tan J. Arendash GW. Bradshaw PC. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer's transgenic mice. J Alzheimers Dis. 2010;20:S535–550. doi: 10.3233/JAD-2010-100342. [DOI] [PubMed] [Google Scholar]
- 75.Du H. Guo L. Fang F. Chen D. Sosunov AA. McKhann GM. Yan Y. Wang C. Zhang H. Molkentin JD. Gunn-Moore FJ. Vonsattel JP. Arancio O. Chen JX. Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Du H. Guo L. Yan S. Sosunov AA. McKhann GM. Yan SS. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duara R. Lopez-Alberola RF. Barker WW. Loewenstein DA. Zatinsky M. Eisdorfer CE. Weinberg GB. A comparison of familial and sporadic Alzheimer's disease. Neurology. 1993;43:1377–1384. doi: 10.1212/wnl.43.7.1377. [DOI] [PubMed] [Google Scholar]
- 78.Dumont M. Wille E. Stack C. Calingasan NY. Beal MF. Lin MT. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer's disease. FASEB J. 2009;23:2459–2466. doi: 10.1096/fj.09-132928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Edland SD. Silverman JM. Peskind ER. Tsuang D. Wijsman E. Morris JC. Increased risk of dementia in mothers of Alzheimer's disease cases: Evidence for maternal inheritance. Neurology. 1996;47:254–256. doi: 10.1212/wnl.47.1.254. [DOI] [PubMed] [Google Scholar]
- 80.Edland SD. Tobe VO. Rieder MJ. Bowen JD. McCormick W. Teri L. Schellenberg GD. Larson EB. Nickerson DA. Kukull WA. Mitochondrial genetic variants and Alzheimer disease: A case-control study of the T4336C and G5460A variants. Alzheimer Dis Assoc Disord. 2002;16:1–7. doi: 10.1097/00002093-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 81.Egensperger R. Kosel S. Schnopp NM. Mehraein P. Graeber MB. Association of the mitochondrial tRNA(A4336G) mutation with Alzheimer's and Parkinson's diseases. Neuropathol Appl Neurobiol. 1997;23:315–321. [PubMed] [Google Scholar]
- 82.Ekstrand MI. Falkenberg M. Rantanen A. Park CB. Gaspari M. Hultenby K. Rustin P. Gustafsson CM. Larsson NG. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–944. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- 83.Ephrussi B. Hottinger H. Chimenes A. Action de l'acriflavine sur les levures, I: la mutation “petite clonie.”. Ann Inst Pasteur. 1949;76:531. [Google Scholar]
- 84.Escobar-Khondiker M. Hollerhage M. Muriel MP. Champy P. Bach A. Depienne C. Respondek G. Yamada ES. Lannuzel A. Yagi T. Hirsch EC. Oertel WH. Jacob R. Michel PP. Ruberg M. Hoglinger GU. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J Neurosci. 2007;27:7827–7837. doi: 10.1523/JNEUROSCI.1644-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Evans DA. Funkenstein HH. Albert MS. Scherr PA. Cook NR. Chown MJ. Hebert LE. Hennekens CH. Taylor JO. Prevalence of Alzheimer's disease in a community population of older persons. Higher than previously reported. JAMA. 1989;262:2551–2556. [PubMed] [Google Scholar]
- 86.Fesahat F. Houshmand M. Panahi MS. Gharagozli K. Mirzajani F. Do haplogroups H and U act to increase the penetrance of Alzheimer's disease? Cell Mol Neurobiol. 2007;27:329–334. doi: 10.1007/s10571-006-9126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Foster NL. Chase TN. Fedio P. Patronas NJ. Brooks RA. Di Chiro G. Alzheimer's disease: Focal cortical changes shown by positron emission tomography. Neurology. 1983;33:961–965. doi: 10.1212/wnl.33.8.961. [DOI] [PubMed] [Google Scholar]
- 88.Frackowiak RS. Pozzilli C. Legg NJ. Du Boulay GH. Marshall J. Lenzi GL. Jones T. Regional cerebral oxygen supply and utilization in dementia. A clinical and physiological study with oxygen-15 and positron tomography. Brain. 1981;104:753–778. doi: 10.1093/brain/104.4.753. [DOI] [PubMed] [Google Scholar]
- 89.Friede RL. Enzyme histochemical studies of senile plaques. J Neuropathol Exp Neurol. 1965;24:477–491. doi: 10.1097/00005072-196410000-00003. [DOI] [PubMed] [Google Scholar]
- 90.Friedland RP. Budinger TF. Ganz E. Yano Y. Mathis CA. Koss B. Ober BA. Huesman RH. Derenzo SE. Regional cerebral metabolic alterations in dementia of the Alzheimer type: Positron emission tomography with [18F]fluorodeoxyglucose. J Comput Assist Tomogr. 1983;7:590–598. doi: 10.1097/00004728-198308000-00003. [DOI] [PubMed] [Google Scholar]
- 91.Fukui H. Diaz F. Garcia S. Moraes CT. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2007;104:14163–14168. doi: 10.1073/pnas.0705738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fukuyama H. Ogawa M. Yamauchi H. Yamaguchi S. Kimura J. Yonekura Y. Konishi J. Altered cerebral energy metabolism in Alzheimer's disease: A PET study. J Nucl Med. 1994;35:1–6. [PubMed] [Google Scholar]
- 93.Gabuzda D. Busciglio J. Chen LB. Matsudaira P. Yankner BA. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem. 1994;269:13623–13628. [PubMed] [Google Scholar]
- 94.Gasparini L. Racchi M. Benussi L. Curti D. Binetti G. Bianchetti A. Trabucchi M. Govoni S. Effect of energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neurosci Lett. 1997;231:113–117. doi: 10.1016/s0304-3940(97)00536-3. [DOI] [PubMed] [Google Scholar]
- 95.Ghosh SS. Swerdlow RH. Miller SW. Sheeman B. Parker WD., Jr. Davis RE. Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimer's disease and Parkinson's disease. Ann NY Acad Sci. 1999;893:176–191. doi: 10.1111/j.1749-6632.1999.tb07825.x. [DOI] [PubMed] [Google Scholar]
- 96.Gibson GE. Nielsen P. Sherman KA. Blass JP. Diminished mitogen-induced calcium uptake by lymphocytes from Alzheimer patients. Biol Psychiatry. 1987;22:1079–1086. doi: 10.1016/0006-3223(87)90050-3. [DOI] [PubMed] [Google Scholar]
- 97.Gibson GE. Sheu KF. Blass JP. Baker A. Carlson KC. Harding B. Perrino P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer's disease. Arch Neurol. 1988;45:836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
- 98.Giles RE. Blanc H. Cann HM. Wallace DC. Maternal inheritance of human mitochondrial DNA. Proc Natl Acad Sci USA. 1980;77:6715–6719. doi: 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Glenner GG. Wong CW. Alzheimer's disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 100.Goate A. Chartier-Harlin MC. Mullan M. Brown J. Crawford F. Fidani L. Giuffra L. Haynes A. Irving N. James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 101.Griffin RJ. Moloney A. Kelliher M. Johnston JA. Ravid R. Dockery P. O'Connor R. O'Neill C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem. 2005;93:105–117. doi: 10.1111/j.1471-4159.2004.02949.x. [DOI] [PubMed] [Google Scholar]
- 102.Gunther C. von Hadeln K. Muller-Thomsen T. Alberici A. Binetti G. Hock C. Nitsch RM. Stoppe G. Reiss J. Gal A. Finckh U. Possible association of mitochondrial transcription factor A (TFAM) genotype with sporadic Alzheimer disease. Neurosci Lett. 2004;369:219–223. doi: 10.1016/j.neulet.2004.07.070. [DOI] [PubMed] [Google Scholar]
- 103.Haan MN. Therapy Insight: Type 2 diabetes mellitus and the risk of late-onset Alzheimer's disease. Nat Clin Pract Neurol. 2006;2:159–166. doi: 10.1038/ncpneuro0124. [DOI] [PubMed] [Google Scholar]
- 104.Hamblet NS. Castora FJ. Elevated levels of the Kearns-Sayre syndrome mitochondrial DNA deletion in temporal cortex of Alzheimer's patients. Mutat Res. 1997;379:253–262. doi: 10.1016/s0027-5107(97)00158-9. [DOI] [PubMed] [Google Scholar]
