

Abstract
Aims: The basal oxidative and nitrosative stress levels measured in cytosol, mitochondria, and nuclei as well as in the whole homogenate obtained from the brain of wild type (wt) and p53 knockout [p53(−/−)] mice were evaluated. We hypothesized that the loss of p53 could trigger the activation of several protective mechanisms such as those involving thioredoxin-1 (Thio-1), the heme-oxygenase-1/biliverdin reductase-A (HO-1/BVR-A) system, manganese superoxide dismutase (MnSOD), the IkB kinase type β (IKKβ)/nuclear factor kappa-B (NF-kB), and the nuclear factor-erythroid 2 (NF-E2) related factor 2 (Nrf-2). Results: A decrease of protein carbonyls, protein-bound 4-hydroxy-2-nonenal (HNE), and 3-nitrotyrosine (3-NT) was observed in the brain from p53(−/−) mice compared with wt. Furthermore, we observed a significant increase of the expression levels of Thio-1, BVR-A, MnSOD, IKKβ, and NF-kB. Conversely a significant decrease of Nrf-2 protein levels was observed in the nuclear fraction isolated from p53(−/−) mice. No changes were found for HO-1. Innovation: This is the first study of basal oxidative/nitrosative stress in in vivo conditions of brain obtained from p53(−/−) mice. New insights into the role of p53 in oxidative stress have been gained. Conclusion: We demonstrated, for the first time, that the lack of p53 reduces basal oxidative stress levels in mice brain. Due to the pivotal role that p53 plays during cellular stress response our results provide new insights into novel therapeutic strategies to modulate protein oxidation and lipid peroxidation having p53 as a target. The implications of this work are profound, particularly for neurodegenerative disorders. Antioxid. Redox Signal. 16, 1407–1420.
Introduction
P53, a sequence-specific transcription factor localized predominantly in the nucleus, plays a main role during cellular stress response due to its involvement in several processes (17). As a transcription factor, p53 consists of two N-terminal transactivation domains, a core DNA-binding domain, and a C-terminal oligomerization domain (17). In unstressed cells, p53 is constitutively restrained by Mdm2, an E3 ubiquitin ligase that promotes p53 degradation; the Mdm2 gene is positively regulated by p53, defining a negative feedback loop that controls p53 activity. Cellular stress relieves inhibitory effects of Mdm2, triggering p53 stabilization and activation. Once activated p53 binds to DNA and transactivates genes encoding proteins responsible for DNA repair, cell cycle arrest, and/or the induction of apoptosis in cells harboring nonrepairable damaged DNA, as well as senescence (42). In this light, it represents a “guardian” of genomic integrity preventing the passage of damaged DNA from cell to cell (41).
In the central nervous system (CNS), p53 is responsible for the elimination of newly born postmitotic neurons that do not appropriately differentiate (15). Since to direct cells toward apoptosis or senescence is a process that has to be rapidly addressed, p53 stability and activity were not modulated at a transcriptional level, which would require long time, and therefore they are tightly regulated by posttranslational mechanisms selectively activated by different stress signals. Phosphorylation of serine and/or threonine residues, acetylation, ubiquitylation, methylation of lysine or arginine residues, and glutathionylation represent some of the mechanisms trough which p53-related functions are modulated (17, 21).
Innovation.
This investigation represents the first study of oxidative and nitrosative stress in the brain from p53(−/−) mice and provides new insights into the interconnectivity of p53 and oxidative stress in the brain.