- 105.Hansson CA. Frykman S. Farmery MR. Tjernberg LO. Nilsberth C. Pursglove SE. Ito A. Winblad B. Cowburn RF. Thyberg J. Ankarcrona M. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J Biol Chem. 2004;279:51654–51660. doi: 10.1074/jbc.M404500200. [DOI] [PubMed] [Google Scholar]
- 106.Hansson Petersen CA. Alikhani N. Behbahani H. Wiehager B. Pavlov PF. Alafuzoff I. Leinonen V. Ito A. Winblad B. Glaser E. Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci USA. 2008;105:13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hardy J. Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 108.Hardy J. Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 109.Hardy JA. Higgins GA. Alzheimer's disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 110.Harold D. Abraham R. Hollingworth P. Sims R. Gerrish A. Hamshere ML. Pahwa JS. Moskvina V. Dowzell K. Williams A. Jones N. Thomas C. Stretton A. Morgan AR. Lovestone S. Powell J. Proitsi P. Lupton MK. Brayne C. Rubinsztein DC. Gill M. Lawlor B. Lynch A. Morgan K. Brown KS. Passmore PA. Craig D. McGuinness B. Todd S. Holmes C. Mann D. Smith AD. Love S. Kehoe PG. Hardy J. Mead S. Fox N. Rossor M. Collinge J. Maier W. Jessen F. Schurmann B. van den Bussche H. Heuser I. Kornhuber J. Wiltfang J. Dichgans M. Frolich L. Hampel H. Hull M. Rujescu D. Goate AM. Kauwe JS. Cruchaga C. Nowotny P. Morris JC. Mayo K. Sleegers K. Bettens K. Engelborghs S. De Deyn PP. Van Broeckhoven C. Livingston G. Bass NJ. Gurling H. McQuillin A. Gwilliam R. Deloukas P. Al-Chalabi A. Shaw CE. Tsolaki M. Singleton AB. Guerreiro R. Muhleisen TW. Nothen MM. Moebus S. Jockel KH. Klopp N. Wichmann HE. Carrasquillo MM. Pankratz VS. Younkin SG. Holmans PA. O'Donovan M. Owen MJ. Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hatanpaa K. Brady DR. Stoll J. Rapoport SI. Chandrasekaran K. Neuronal activity and early neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1996;40:411–420. doi: 10.1002/ana.410400310. [DOI] [PubMed] [Google Scholar]
- 112.Hauptmann S. Scherping I. Drose S. Brandt U. Schulz KL. Jendrach M. Leuner K. Eckert A. Muller WE. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging. 2009;30:1574–1586. doi: 10.1016/j.neurobiolaging.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 113.Hirai K. Aliev G. Nunomura A. Fujioka H. Russell RL. Atwood CS. Johnson AB. Kress Y. Vinters HV. Tabaton M. Shimohama S. Cash AD. Siedlak SL. Harris PL. Jones PK. Petersen RB. Perry G. Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hirsch T. Marzo I. Kroemer G. Role of the mitochondrial permeability transition pore in apoptosis. Biosci Rep. 1997;17:67–76. doi: 10.1023/a:1027339418683. [DOI] [PubMed] [Google Scholar]
- 115.Hockenbery D. Nunez G. Milliman C. Schreiber RD. Korsmeyer SJ. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature. 1990;348:334–336. doi: 10.1038/348334a0. [DOI] [PubMed] [Google Scholar]
- 116.Hoglinger GU. Lannuzel A. Khondiker ME. Michel PP. Duyckaerts C. Feger J. Champy P. Prigent A. Medja F. Lombes A. Oertel WH. Ruberg M. Hirsch EC. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J Neurochem. 2005;95:930–939. doi: 10.1111/j.1471-4159.2005.03493.x. [DOI] [PubMed] [Google Scholar]
- 117.Holt IJ. Zen and the art of mitochondrial DNA maintenance. Trends Genet. 2010;26:103–109. doi: 10.1016/j.tig.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 118.Honea RA. Swerdlow RH. Vidoni E. Burns JM. Progressive regional atrophy in normaladults with a maternal history of Alzheimer disease. Neurology. 2011;76:822–829. doi: 10.1212/WNL.0b013e31820e7b74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Honea RA. Swerdlow RH. Vidoni ED. Goodwin J. Burns JM. Reduced gray matter volume in normal adults with a maternal family history of Alzheimer disease. Neurology. 2010;74:113–120. doi: 10.1212/WNL.0b013e3181c918cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hoyer S. Brain oxidative energy and related metabolism, neuronal stress, and Alzheimer's disease: A speculative synthesis. J Geriatr Psychiatry Neurol. 1993;6:3–13. doi: 10.1177/002383099300600101. [DOI] [PubMed] [Google Scholar]
- 121.Hoyer S. Brain glucose and energy metabolism abnormalities in sporadic Alzheimer disease. Causes and consequences: An update. Exp Gerontol. 2000;35:1363–1372. doi: 10.1016/s0531-5565(00)00156-x. [DOI] [PubMed] [Google Scholar]
- 122.Hutchin T. Cortopassi G. A mitochondrial DNA clone is associated with increased risk for Alzheimer disease. Proc Natl Acad Sci USA. 1995;92:6892–6895. doi: 10.1073/pnas.92.15.6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ienco EC. Simoncini C. Orsucci D. Petrucci L. Filosto M. Mancuso M. Siciliano G. May “mitochondrial eve” and mitochondrial haplogroups play a role in neurodegeneration and Alzheimer's disease? Int J Alzheimers Dis. 2011;2011:709061. doi: 10.4061/2011/709061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ito E. Oka K. Etcheberrigaray R. Nelson TJ. McPhie DL. Tofel-Grehl B. Gibson GE. Alkon DL. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:534–538. doi: 10.1073/pnas.91.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ito S. Ohta S. Nishimaki K. Kagawa Y. Soma R. Kuno SY. Komatsuzaki Y. Mizusawa H. Hayashi J. Functional integrity of mitochondrial genomes in human platelets and autopsied brain tissues from elderly patients with Alzheimer's disease. Proc Natl Acad Sci USA. 1999;96:2099–2103. doi: 10.1073/pnas.96.5.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Iwangoff P. Armbruster R. Enz A. and Meier-Ruge W. Glycolytic enzymes from human autoptic brain cortex: Normal aged and demented cases. Mech Ageing Dev. 1980;14:203–209. doi: 10.1016/0047-6374(80)90120-7. [DOI] [PubMed] [Google Scholar]
- 127.Jia Z. Misra HP. Reactive oxygen species in in vitro pesticide-induced neuronal cell (SH-SY5Y) cytotoxicity: Role of NFkappaB and caspase-3. Free Radic Biol Med. 2007;42:288–298. doi: 10.1016/j.freeradbiomed.2006.10.047. [DOI] [PubMed] [Google Scholar]
- 128.Johnson AB. Blum NR. Nucleoside phosphatase activities associated with the tangles and plaques of Alzheimer's disease: A histochemical study of natural and experimental neurofibrillary tangles. J Neuropathol Exp Neurol. 1970;29:463–478. doi: 10.1097/00005072-197007000-00009. [DOI] [PubMed] [Google Scholar]
- 129.Jope RS. Cholinergic muscarinic receptor signaling by the phosphoinositide signal transduction system in Alzheimer's disease. J Alzheimers Dis. 1999;1:231–247. doi: 10.3233/jad-1999-14-505. [DOI] [PubMed] [Google Scholar]
- 130.Jorm AF. Jolley D. The incidence of dementia: A meta-analysis. Neurology. 1998;51:728–733. doi: 10.1212/wnl.51.3.728. [DOI] [PubMed] [Google Scholar]