Interesting is the interplay between p53 and reactive oxygen species/reactive nitrogen species (ROS/RNS) production, particularly for the “Janus” face of this interaction, since p53 can either increase or decrease ROS/RNS generation and, at the same time, these latter can modulate selective transactivation of p53 target gene (42). The differential effects of p53 on cellular redox status depend on the single cell type (43). Most of the evidence about the link between p53 and oxidative stress derive from in vitro and in vivo studies in which p53 is modulated by using stimulatory or inhibitory tools (27, 38). However, previous studies have also demonstrated an opposite effect of p53 deficiency on oxidative stress levels measured in neurons, cardiac tissues, and lung fibroblasts: pro-oxidant in the former, overall antioxidant in the latter (16, 49, 66). Similarly, in an in vivo model of p53-null mice, opposite results were found toward the extent of lipid peroxidation, measured as malondialdheyde adducts, in liver, lung, and cortex of these mice (16). One proposed explication of this paradigm was that the effect of p53 depends on the threshold levels of oxidative stress required for its activation (15). In particular, in cells different from neurons, and in the absence of exogenous stress, low levels of p53 are sufficient to induce expression of antioxidant genes such as sestrins that contribute to cellular homeostasis (56). On the other hand, an increase of the oxidative stress levels in the same cells lead to the activation of p53 that became pro-oxidant and pro-apoptotic (56). Conversely, in neurons, the threshold levels of oxidative stress required for p53-induced pro-oxidant effects are lower than those observed in other cell lines (15) and so p53 remains pro-oxidant even under normal conditions. These basal pro-oxidant effects, in any case, should not be considered necessarily deleterious for neurons, because they seem to be linked to still uncharacterized mechanisms for which ROS/RNS are required for synaptic plasticity, long-term potentiation, or neuronal plasticity (15). Growing experimental findings in fact underscore ROS and RNS, produced in low amounts and in a controlled manner, as physiological components of the signaling generated by cytokines (57), growth factors (24), and neurotrophic peptides (6).
However, despite these physiological activities, the roles of (i) ROS/RNS, (ii) p53, and (iii) their interaction in the CNS raise a lot of questions due to their involvement in the pathogenesis of neurodegenerative disorders such as amnestic mild cognitive impairment (MCI) and Alzheimer disease (AD) (9, 39). Generally, an increased production of ROS or RNS triggers an array of signal transduction pathways, resulting in stimulatory or inhibitory output signals and causing cellular damage (47, 48) often linked to lipid and protein oxidative/nitrosative modifications according to the mechanisms reviewed by us (62) (Fig. 1). As previously reported, even p53 represents a target for oxidative and nitrosative posttranslational modifications in MCI and AD (13, 14). In particular, an increase of p53 protein levels was observed in MCI and AD brain compared with control samples (14), paralleled by an increase of oxidative/nitrosative modifications, which, in turn, could affect its activity (13, 14). These results are consistent with the notion of an involvement of p53 in neurodegenerative conditions and its special link with oxidative stress, but the impact of cellular redox changes on p53 and vice versa remain elusive.
FIG. 1.

Reaction mechanisms to identify the products of protein oxidation, lipid peroxidation, and protein nitration. (A) Reaction mechanism to identify oxidized proteins by using DNPH. DNPH can be used to detect the carbonyl functionality of a ketone or aldehyde functional group. This process is an addition–elimination reaction in which a nucleophilic addition of the -NH2 group of a molecule of dinitrophenylhydrazine to the C=O carbonyl group of the protein is followed by the removal of a molecule of water (H2O) with the formation of a dinitrophenylhydrazone adduct. This latter can be recognized by using specific antibody antidinitrophenylhydrazone adduct and then quantified by using specific software (see Materials and Methods section). (B) Protein-bound 4-hydroxy-2-nonenals (HNE) adducts. Following lipid peroxidation the formation of HNE occurs. The reactive alkenal is capable of easily attaching covalently to proteins by forming Michael adducts with, for example, histidine or cysteine residues. (C) Formation of 3-nitrotyrosine (3-NT). Nitric oxide (NO) can react with superoxide anions (O2−•)—produced, for example, by complexes I and III of mitochondrial respiration chain—to form peroxynitrite (ONOO−), an anion with strong oxidant properties. As a consequence of the subsequent reactions, nitration of tyrosine residues of cellular proteins, 3-NT, takes place, resulting in damage to cellular components. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
The aim of this work was to evaluate the effects that the lack of p53 had on the basal levels of oxidative and nitrosative stress in the mice brain. Furthermore, we hypothesized that the loss of p53 could trigger the activation of an integrated network of mechanisms that are under control of genes strictly involved in preserving cellular homeostasis during stressful conditions, such as those encoding for thioredoxin-1 (Thio-1), members of heme-oxygenase-1/biliverdin reductase-A (HO-1/BVR-A) system, manganese superoxide dismutase (MnSOD), the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), the IkB kinase type β (IKKβ), and nuclear factor-erythroid 2 (NF-E2) related factor 2 (Nrf-2). Due to the different localization of these proteins into the whole cell, for instance membrane-bound proteins, cytosolic proteins, mitochondrial proteins, or nuclear proteins, their expression levels were measured in the related fraction isolated from the brain of wild-type (wt) and p53 knockout [p53(−/−)] mice.
Results
Oxidative and nitrosative stress levels in the brain from wt and p53(−/−) mice
To clarify the contribution of p53 on brain oxidative and nitrosative stress, protein carbonyls (PCs), protein-bound 4-hydroxy-2-nonenal (HNE), and 3-nitrotyrosine (3-NT) levels were assayed in different cellular fractions such as cytosol, mitochondria, and nucleus as well as in brain whole homogenate isolated from brain from wt and p53(−/−) mice.
As shown in Figure 2, only 3-NT levels were significantly reduced by about 15% in the cytosolic fraction from p53(−/−) mice with respect to wt mice (Fig. 2C). No changes were observed for PC and HNE levels (Fig. 2A, B, respectively). Isolated brain mitochondria from p53(−/−) mice displayed a significant reduction of about 20% in PCs with respect to wt mice (Fig. 3A). Similarly, protein-bound HNE levels were significantly decreased by 18% in brain mitochondria from p53(−/−) mice (Fig. 3B). No significant changes were observed for 3-NT (Fig. 3C). For the nuclear fraction, we observed a significant reduction of all the biomarkers measured. In particular, PCs were reduced by 19% (Fig. 4A), HNE by 17% (Fig. 4B), and 3-NT by 17% (Fig. 4C) in the nuclei obtained from p53(−/−) mice.
FIG. 2.

In vivo oxidative and nitrosative modifications observed in brain-isolated cytosol from wild-type (wt) and p53 knockout [p53(−/−)] mice. (A) Protein carbonyl (PC) levels, (B) protein-bound HNE levels, and (C) 3-NT levels were measured in the cytosolic fraction obtained from the brain of wt and p53(−/−) mice. Densitometric values shown are given as percentage of the wt group, set as 100%. Data are expressed as mean±SD of three replicates of each individual sample (n=6) per group. *p<0.05 versus wt (Student's t-test).
FIG. 3.

In vivo oxidative and nitrosative modifications observed in brain-isolated mitochondria from wt and p53(−/−) mice. (A) PC levels, (B) protein-bound HNE levels, and (C) 3-NT levels were measured in the mitochondrial fraction obtained from the brain of wt and p53(−/−) mice. Densitometric values shown are given as percentage of the wt group, set as 100%. Data are expressed as mean±SD of three replicates of each individual sample (n=6) per group. *p<0.05 and **p<0.01 versus wt (Student's t-test).
FIG. 4.

In vivo oxidative and nitrosative modifications observed in brain-isolated nuclei from wt and p53(−/−) mice. (A) PC levels, (B) protein-bound HNE levels, and (C) 3-NT levels were measured in the nuclear fraction obtained from the brain of wt and p53(−/−) mice. Densitometric values shown are given as percentage of the wt group, set as 100%. Data are expressed as mean±SD of three replicates of each individual sample (n=6) per group. **p<0.01 versus wt (Student's t-test).
Finally, to better understand whether changes observed in each fraction isolated from the brain of p53(−/−) mice contributed to an overall decrease of oxidative stress levels, we measured the extent of PC, HNE, and 3-NT in the total homogenate obtained from the brain of wt and p53(−/−) mice. As shown in Figure 5 we found a significant reduction of PC (Fig. 5A), HNE (Fig. 5B), and 3-NT (Fig. 5C) of 19%, 20%, and 28%, respectively, in the whole homogenate obtained from the brain of p53(−/−) mice with respect to wt mice.
FIG. 5.

In vivo oxidative and nitrosative modifications observed in the whole homogenate obtained from the brain of wt and p53(−/−) mice. (A) PC levels, (B) protein-bound HNE levels, and (C) 3-NT levels were measured in the whole homogenate obtained from the brain of wt and p53−/− mice. Densitometric values shown are given as percentage of the wt group, set as 100%. Data are expressed as mean±SD of three replicates of each individual sample (n=6) per group. *p<0.05 and **p<0.01 versus wt (Student's t-test).