- 131.Jun G. Naj AC. Beecham GW. Wang LS. Buros J. Gallins PJ. Buxbaum JD. Ertekin-Taner N. Fallin MD. Friedland R. Inzelberg R. Kramer P. Rogaeva E. St George-Hyslop P. Cantwell LB. Dombroski BA. Saykin AJ. Reiman EM. Bennett DA. Morris JC. Lunetta KL. Martin ER. Montine TJ. Goate AM. Blacker D. Tsuang DW. Beekly D. Cupples LA. Hakonarson H. Kukull W. Foroud TM. Haines J. Mayeux R. Farrer LA. Pericak-Vance MA. Schellenberg GD. Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch Neurol. 2010;67:1473–1484. doi: 10.1001/archneurol.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kaltschmidt B. Uherek M. Volk B. Baeuerle PA. Kaltschmidt C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:2642–2647. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kamata H. Hirata H. Redox regulation of cellular signalling. Cell Signal. 1999;11:1–14. doi: 10.1016/s0898-6568(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 134.Kang D. Kim SH. Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7:39–44. doi: 10.1016/j.mito.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 135.Kang J. Lemaire HG. Unterbeck A. Salbaum JM. Masters CL. Grzeschik KH. Multhaup G. Beyreuther K. Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 136.Kang JE. Lim MM. Bateman RJ. Lee JJ. Smyth LP. Cirrito JR. Fujiki N. Nishino S. Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Katzman R. Alzheimer's disease. N Engl J Med. 1986;314:964–973. doi: 10.1056/NEJM198604103141506. [DOI] [PubMed] [Google Scholar]
- 138.Keller JN. Interplay between oxidative damage, protein synthesis, and protein degradation in Alzheimer's disease. J Biomed Biotechnol. 2006;2006:12129. doi: 10.1155/JBB/2006/12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Keller JN. Schmitt FA. Scheff SW. Ding Q. Chen Q. Butterfield DA. Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 140.Khachaturian ZS. Diagnosis of Alzheimer's disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- 141.Khan SM. Cassarino DS. Abramova NN. Keeney PM. Borland MK. Trimmer PA. Krebs CT. Bennett JC. Parks JK. Swerdlow RH. Parker WD., Jr. Bennett JP., Jr Alzheimer's disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann Neurol. 2000;48:148–155. [PubMed] [Google Scholar]
- 142.Kimchi-Sarfaty C. Oh JM. Kim IW. Sauna ZE. Calcagno AM. Ambudkar SV. Gottesman MM. A "silent" polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 143.King MP. Attardi G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 144.Kish SJ. Bergeron C. Rajput A. Dozic S. Mastrogiacomo F. Chang LJ. Wilson JM. DiStefano LM. Nobrega JN. Brain cytochrome oxidase in Alzheimer's disease. J Neurochem. 1992;59:776–779. doi: 10.1111/j.1471-4159.1992.tb09439.x. [DOI] [PubMed] [Google Scholar]
- 145.Kish SJ. Mastrogiacomo F. Guttman M. Furukawa Y. Taanman JW. Dozic S. Pandolfo M. Lamarche J. DiStefano L. Chang LJ. Decreased brain protein levels of cytochrome oxidase subunits in Alzheimer's disease and in hereditary spinocerebellar ataxia disorders: A nonspecific change? J Neurochem. 1999;72:700–707. doi: 10.1046/j.1471-4159.1999.0720700.x. [DOI] [PubMed] [Google Scholar]
- 146.Komar AA. Guillemet E. Reiss C. Cullin C. Enhanced expression of the yeast Ure2 protein in Escherichia coli: The effect of synonymous codon substitutions at a selected place in the gene. Biol Chem. 1998;379:1295–1300. [PubMed] [Google Scholar]
- 147.Komar AA. Lesnik T. Reiss C. Synonymous codon substitutions affect ribosome traffic and protein folding during in vitro translation. FEBS Lett. 1999;462:387–391. doi: 10.1016/s0014-5793(99)01566-5. [DOI] [PubMed] [Google Scholar]
- 148.Kroemer G. Petit P. Zamzami N. Vayssiere JL. Mignotte B. The biochemistry of programmed cell death. FASEB J. 1995;9:1277–1287. doi: 10.1096/fasebj.9.13.7557017. [DOI] [PubMed] [Google Scholar]
- 149.Kujoth GC. Hiona A. Pugh TD. Someya S. Panzer K. Wohlgemuth SE. Hofer T. Seo AY. Sullivan R. Jobling WA. Morrow JD. Van Remmen H. Sedivy JM. Yamasoba T. Tanokura M. Weindruch R. Leeuwenburgh C. Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 150.Kumar U. Dunlop DM. Richardson JS. Mitochondria from Alzheimer's fibroblasts show decreased uptake of calcium and increased sensitivity to free radicals. Life Sci. 1994;54:1855–1860. doi: 10.1016/0024-3205(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 151.Kusiak JW. Izzo JA. Zhao B. Neurodegeneration in Alzheimer disease. Is apoptosis involved? Mol Chem Neuropathol. 1996;28:153–162. doi: 10.1007/BF02815217. [DOI] [PubMed] [Google Scholar]
- 152.Lakatos A. Derbeneva O. Younes D. Keator D. Bakken T. Lvova M. Brandon M. Guffanti G. Reglodi D. Saykin A. Weiner M. Macciardi F. Schork N. Wallace DC. Potkin SG. Association between mitochondrial DNA variations and Alzheimer's disease in the ADNI cohort. Neurobiol Aging. 2010;31:1355–1363. doi: 10.1016/j.neurobiolaging.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lambert JC. Heath S. Even G. Campion D. Sleegers K. Hiltunen M. Combarros O. Zelenika D. Bullido MJ. Tavernier B. Letenneur L. Bettens K. Berr C. Pasquier F. Fievet N. Barberger-Gateau P. Engelborghs S. De Deyn P. Mateo I. Franck A. Helisalmi S. Porcellini E. Hanon O. de Pancorbo MM. Lendon C. Dufouil C. Jaillard C. Leveillard T. Alvarez V. Bosco P. Mancuso M. Panza F. Nacmias B. Bossu P. Piccardi P. Annoni G. Seripa D. Galimberti D. Hannequin D. Licastro F. Soininen H. Ritchie K. Blanche H. Dartigues JF. Tzourio C. Gut I. Van Broeckhoven C. Alperovitch A. Lathrop M. Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 154.Levy-Lahad E. Wasco W. Poorkaj P. Romano DM. Oshima J. Pettingell WH. Yu CE. Jondro PD. Schmidt SD. Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 155.Li F. Calingasan NY. Yu F. Mauck WM. Toidze M. Almeida CG. Takahashi RH. Carlson GA. Flint Beal M. Lin MT. Gouras GK. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- 156.Li X. Song L. Jope RS. Cholinergic stimulation of AP-1 and NF kappa B transcription factors is differentially sensitive to oxidative stress in SH-SY5Y neuroblastoma: Relationship to phosphoinositide hydrolysis. J Neurosci. 1996;16:5914–5922. doi: 10.1523/JNEUROSCI.16-19-05914.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lin MT. Beal MF. Alzheimer's APP mangles mitochondria. Nat Med. 2006;12:1241–1243. doi: 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- 158.Lin MT. Simon DK. Ahn CH. Kim LM. Beal MF. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer's disease brain. Hum Mol Genet. 2002;11:133–145. doi: 10.1093/hmg/11.2.133. [DOI] [PubMed] [Google Scholar]
- 159.Liu Y. Liu F. Grundke-Iqbal I. Iqbal K. Gong CX. Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J Pathol. 2011;225:54–62. doi: 10.1002/path.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lovell MA. Ehmann WD. Butler SM. Markesbery WR. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer's disease. Neurology. 1995;45:1594–1601. doi: 10.1212/wnl.45.8.1594. [DOI] [PubMed] [Google Scholar]