Effect of lack of p53 on the expression of membrane-bound protein primarily involved in the cell stress response: thioredoxin-1 and HO-1
HO, also known as heat-shock protein (Hsp)-32, is a microsomal protein existing in two isoforms: the constitutive isoform (HO-2) and the inducible isoform (HO-1). This latter is induced by various stimuli, including oxidative and nitrosative stress, ischemia, heat shock, bacterial lipopolysaccharide (LPS), hemin, and the neuroprotective agent leteprinim potassium (Neotrofin). HO-1 catalyzes the degradation of the intracellular levels of pro-oxidant iron-protoporphyrin-IX-alpha (heme) into ferrous iron, carbon monoxide, and biliverdin-IX-alpha (BV) (44) the precursor of bilirubin-IX-alpha (BR), this latter being an endogenous molecule with powerful antioxidant and antinitrosative features (5, 45). Thio-1 is constitutively present as a surface-associated sulfhydryl protein in plasma membrane of a wide range of cells (31) and is induced under pro-oxidant conditions, such as UV irradiation and hydrogen peroxide (H2O2), in order to prevent cell injury against oxidative stress (3). Together with reduced glutathione, Thio-1 forms a powerful system involved in a variety of redox-dependent pathways such as supplying reducing equivalent for ribonucleotide reductase, and peptide methionine sulfoxide reductase, the latter being involved in antioxidant defense (3).
To elucidate the effect that the lack of p53 had on the expression levels of both HO-1 and Thio-1, Western blot analyses for HO-1 and Thio-1 were performed in the membrane fraction isolated from brain of p53(−/−) and wt mice. As shown in Figure 6A, no significant changes were observed for HO-1 protein levels. However, Thio-1 protein levels were significantly increased in the membrane fraction from p53(−/−) mice with respect to the control group (Fig. 6B).
FIG. 6.

Effect of lack of p53 on the expression levels of thioredoxin-1 (Thio-1) and heme oxygenase-1 (HO-1) in mice brain. (A) Thio-1 and (B) HO-1 protein levels were measured in the membrane fraction isolated from the brain of wt and p53(−/−) mice as described under Materials and Methods section. Representative gels are shown. Data are expressed as mean±SD (n=6 animals per group). *p<0.05 versus wt group.
Effect of lack of p53 on the expression of cytosolic and nuclear proteins primarily involved in cell stress response: BVR-A, Nrf-2, IKKβ, and NF-kB
In addition to membrane-bound proteins we analyzed the effects of the loss of p53 on the expression levels of some cytosolic proteins involved in the cell stress response. BVR-A, the main isoform of BVR (26, 36), catalyzes the reduction of BV into BR and is coexpressed with HO-1 in cells of the rat brain that express these enzymes under normal conditions (23). Furthermore, BVR-A is also a serine/threonine/tyrosine kinase that interacts with members of the mitogen-activated protein kinase family, in particular, the extracellular signal–regulated kinases 1/2, and once translocated into the nucleus regulates the expression of oxidative stress–responsive genes such as HO-1 or inducible nitric oxide synthase (iNOS) (36). As shown in Figure 7A, BVR-A protein levels, measured in the cytosolic fraction isolated from mice brain, were significantly increased in the p53(−/−) group with respect to wt. Likewise, the absence of p53 promotes the translocation of BVR-A from the cytosol into the nucleus as demonstrated by the significant increase of BVR-A protein levels (∼36%) in the nuclear fraction isolated from the brain of p53(−/−) mice with respect to wt group (Fig. 8A).
FIG. 7.

Effect of lack of p53 on the cytosolic levels of biliverdin reductase-A (BVR-A), nuclear factor-erythroid 2 (NF-E2) related factor 2 (Nrf-2), nuclear factor kappa-B (NF-kB), and IkB kinase type β (IKKβ) in mice brain. (A) BVR-A, (B) Nrf-2, (C) NF-kB, and (D) IKKβ protein levels were measured in the cytosolic fraction isolated from the brain of wt and p53(−/−) mice as described under Materials and Methods section. Representative gels are shown. Data are expressed as mean±SD (n=6 animals per group). *p<0.05, **p<0.01, and ***p<0.001 versus wt group.
FIG. 8.

Effect of lack of p53 on the nuclear levels of BVR-A, Nrf-2, and NF-kB in mice brain. (A) BVR-A, (B) Nrf-2, and (C) NF-kB protein levels were measured in the nuclear fraction isolated from the brain of wt and p53(−/−) mice as described under Materials and Methods section. Representative gels are shown. Data are expressed as mean±SD (n=6 animals per group). *p<0.05, and **p<0.01 versus wt group.
The transcription factor NF-E2 (Nrf-2) was demonstrated to regulate the expression of both HO-1 (63) and Thio-1 (37). Under normal conditions it is bound to its cytoplasmic repressor partner keap1, while during conditions of increased oxidative stress, Nrf-2 translocates into the nucleus where it recognizes a region called antioxidant-response element (ARE) on the promoter of the genes encoding for HO-1 or Thio-1 promoting their expression (10, 37, 63). The loss of p53 did not produce any significant effect on the cytosolic levels of Nrf-2 (Fig. 7B). Conversely, as shown in Figure 8B, Nrf-2 protein levels were found to be decreased (∼62%) in a significant manner in the nuclear fraction isolated from the brain of p53(−/−) mice with respect to wt mice.
NF-kB is a critical mediator of the cellular response to inflammatory cytokines, developmental signals, pathogens, and cellular stresses (7). NF-kB exists in a latent form in the cytoplasm of unstimulated cells comprising a transcriptionally active heterodimer (p65/p50 or 52) bound to an inhibitor protein, IkB (7). Following stimulation with many NF-kB inducers, IkB is rapidly phosphorylated by the IKK, such as IKKβ, which promotes IkB degradation and NF-kB activation and nuclear translocation (7). We found a significant increase of both NF-kB and Ikkβ protein levels, of 43% and 20%, respectively, in the cytosolic fraction isolated from the brain of p53(−/−) mice (Fig. 7C, D) with respect to wt. In agreement with the role of Ikkβ, NF-kB protein levels, measured in the nuclear fraction isolated from the brain of p53(−/−) mice, were significantly increased by 103% with respect to wt mice (Fig. 8C).
Effect of lack of p53 on the expression of mitochondrial proteins primarily involved in cell stress response: MnSOD
SODs, primary ROS detoxification enzymes in the cell, catalyze the dismutation of superoxide radicals to molecular oxygen and H2O2. The SOD family of enzymes is made up of three structurally unrelated proteins encoded by different genes (34). Copper- and zinc-containing SOD (CuZnSOD, SOD1) is a homodimeric enzyme found primarily in the cytoplasm (with small amounts within the mitochondria) (34). Extracellular SOD (ECSOD, SOD3) shares 40%–60% homology with CuZnSOD but resides in the extracellular compartment of the cell (34). Manganese-containing SOD (MnSOD, SOD2) is a homotetramer found exclusively in the mitochondrial matrix (68). MnSOD is the only SOD indispensable to aerobic life and is not compensated by the presence of CuZnSOD. The importance of MnSOD is due to its strategic location in the mitochondrial matrix, since SOD offers a primary antioxidant defense through rapid conversion of superoxide radicals into H2O2 and oxygen (34).
Relevant to the current study, proteomic analysis on the same mitochondrial samples for another study ongoing in our laboratory identified a significant increase of MnSOD protein levels of 1330% (p<0.01) in the mitochondrial fraction isolated from the brain of p53(−/−) mice (Fig. 9) with respect to wt.
FIG. 9.
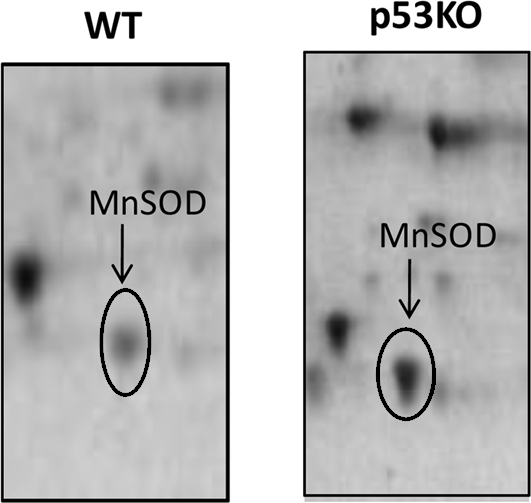
Effect of lack of p53 on the levels of mitochondrial-resident manganese superoxide dismutase (MnSOD) in mice brain. MnSOD protein levels were measured in the mitochondrial fraction isolated from the brain of wt and p53(−/−) mice as described under Materials and Methods section. Representative gels are shown.