- 161.Lu J. Wang K. Rodova M. Esteves R. Berry D. E L. Crafter A. Barrett M. Cardoso SM. Onyango I. Parker WD. Fontes J. Burns JM. Swerdlow RH. Polymorphic variation in cytochrome oxidase subunit genes. J Alzheimers Dis. 2010;21:141–154. doi: 10.3233/JAD-2010-100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lustbader JW. Cirilli M. Lin C. Xu HW. Takuma K. Wang N. Caspersen C. Chen X. Pollak S. Chaney M. Trinchese F. Liu S. Gunn-Moore F. Lue LF. Walker DG. Kuppusamy P. Zewier ZL. Arancio O. Stern D. Yan SS. Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 163.Magistretti PJ. Pellerin L. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond B Biol Sci. 1999;354:1155–1163. doi: 10.1098/rstb.1999.0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Maliarchuk BA. Derenko MV. [T4336C variant—A marker of mitochondrial subgroup H1, a common component of the Russian and German gene pool] Genetika. 2001;37:1578–1580. [PubMed] [Google Scholar]
- 165.Mancuso M. Filosto M. Bosetti F. Ceravolo R. Rocchi A. Tognoni G. Manca ML. Solaini G. Siciliano G. Murri L. Decreased platelet cytochrome c oxidase activity is accompanied by increased blood lactate concentration during exercise in patients with Alzheimer disease. Exp Neurol. 2003;182:421–426. doi: 10.1016/s0014-4886(03)00092-x. [DOI] [PubMed] [Google Scholar]
- 166.Mancuso M. Siciliano G. Filosto M. Murri L. Mitochondrial dysfunction and Alzheimer's disease: New developments. J Alzheimers Dis. 2006;9:111–117. doi: 10.3233/jad-2006-9203. [DOI] [PubMed] [Google Scholar]
- 167.Manczak M. Anekonda TS. Henson E. Park BS. Quinn J. Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 168.Manczak M. Calkins MJ. Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: Implications for neuronal damage. Hum Mol Genet. 2011;220:2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Manczak M. Park BS. Jung Y. Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer's disease: Implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004;5:147–162. doi: 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- 170.Markesbery WR. The role of oxidative stress in Alzheimer disease. Arch Neurol. 1999;56:1449–1452. doi: 10.1001/archneur.56.12.1449. [DOI] [PubMed] [Google Scholar]
- 171.Martinez-Redondo D. Marcuello A. Casajus JA. Ara I. Dahmani Y. Montoya J. Ruiz-Pesini E. Lopez-Perez MJ. Diez-Sanchez C. Human mitochondrial haplogroup H: the highest VO2max consumer—Is it a paradox? Mitochondrion. 2010;10:102–107. doi: 10.1016/j.mito.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 172.Martins RN. Harper CG. Stokes GB. Masters CL. Increased cerebral glucose-6-phosphate dehydrogenase activity in Alzheimer's disease may reflect oxidative stress. J Neurochem. 1986;46:1042–1045. doi: 10.1111/j.1471-4159.1986.tb00615.x. [DOI] [PubMed] [Google Scholar]
- 173.Maruszak A. Canter JA. Styczynska M. Zekanowski C. Barcikowska M. Mitochondrial haplogroup H and Alzheimer's disease—Is there a connection? Neurobiol Aging. 2009;30:1749–1755. doi: 10.1016/j.neurobiolaging.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 174.Maruszak A. Safranow K. W B. Gaweda-Walerych K. Pospiech E. Gabryelewicz T. Canter JA. Barcikowska M. Zekanowski C. The impact of mitochondrial and nuclear DNA variants in late-onset Alzheimer's disease risk. JAD. doi: 10.3233/JAD-2011-110710. in press. [DOI] [PubMed] [Google Scholar]
- 175.Masliah E. Cole GM. Hansen LA. Mallory M. Albright T. Terry RD. Saitoh T. Protein kinase C alteration is an early biochemical marker in Alzheimer's disease. J Neurosci. 1991;11:2759–2767. doi: 10.1523/JNEUROSCI.11-09-02759.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Maurer I. Zierz S. Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455–462. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 177.McKhann G. Drachman D. Folstein M. Katzman R. Price D. Stadlan EM. Clinical diagnosis of Alzheimer's disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 178.McKhann GM. Knopman DS. Chertkow H. Hyman BT. Jack CR., Jr. Kawas CH. Klunk WE. Koroshetz WJ. Manly JJ. Mayeux R. Mohs RC. Morris JC. Rossor MN. Scheltens P. Carrillo MC. Thies B. Weintraub S. Phelps CH. The diagnosis of dementia due to Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mecocci P. MacGarvey U. Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 180.Mirra SS. Heyman A. McKeel D. Sumi SM. Crain BJ. Brownlee LM. Vogel FS. Hughes JP. van Belle G. Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 181.Morais R. Desjardins P. Turmel C. Zinkewich-Peotti K. Development and characterization of continuous avian cell lines depleted of mitochondrial DNA. In Vitro Cell Dev Biol. 1988;24:649–658. doi: 10.1007/BF02623602. [DOI] [PubMed] [Google Scholar]
- 182.Moreira PI. Carvalho C. Zhu X. Smith MA. Perry G. Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim Biophys Acta. 2010;1802:2–10. doi: 10.1016/j.bbadis.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 183.Mosconi L. Berti V. Swerdlow RH. Pupi A. Duara R. de Leon M. Maternal transmission of Alzheimer's disease: Prodromal metabolic phenotype and the search for genes. Hum Genomics. 2010;4:170–193. doi: 10.1186/1479-7364-4-3-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mosconi L. Brys M. Switalski R. Mistur R. Glodzik L. Pirraglia E. Tsui W. De Santi S. de Leon MJ. Maternal family history of Alzheimer's disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci USA. 2007;104:19067–19072. doi: 10.1073/pnas.0705036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mosconi L. de Leon M. Murray J. E L. Lu J. Javier E. McHugh P. Swerdlow RH. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer's disease. JAD. doi: 10.3233/JAD-2011-110866. [Epub ahead of print, 2011]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mosconi L. Glodzik L. Mistur R. McHugh P. Rich KE. Javier E. Williams S. Pirraglia E. De Santi S. Mehta PD. Zinkowski R. Blennow K. Pratico D. de Leon MJ. Oxidative stress and amyloid-beta pathology in normal individuals with a maternal history of Alzheimer's. Biol Psychiatry. 2010;68:913–921. doi: 10.1016/j.biopsych.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mosconi L. Mistur R. Switalski R. Brys M. Glodzik L. Rich K. Pirraglia E. Tsui W. De Santi S. de Leon MJ. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology. 2009;72:513–520. doi: 10.1212/01.wnl.0000333247.51383.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mosconi L. Rinne JO. Tsui WH. Berti V. Li Y. Wang H. Murray J. Scheinin N. Nagren K. Williams S. Glodzik L. De Santi S. Vallabhajosula S. de Leon MJ. Increased fibrillar amyloid-{beta} burden in normal individuals with a family history of late-onset Alzheimer's. Proc Natl Acad Sci USA. 2010;107:5949–5954. doi: 10.1073/pnas.0914141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mutisya EM. Bowling AC. Beal MF. Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. J Neurochem. 1994;63:2179–2184. doi: 10.1046/j.1471-4159.1994.63062179.x. [DOI] [PubMed] [Google Scholar]
- 190.Nagy Z. Esiri MM. LeGris M. Matthews PM. Mitochondrial enzyme expression in the hippocampus in relation to Alzheimer-type pathology. Acta Neuropathol. 1999;97:346–354. doi: 10.1007/s004010050997. [DOI] [PubMed] [Google Scholar]
- 191.Nakamura T. Watanabe A. Fujino T. Hosono T. Michikawa M. Apolipoprotein E4 (1-272) fragment is associated with mitochondrial proteins and affects mitochondrial function in neuronal cells. Mol Neurodegener. 2009;4:35. doi: 10.1186/1750-1326-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Nakano H. Nakajima A. Sakon-Komazawa S. Piao JH. Xue X. Okumura K. Reactive oxygen species mediate crosstalk between NF-kappaB and JNK. Cell Death Differ. 2006;13:730–737. doi: 10.1038/sj.cdd.4401830. [DOI] [PubMed] [Google Scholar]