Discussion
Several lines of evidence highlight the pivotal role played by p53 in the maintenance of genomic integrity following a stressful insult through the differential activation of target genes that direct cells to cycle arrest, senescence, or apoptosis (67). Given that p53 and ROS/RNS participate and share overlapping signal transduction pathways, we investigated the connections that link p53 to ROS/RNS levels as well as the possible mechanisms involved. In this light, our work represents a step forward in the comprehension of the mechanisms that regulate cellular redox status in the CNS, since, for the first time, we provide new insights on the in vivo effects produced by the loss of p53 on (i) the levels of oxidative and nitrosative stress in different cellular compartments (cytosol, mitochondria, and nucleus) as well as in the whole cell in brain and (ii) the modulation of Thio-1, HO-1, BVR-A, IKKβ, NF-kB, and Nrf-2 protein levels as possible mechanisms useful to explain the differences observed as regard the levels of oxidative and nitrosative stress between wt and p53(−/−) mice.
Cellular ROS production is tightly linked to the p53 levels (42). Physiological levels of p53 serve to contain ROS at nontoxic levels through the transactivation of genes encoding for proteins with antioxidant activity such as glutathione peroxidase-1 (GPX-1) (56) (Fig. 10, left side). In contrast, hyper-physiological levels of p53 increase ROS production at least by two mechanisms: (i) transactivation of target genes that encode for proteins whose activity promote the formation of ROS such as quinine oxidoreductase (NQO1) (42) and proline oxidase (POX) (42) as well as for proteins that perturb mitochondrial electron transport chain such as Bcl-2–associated X protein (BAX) (42) and p53 upregulated modulator of apoptosis (PUMA) (42) (Fig. 10, left side); (ii) suppression of genes encoding for proteins with antioxidant activity such as the mitochondrial isoform of SOD, MnSOD (42) (Fig. 10, left side).
FIG. 10.

Model of p53 leading to reactive oxygen species/reactive nitrogen species (ROS/RNS) in the brain. Physiological levels of p53 (black thin arrows) serve to contain ROS and RNS at nontoxic levels through the transactivation of genes encoding for proteins with antioxidant activity such as glutathione peroxidase-1 (GPX-1) (56, 60, 64). Furthermore, basal levels (black thin arrows) of p53 are required to balance energy metabolism among mitochondrial respiration, glycolysis, and the pentose phosphate shunt, with mitochondrial respiration that is the major source of ROS production (42). In addition, p53 is able to mediate the transcriptional transrepression of the mRNA of the inducible nitric oxide synthase (iNOS) by a negative feedback loop, thus reducing (black dotted thin line) NO accumulation (12, 25). Conversely, hyper-physiological (black full arrows or dotted full line) levels of p53 increase ROS production at least by two mechanisms: (i) transactivation (black full arrows) of targets genes that encode for proteins whose activity promote the formation of ROS such as quinine oxidoreductase (NQO1) (54) and proline oxidase (POX) (55); (ii) suppression (dotted full line) of genes encoding for proteins with antioxidant activity such as the mitochondrial isoform of superoxide dismutase, MnSOD (18). Lack of p53 decreases (dotted red lines) lipid peroxidation and protein oxidation/nitration in mice brain by several mechanisms including increase (red arrows) of (i) Thio-1, (ii) BVR-A, (iii) MnSOD, and (iv) IKKβ/NF-kB. In addition, the reduced levels of PC, HNE, and 3-NT could be due to a shift toward the use of glycolysis instead of mitochondrial respiration to produce ATP (8, 42). Moreover, in the absence of p53, there could be an upregulation of iNOS, thus an increase (red arrow) in NO production (2), although an overall decrease of 3-NT was observed because of the reasons explained in the main text. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
In addition, basal levels of p53 are required to balance energy metabolism among mitochondrial respiration, glycolysis, and the pentose phosphate shunt, with mitochondrial respiration being the major source of ROS production (Fig. 10, left side) (42).