- 193.Nunomura A. Perry G. Aliev G. Hirai K. Takeda A. Balraj EK. Jones PK. Ghanbari H. Wataya T. Shimohama S. Chiba S. Atwood CS. Petersen RB. Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 194.O'Barr S. Schultz J. Rogers J. Expression of the protooncogene bcl-2 in Alzheimer's disease brain. Neurobiol Aging. 1996;17:131–136. doi: 10.1016/0197-4580(95)02024-1. [DOI] [PubMed] [Google Scholar]
- 195.Onyango IG. Ahn JY. Tuttle JB. Bennett JP., Jr. Swerdlow RH. Nerve growth factor attenuates oxidant-induced beta-amyloid neurotoxicity in sporadic Alzheimer's disease cybrids. J Neurochem. 2010;114:1605–1618. doi: 10.1111/j.1471-4159.2010.06871.x. [DOI] [PubMed] [Google Scholar]
- 196.Onyango IG. Bennett JP., Jr. Tuttle JB. Endogenous oxidative stress in sporadic Alzheimer's disease neuronal cybrids reduces viability by increasing apoptosis through pro-death signaling pathways and is mimicked by oxidant exposure of control cybrids. Neurobiol Dis. 2005;19:312–322. doi: 10.1016/j.nbd.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 197.Onyango IG. Tuttle JB. Bennett JP., Jr Altered intracellular signaling and reduced viability of Alzheimer's disease neuronal cybrids is reproduced by beta-amyloid peptide acting through receptor for advanced glycation end products (RAGE) Mol Cell Neurosci. 2005;29:333–343. doi: 10.1016/j.mcn.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 198.Ott A. Stolk RP. van Harskamp F. Pols HA. Hofman A. Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 199.Pappolla MA. Omar RA. Kim KS. Robakis NK. Immunohistochemical evidence of oxidative [corrected] stress in Alzheimer's disease. Am J Pathol. 1992;140:621–628. [PMC free article] [PubMed] [Google Scholar]
- 200.Parisi MA. Clayton DA. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science. 1991;252:965–969. doi: 10.1126/science.2035027. [DOI] [PubMed] [Google Scholar]
- 201.Parker WD. Sporadic neurologic disease and the electron transport chain: A hypothesis. Proceedings of the 1989 Scientfic Meeting of the American Society for Neurological Investigation: New Developments in Neuromuscular Disease edited by Pascuzzi R; Bloomington, Indiana: Indiana University Printing Services; 1990. [Google Scholar]
- 202.Parker WD., Jr. Boyson SJ. Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol. 1989;26:719–23. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- 203.Parker WD., Jr. Filley CM. Parks JK. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology. 1990;40:1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 204.Parker WD., Jr. Mahr NJ. Filley CM. Parks JK. Hughes D. Young DA. Cullum CM. Reduced platelet cytochrome c oxidase activity in Alzheimer's disease. Neurology. 1994;44:1086–1090. doi: 10.1212/wnl.44.6.1086. [DOI] [PubMed] [Google Scholar]
- 205.Parker WD., Jr. Parks J. Filley CM. Kleinschmidt-DeMasters BK. Electron transport chain defects in Alzheimer's disease brain. Neurology. 1994;44:1090–1096. doi: 10.1212/wnl.44.6.1090. [DOI] [PubMed] [Google Scholar]
- 206.Parker WD., Jr. Parks JK. Cytochrome c oxidase in Alzheimer's disease brain: Purification and characterization. Neurology. 1995;45:482–486. doi: 10.1212/wnl.45.3.482. [DOI] [PubMed] [Google Scholar]
- 207.Parks JK. Smith TS. Trimmer PA. Bennett JP., Jr. Parker WD., Jr Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J Neurochem. 2001;76:1050–1056. doi: 10.1046/j.1471-4159.2001.00112.x. [DOI] [PubMed] [Google Scholar]
- 208.Pei JJ. Khatoon S. An WL. Nordlinder M. Tanaka T. Braak H. Tsujio I. Takeda M. Alafuzoff I. Winblad B. Cowburn RF. Grundke-Iqbal I. Iqbal K. Role of protein kinase B in Alzheimer's neurofibrillary pathology. Acta Neuropathol. 2003;105:381–392. doi: 10.1007/s00401-002-0657-y. [DOI] [PubMed] [Google Scholar]
- 209.Pellerin L. Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Pereira C. Santos MS. Oliveira C. Mitochondrial function impairment induced by amyloid beta-peptide on PC12 cells. Neuroreport. 1998;9:1749–1755. doi: 10.1097/00001756-199806010-00015. [DOI] [PubMed] [Google Scholar]
- 211.Pericak-Vance MA. Bebout JL. Gaskell PC., Jr. Yamaoka LH. Hung WY. Alberts MJ. Walker AP. Bartlett RJ. Haynes CA. Welsh KA, et al. Linkage studies in familial Alzheimer disease: Evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48:1034–1050. [PMC free article] [PubMed] [Google Scholar]
- 212.Perry EK. Perry RH. Tomlinson BE. Blessed G. Gibson PH. Coenzyme A-acetylating enzymes in Alzheimer's disease: Possible cholinergic 'compartment' of pyruvate dehydrogenase. Neurosci Lett. 1980;18:105–110. doi: 10.1016/0304-3940(80)90220-7. [DOI] [PubMed] [Google Scholar]
- 213.Perry G. Nunomura A. Hirai K. Zhu X. Perez M. Avila J. Castellani RJ. Atwood CS. Aliev G. Sayre LM. Takeda A. Smith MA. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer's and other neurodegenerative diseases? Free Radic Biol Med. 2002;33:1475–1479. doi: 10.1016/s0891-5849(02)01113-9. [DOI] [PubMed] [Google Scholar]
- 214.Peterson C. Gibson GE. Blass JP. Altered calcium uptake in cultured skin fibroblasts from patients with Alzheimer's disease. N Engl J Med. 1985;312:1063–1065. doi: 10.1056/NEJM198504183121618. [DOI] [PubMed] [Google Scholar]
- 215.Peterson C. Goldman JE. Alterations in calcium content and biochemical processes in cultured skin fibroblasts from aged and Alzheimer donors. Proc Natl Acad Sci USA. 1986;83:2758–2762. doi: 10.1073/pnas.83.8.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Petit PX. Lecoeur H. Zorn E. Dauguet C. Mignotte B. Gougeon ML. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J Cell Biol. 1995;130:157–167. doi: 10.1083/jcb.130.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Potkin SG. Guffanti G. Lakatos A. Turner JA. Kruggel F. Fallon JH. Saykin AJ. Orro A. Lupoli S. Salvi E. Weiner M. Macciardi F. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer's disease. PLoS One. 2009;4:e6501. doi: 10.1371/journal.pone.0006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Reddy PH. McWeeney S. Park BS. Manczak M. Gutala RV. Partovi D. Jung Y. Yau V. Searles R. Mori M. Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer's disease. Hum Mol Genet. 2004;13:1225–1240. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- 219.Rickle A. Bogdanovic N. Volkman I. Winblad B. Ravid R. Cowburn RF. Akt activity in Alzheimer's disease and other neurodegenerative disorders. Neuroreport. 2004;15:955–959. doi: 10.1097/00001756-200404290-00005. [DOI] [PubMed] [Google Scholar]
- 220.Rodriguez-Santiago B. Casademont J. Nunes V. Is mitochondrial DNA depletion involved in Alzheimer's disease? Eur J Hum Genet. 2001;9:279–285. doi: 10.1038/sj.ejhg.5200629. [DOI] [PubMed] [Google Scholar]
- 221.Rodriguez Santiago B. Casademont J. Nunes V. [Is there a relation between Alzheimer s disease and defects of mitochondrial DNA?] Rev Neurol. 2001;33:301–305. [PubMed] [Google Scholar]
- 222.Rohn TT. Head E. Su JH. Anderson AJ. Bahr BA. Cotman CW. Cribbs DH. Correlation between caspase activation and neurofibrillary tangle formation in Alzheimer's disease. Am J Pathol. 2001;158:189–198. doi: 10.1016/S0002-9440(10)63957-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Roses AD. An inherited variable poly-T repeat genotype in TOMM40 in Alzheimer disease. Arch Neurol. 2010;67:536–541. doi: 10.1001/archneurol.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Roses AD. Lutz MW. Amrine-Madsen H. Saunders AM. Crenshaw DG. Sundseth SS. Huentelman MJ. Welsh-Bohmer KA. Reiman EM. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer's disease. Pharmacogenomics J. 2010;10:375–384. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Roses AD. Saunders AM. Huang Y. Strum J. Weisgraber KH. Mahley RW. Complex disease-associated pharmacogenetics: Drug efficacy, drug safety, and confirmation of a pathogenetic hypothesis (Alzheimer's disease) Pharmacogenomics J. 2007;7:10–28. doi: 10.1038/sj.tpj.6500397. [DOI] [PubMed] [Google Scholar]
- 226.Salehi A. Verhaagen J. Dijkhuizen PA. Swaab DF. Co-localization of high-affinity neurotrophin receptors in nucleus basalis of Meynert neurons and their differential reduction in Alzheimer's disease. Neuroscience. 1996;75:373–387. doi: 10.1016/0306-4522(96)00273-4. [DOI] [PubMed] [Google Scholar]
- 227.Santoro A. Balbi V. Balducci E. Pirazzini C. Rosini F. Tavano F. Achilli A. Siviero P. Minicuci N. Bellavista E. Mishto M. Salvioli S. Marchegiani F. Cardelli M. Olivieri F. Nacmias B. Chiamenti AM. Benussi L. Ghidoni R. Rose G. Gabelli C. Binetti G. Sorbi S. Crepaldi G. Passarino G. Torroni A. Franceschi C. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer's disease. PLoS One. 2010;5:e12037. doi: 10.1371/journal.pone.0012037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Satoh T. Nakatsuka D. Watanabe Y. Nagata I. Kikuchi H. Namura S. Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons. Neurosci Lett. 2000;288:163–166. doi: 10.1016/s0304-3940(00)01229-5. [DOI] [PubMed] [Google Scholar]
- 229.Schreck R. Albermann K. Baeuerle PA. Nuclear factor kappa B: An oxidative stress-responsive transcription factor of eukaryotic cells (a review) Free Radic Res Commun. 1992;17:221–237. doi: 10.3109/10715769209079515. [DOI] [PubMed] [Google Scholar]
- 230.Schreck R. Rieber P. Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Selznick LA. Holtzman DM. Han BH. Gokden M. Srinivasan AN. Johnson EM., Jr. Roth KA. In situ immunodetection of neuronal caspase-3 activation in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58:1020–1026. doi: 10.1097/00005072-199909000-00012. [DOI] [PubMed] [Google Scholar]
- 232.Seshadri S. Fitzpatrick AL. Ikram MA. DeStefano AL. Gudnason V. Boada M. Bis JC. Smith AV. Carassquillo MM. Lambert JC. Harold D. Schrijvers EM. Ramirez-Lorca R. Debette S. Longstreth WT., Jr. Janssens AC. Pankratz VS. Dartigues JF. Hollingworth P. Aspelund T. Hernandez I. Beiser A. Kuller LH. Koudstaal PJ. Dickson DW. Tzourio C. Abraham R. Antunez C. Du Y. Rotter JI. Aulchenko YS. Harris TB. Petersen RC. Berr C. Owen MJ. Lopez-Arrieta J. Varadarajan BN. Becker JT. Rivadeneira F. Nalls MA. Graff-Radford NR. Campion D. Auerbach S. Rice K. Hofman A. Jonsson PV. Schmidt H. Lathrop M. Mosley TH. Au R. Psaty BM. Uitterlinden AG. Farrer LA. Lumley T. Ruiz A. Williams J. Amouyel P. Younkin SG. Wolf PA. Launer LJ. Lopez OL. van Duijn CM. Breteler MM. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sheehan JP. Swerdlow RH. Miller SW. Davis RE. Parks JK. Parker WD. Tuttle JB. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer's disease. J Neurosci. 1997;17:4612–4622. doi: 10.1523/JNEUROSCI.17-12-04612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Sheehan JP. Swerdlow RH. Parker WD. Miller SW. Davis RE. Tuttle JB. Altered calcium homeostasis in cells transformed by mitochondria from individuals with Parkinson's disease. J Neurochem. 1997;68:1221–1233. doi: 10.1046/j.1471-4159.1997.68031221.x. [DOI] [PubMed] [Google Scholar]
- 235.Sherrington R. Rogaev EI. Liang Y. Rogaeva EA. Levesque G. Ikeda M. Chi H. Lin C. Li G. Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 236.Shimohama S. Narita M. Matsushima H. Kimura J. Kameyama M. Hagiwara M. Hidaka H. Taniguchi T. Assessment of protein kinase C isozymes by two-site enzyme immunoassay in human brains and changes in Alzheimer's disease. Neurology. 1993;43:1407–1413. doi: 10.1212/wnl.43.7.1407. [DOI] [PubMed] [Google Scholar]
- 237.Shoffner JM. Brown MD. Torroni A. Lott MT. Cabell MF. Mirra SS. Beal MF. Yang CC. Gearing M. Salvo R, et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics. 1993;17:171–184. doi: 10.1006/geno.1993.1299. [DOI] [PubMed] [Google Scholar]
- 238.Silva DF. Esteves AR. Arduino DM. Oliveira CR. Cardoso SM. Amyloid-beta-induced mitochondrial dysfunction impairs the autophagic lysosomal pathway in a tubulin dependent pathway. J Alzheimers Dis. 2011 doi: 10.3233/JAD-2011-110423. in press. [DOI] [PubMed] [Google Scholar]
- 239.Silva DFF. Esteves AR. Oliveira CR. Cardoso SM. Mitochondria: The common upstream driver of amyloid-beta and tau pathology in Alzheimer's disease. Curr Alzheimer Res. 2011 doi: 10.2174/156720511796391872. in press. [DOI] [PubMed] [Google Scholar]
- 240.Simonian NA. Hyman BT. Functional alterations in Alzheimer's disease: Diminution of cytochrome oxidase in the hippocampal formation. J Neuropathol Exp Neurol. 1993;52:580–585. [PubMed] [Google Scholar]
- 241.Sims NR. Finegan JM. Blass JP. Altered glucose metabolism in fibroblasts from patients with Alzheimer's disease. N Engl J Med. 1985;313:638–639. doi: 10.1056/NEJM198509053131013. [DOI] [PubMed] [Google Scholar]
- 242.Sims NR. Finegan JM. Blass JP. Altered metabolic properties of cultured skin fibroblasts in Alzheimer's disease. Ann Neurol. 1987;21:451–457. doi: 10.1002/ana.410210507. [DOI] [PubMed] [Google Scholar]
- 243.Sims NR. Finegan JM. Blass JP. Bowen DM. Neary D. Mitochondrial function in brain tissue in primary degenerative dementia. Brain Res. 1987;436:30–38. doi: 10.1016/0006-8993(87)91553-8. [DOI] [PubMed] [Google Scholar]
- 244.Smale G. Nichols NR. Brady DR. Finch CE. Horton WE., Jr Evidence for apoptotic cell death in Alzheimer's disease. Exp Neurol. 1995;133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- 245.Smith MA. Kutty RK. Richey PL. Yan SD. Stern D. Chader GJ. Wiggert B. Petersen RB. Perry G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer's disease. Am J Pathol. 1994;145:42–47. [PMC free article] [PubMed] [Google Scholar]
- 246.Smith MA. Rudnicka-Nawrot M. Richey PL. Praprotnik D. Mulvihill P. Miller CA. Sayre LM. Perry G. Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer's disease. J Neurochem. 1995;64:2660–2666. doi: 10.1046/j.1471-4159.1995.64062660.x. [DOI] [PubMed] [Google Scholar]
- 247.Sorbi S. Bird ED. Blass JP. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol. 1983;13:72–78. doi: 10.1002/ana.410130116. [DOI] [PubMed] [Google Scholar]
- 248.Sperling RA. Aisen PS. Beckett LA. Bennett DA. Craft S. Fagan AM. Iwatsubo T. Jack CR., Jr. Kaye J. Montine TJ. Park DC. Reiman EM. Rowe CC. Siemers E. Stern Y. Yaffe K. Carrillo MC. Thies B. Morrison-Bogorad M. Wagster MV. Phelps CH. Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Strazielle C. Jazi R. Verdier Y. Qian S. Lalonde R. Regional brain metabolism with cytochrome c oxidase histochemistry in a PS1/A246E mouse model of autosomal dominant Alzheimer's disease: Correlations with behavior and oxidative stress. Neurochem Int. 2009;55:806–814. doi: 10.1016/j.neuint.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 250.Su B. Wang X. Nunomura A. Moreira PI. Lee HG. Perry G. Smith MA. Zhu X. Oxidative stress signaling in Alzheimer's disease. Curr Alzheimer Res. 2008;5:525–532. doi: 10.2174/156720508786898451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Su JH. Anderson AJ. Cummings BJ. Cotman CW. Immunohistochemical evidence for apoptosis in Alzheimer's disease. Neuroreport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 252.Su JH. Deng G. Cotman CW. Bax protein expression is increased in Alzheimer's brain: Correlations with DNA damage, Bcl-2 expression, and brain pathology. J Neuropathol Exp Neurol. 1997;56:86–93. doi: 10.1097/00005072-199701000-00009. [DOI] [PubMed] [Google Scholar]
- 253.Suzuki A. Stern SA. Bozdagi O. Huntley GW. Walker RH. Magistretti PJ. Alberini CM. Astrocyte–neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–823. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Suzuki K. Terry RD. Fine structural localization of acid phosphatase in senile plaques in Alzheimer's presenile dementia. Acta Neuropathol. 1967;8:276–284. doi: 10.1007/BF00688828. [DOI] [PubMed] [Google Scholar]
- 255.Swerdlow R. Marcus DL. Landman J. Kooby D. Frey W., 2nd Freedman ML. Brain glucose metabolism in Alzheimer's disease. Am J Med Sci. 1994;308:141–144. doi: 10.1097/00000441-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 256.Swerdlow RH. Is aging part of Alzheimer's disease, or is Alzheimer's disease part of aging? Neurobiol Aging. 2007;28:1465–1480. doi: 10.1016/j.neurobiolaging.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 257.Swerdlow RH. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J Neurosci Res. 2007;85:3416–3428. doi: 10.1002/jnr.21167. [DOI] [PubMed] [Google Scholar]
- 258.Swerdlow RH. Pathogenesis of Alzheimer's disease. Clin Interv Aging. 2007;2:347–359. [PMC free article] [PubMed] [Google Scholar]
- 259.Swerdlow RH. The neurodegenerative mitochondriopathies. J Alzheimers Dis. 2009;17:737–751. doi: 10.3233/JAD-2009-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Swerdlow RH. Burns JM. Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis. J Alzheimers Dis. 2010;20:S265–279. doi: 10.3233/JAD-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Swerdlow RH. Khan SM. A "mitochondrial cascade hypothesis" for sporadic Alzheimer's disease. Med Hypotheses. 2004;63:8–20. doi: 10.1016/j.mehy.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 262.Swerdlow RH. Khan SM. The Alzheimer's disease mitochondrial cascade hypothesis: An update. Exp Neurol. 2009;218:308–315. doi: 10.1016/j.expneurol.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Swerdlow RH. Kish SJ. Mitochondria in Alzheimer's disease. Int Rev Neurobiol. 2002;53:341–385. doi: 10.1016/s0074-7742(02)53013-0. [DOI] [PubMed] [Google Scholar]
- 264.Swerdlow RH. Parks JK. Cassarino DS. Maguire DJ. Maguire RS. Bennett JP., Jr. Davis RE. Parker WD., Jr Cybrids in Alzheimer's disease: A cellular model of the disease? Neurology. 1997;49:918–925. doi: 10.1212/wnl.49.4.918. [DOI] [PubMed] [Google Scholar]
- 265.Szabados T. Dul C. Majtenyi K. Hargitai J. Penzes Z. Urbanics R. A chronic Alzheimer's model evoked by mitochondrial poison sodium azide for pharmacological investigations. Behav Brain Res. 2004;154:31–40. doi: 10.1016/j.bbr.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 266.Takasaki S. Mitochondrial haplogroups associated with Japanese Alzheimer's patients. J Bioenerg Biomembr. 2009;41:407–410. doi: 10.1007/s10863-009-9240-8. [DOI] [PubMed] [Google Scholar]
- 267.Takei N. Miyashita A. Tsukie T. Arai H. Asada T. Imagawa M. Shoji M. Higuchi S. Urakami K. Kimura H. Kakita A. Takahashi H. Tsuji S. Kanazawa I. Ihara Y. Odani S. Kuwano R. Genetic association study on in and around the APOE in late-onset Alzheimer disease in Japanese. Genomics. 2009;93:441–448. doi: 10.1016/j.ygeno.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 268.Tamagno E. Bardini P. Obbili A. Vitali A. Borghi R. Zaccheo D. Pronzato MA. Danni O. Smith MA. Perry G. Tabaton M. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis. 2002;10:279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- 269.Tamagno E. Guglielmotto M. Giliberto L. Vitali A. Borghi R. Autelli R. Danni O. Tabaton M. JNK and ERK1/2 pathways have a dual opposite effect on the expression of BACE1. Neurobiol Aging. 2009;30:1563–1573. doi: 10.1016/j.neurobiolaging.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 270.Tanaka N. Goto Y. Akanuma J. Kato M. Kinoshita T. Yamashita F. Tanaka M. Asada T. Mitochondrial DNA variants in a Japanese population of patients with Alzheimer's disease. Mitochondrion. 2010;10:32–37. doi: 10.1016/j.mito.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 271.Teng FY. Tang BL. Widespread gamma-secretase activity in the cell, but do we need it at the mitochondria? Biochem Biophys Res Commun. 2005;328:1–5. doi: 10.1016/j.bbrc.2004.12.131. [DOI] [PubMed] [Google Scholar]
- 272.Thiffault C. Bennett JP., Jr Cyclical mitochondrial deltapsiM fluctuations linked to electron transport, F0F1 ATP-synthase and mitochondrial Na+/Ca+2 exchange are reduced in Alzheimer's disease cybrids. Mitochondrion. 2005;5:109–119. doi: 10.1016/j.mito.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 273.Tortosa A. Lopez E. Ferrer I. Bcl-2 and Bax protein expression in Alzheimer's disease. Acta Neuropathol. 1998;95:407–412. doi: 10.1007/s004010050817. [DOI] [PubMed] [Google Scholar]
- 274.Trifunovic A. Wredenberg A. Falkenberg M. Spelbrink JN. Rovio AT. Bruder CE. Bohlooly YM. Gidlof S. Oldfors A. Wibom R. Tornell J. Jacobs HT. Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 275.Trimmer PA. Borland MK. Differentiated Alzheimer's disease transmitochondrial cybrid cell lines exhibit reduced organelle movement. Antioxid Redox Signal. 2005;7:1101–1109. doi: 10.1089/ars.2005.7.1101. [DOI] [PubMed] [Google Scholar]
- 276.Trimmer PA. Keeney PM. Borland MK. Simon FA. Almeida J. Swerdlow RH. Parks JP. Parker WD., Jr. Bennett JP., Jr Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer's disease worsen with passage in culture. Neurobiol Dis. 2004;15:29–39. doi: 10.1016/j.nbd.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 277.Trimmer PA. Swerdlow RH. Parks JK. Keeney P. Bennett JP., Jr. Miller SW. Davis RE. Parker WD., Jr Abnormal mitochondrial morphology in sporadic Parkinson's and Alzheimer's disease cybrid cell lines. Exp Neurol. 2000;162:37–50. doi: 10.1006/exnr.2000.7333. [DOI] [PubMed] [Google Scholar]
- 278.Tsujimoto Y. Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–840. doi: 10.1007/s10495-006-0525-7. [DOI] [PubMed] [Google Scholar]
- 279.Turner CJ. Granycome C. Hurst R. Pohler E. Juhola MK. Juhola MI. Jacobs HT. Sutherland L. Holt IJ. Systematic segregation to mutant mitochondrial DNA and accompanying loss of mitochondrial DNA in human NT2 teratocarcinoma cybrids. Genetics. 2005;170:1879–1885. doi: 10.1534/genetics.105.043653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Valla J. Berndt JD. Gonzalez-Lima F. Energy hypometabolism in posterior cingulate cortex of Alzheimer's patients: Superficial laminar cytochrome oxidase associated with disease duration. J Neurosci. 2001;21:4923–4930. doi: 10.1523/JNEUROSCI.21-13-04923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Valla J. Schneider L. Niedzielko T. Coon KD. Caselli R. Sabbagh MN. Ahern GL. Baxter L. Alexander G. Walker DG. Reiman EM. Impaired platelet mitochondrial activity in Alzheimer's disease and mild cognitive impairment. Mitochondrion. 2006;6:323–330. doi: 10.1016/j.mito.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Valla J. Yaari R. Wolf AB. Kusne Y. Beach TG. Roher AE. Corneveaux JJ. Huentelman MJ. Caselli RJ. Reiman EM. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer's susceptibility gene. J Alzheimers Dis. 2010;22:307–313. doi: 10.3233/JAD-2010-100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.van der Walt JM. Dementieva YA. Martin ER. Scott WK. Nicodemus KK. Kroner CC. Welsh-Bohmer KA. Saunders AM. Roses AD. Small GW. Schmechel DE. Murali Doraiswamy P. Gilbert JR. Haines JL. Vance JM. Pericak-Vance MA. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci Lett. 2004;365:28–32. doi: 10.1016/j.neulet.2004.04.051. [DOI] [PubMed] [Google Scholar]
- 284.Verwer RW. Jansen KA. Sluiter AA. Pool CW. Kamphorst W. Swaab DF. Decreased hippocampal metabolic activity in Alzheimer patients is not reflected in the immunoreactivity of cytochrome oxidase subunits. Exp Neurol. 2000;163:440–451. doi: 10.1006/exnr.2000.7385. [DOI] [PubMed] [Google Scholar]
- 285.Wallace DC. Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science. 1992;256:628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 286.Wallace DC. Bunn CL. Eisenstadt JM. Cytoplasmic transfer of chloramphenicol resistance in human tissue culture cells. J Cell Biol. 1975;67:174–188. doi: 10.1083/jcb.67.1.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Wang X. Su B. Fujioka H. Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer's disease patients. Am J Pathol. 2008;173:470–482. doi: 10.2353/ajpath.2008.071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Wang X. Su B. Lee HG. Li X. Perry G. Smith MA. Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Wang X. Su B. Siedlak SL. Moreira PI. Fujioka H. Wang Y. Casadesus G. Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Webster MT. Pearce BR. Bowen DM. Francis PT. The effects of perturbed energy metabolism on the processing of amyloid precursor protein in PC12 cells. J Neural Transm. 1998;105:839–853. doi: 10.1007/s007020050098. [DOI] [PubMed] [Google Scholar]
- 291.Wisniewski H. Terry RD. Hirano A. Neurofibrillary pathology. J Neuropathol Exp Neurol. 1970;29:163–176. [PubMed] [Google Scholar]
- 292.Wong-Riley M. Antuono P. Ho KC. Egan R. Hevner R. Liebl W. Huang Z. Rachel R. Jones J. Cytochrome oxidase in Alzheimer's disease: Biochemical, histochemical, and immunohistochemical analyses of the visual and other systems. Vision Res. 1997;37:3593–3608. doi: 10.1016/S0042-6989(96)00210-6. [DOI] [PubMed] [Google Scholar]
- 293.Wong-Riley MT. Cytochrome oxidase: An endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989;12:94–101. doi: 10.1016/0166-2236(89)90165-3. [DOI] [PubMed] [Google Scholar]
- 294.Wragg MA. Talbot CJ. Morris JC. Lendon CL. Goate AM. No association found between Alzheimer's disease and a mitochondrial tRNA glutamine gene variant. Neurosci Lett. 1995;201:107–110. doi: 10.1016/0304-3940(95)12146-3. [DOI] [PubMed] [Google Scholar]
- 295.Yaffe K. Middleton LE. Lui LY. Spira AP. Stone K. Racine C. Ensrud KE. Kramer JH. Mild cognitive impairment, dementia, and their subtypes in oldest old women. Arch Neurol. 2011;68:631–636. doi: 10.1001/archneurol.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Yamaguchi H. Yamazaki T. Ishiguro K. Shoji M. Nakazato Y. Hirai S. Ultrastructural localization of Alzheimer amyloid beta/A4 protein precursor in the cytoplasm of neurons and senile plaque-associated astrocytes. Acta Neuropathol. 1992;85:15–22. doi: 10.1007/BF00304629. [DOI] [PubMed] [Google Scholar]
- 297.Yanagisawa M. Planel E. Ishiguro K. Fujita SC. Starvation induces tau hyperphosphorylation in mouse brain: Implications for Alzheimer's disease. FEBS Lett. 1999;461:329–333. doi: 10.1016/s0014-5793(99)01480-5. [DOI] [PubMed] [Google Scholar]
- 298.Yao J. Du H. Yan S. Fang F. Wang C. Lue LF. Guo L. Chen D. Stern DM. Gunn Moore FJ. Xi Chen J. Arancio O. Yan SS. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer's disease. J Neurosci. 2011;31:2313–2320. doi: 10.1523/JNEUROSCI.4717-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Yao J. Irwin RW. Zhao L. Nilsen J. Hamilton RT. Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Yu CE. Seltman H. Peskind ER. Galloway N. Zhou PX. Rosenthal E. Wijsman EM. Tsuang DW. Devlin B. Schellenberg GD. Comprehensive analysis of APOE and selected proximate markers for late-onset Alzheimer's disease: Patterns of linkage disequilibrium and disease/marker association. Genomics. 2007;89:655–665. doi: 10.1016/j.ygeno.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Zamzami N. Marchetti P. Castedo M. Zanin C. Vayssiere JL. Petit PX. Kroemer G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med. 1995;181:1661–1672. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Zhang H. Liu Y. Lao M. Ma Z. Yi X. Puerarin protects Alzheimer's disease neuronal cybrids from oxidant-stress induced apoptosis by inhibiting pro-death signaling pathways. Exp Gerontol. 2011;46:30–37. doi: 10.1016/j.exger.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 303.Zhang Q. Yu JT. Wang P. Chen W. Wu ZC. Jiang H. Tan L. Mitochondrial transcription factor A (TFAM) polymorphisms and risk of late-onset Alzheimer's disease in Han Chinese. Brain Res. 2011;1368:355–360. doi: 10.1016/j.brainres.2010.10.074. [DOI] [PubMed] [Google Scholar]
- 304.Zhu X. Castellani RJ. Takeda A. Nunomura A. Atwood CS. Perry G. Smith MA. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the 'two hit' hypothesis. Mech Ageing Dev. 2001;123:39–46. doi: 10.1016/s0047-6374(01)00342-6. [DOI] [PubMed] [Google Scholar]
- 305.Zhu X. Raina AK. Rottkamp CA. Aliev G. Perry G. Boux H. Smith MA. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem. 2001;76:435–441. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- 306.Zsurka G. Kalman J. Csaszar A. Rasko I. Janka Z. Venetianer P. No mitochondrial haplotype was found to increase risk for Alzheimer's disease. Biol Psychiatry. 1998;44:371–373. doi: 10.1016/s0006-3223(97)00461-7. [DOI] [PubMed] [Google Scholar]