The substantial decrease of protein oxidation/nitration and lipid peroxidation, observed in the brain of p53(−/−) mice (Figs. 2–5) under basal conditions, is in good agreement with previous studies that showed that the deletion of p53, in other cell lines different than those of the CNS, was coupled with a reduction of ROS production (40, 42, 65). One possible mechanism to explain the previous results could be that the absence of p53 forces cells to use glycolysis instead of mitochondrial respiration for energy production (Fig. 10, right side) with a concomitant decrease of ROS (8, 42). However, this mechanism, although plausible, is not be completely exhaustive to explain the differences observed previously, since even cancer cells are characterized by a metabolic shift to glycolysis but at the same time showed high levels of oxidative stress (58).
Here we propose that the loss of p53 could be seen as a signal that perturbs cellular homeostasis, thus induces cells to activate protective machinery in order to prevent cellular damage. However, at this time, we cannot point out whether the decrease of oxidative/nitrosative stress levels observed in each cellular fraction from p53(−/−) mice, happening simultaneously for each fraction or, more likely, is the result of a serial process starting in one of these fractions, for example, nucleus or mitochondria. In our opinion, the second hypothesis could be most appropriate to explain our results, for several reasons based on the analysis of the expression levels of the antioxidant enzymes considered for this study. In this scenario, the increase of Thio-1 protein levels represents the first mechanism through which decreased basal oxidative stress levels could be explained.
Although Thio-1 was demonstrated to be an important partner in the regulation of p53 redox status and biological activity (29), p53 is also a repressor of Thio-1 (28). Our results go in the same direction. Thus, by the deletion of p53, the inhibition could be removed leading to an increase of Thio-1 protein levels which in turn can act not only as cofactor of the earlier cited process, but also as antioxidant or ROS scavenger (31).
In support of this hypothesis is the evidence about the expression levels of Nrf-2 in both cytosolic and nuclear fractions (Figs. 7B and 8B). Nrf-2 is one of the main transcription factors that regulates Thio-1 expression (31), particularly under pro-oxidant condition (10, 37). Contrary to this well-known mechanism, here we found a significant increase of Thio-1 together a significant decrease of Nrf-2 protein levels in the nuclei of p53(−/−) mice (Fig. 8B). The results about Nrf-2 can be easily explained by the low levels of oxidative stress observed in each cellular fraction, which preclude Nrf-2 translocation into the nucleus. The same cannot be the case for Thio-1 since the behavior of Nrf-2 does not allow us to explain why we observed an upregulation of Thio-1, and alternative explanations have to be explored. It is also possible that the expression of Thio-1 in the absence of p53 is independent of Nrf-2. So the possibility that p53 exerts directly or indirectly an inhibitory effect on Thio-1 could be conceivable.
Nrf-2 is also the main transcription factor that regulates HO-1 expression (45). The induction process is regulated principally by two upstream enhancer regions, E1 and E2, on the gene encoding for HO-1, containing multiple AREs recognized by Nrf-2 (45). In this case, the results about Nrf-2 are in good agreement with those about the expression levels of HO-1 showing no changes following the lack of p53 (Fig. 6A). In addition, the stimulation of HO-1 was found to be under control of p53 in thymus and spleen obtained from p53(−/−) mice that showed no increase of HO-1 protein levels in response to γ-irradiation (50) as well as in mice and human cells lacking p53 and exposed to H2O2, a well-known inducer of HO-1 (52). Thus, the lack of activation of Nrf-2 together with the lack of p53 could represent two good reasons to explain the results of HO-1 in the brain of p53(−/−) mice under basal conditions.
For BVR-A, the observed upregulation in both cytosolic and nuclear fractions following the loss of p53 (Figs. 7A and 8A) adds a new element in the picture of the stimuli able to modulate this enzyme. In fact, although it was initially considered a noninducible protein, later studies showed that BVR-A can be induced by substances such as LPS and bromobenzene at the posttranscriptional level, while its expression is unaffected by heat shock (45). Our results, for the first time, propose BVR-A as a target of p53, which probably under basal condition exerts an inhibitory effect on BVR-A. Recalling the pivotal role of BVR-A during cell stress response for the production of the powerful antioxidant BR (5, 61), and for being a protein with a pleiotropic functions as described previously, one possibility could be that in order to maintain the basal levels of ROS/RNS necessary for the modulation of important neuronal processes (synaptic plasticity, long-term potentiation, or neuronal plasticity), p53 acts as controller of BVR-A as well. This does not mean that under basal conditions BVR-A does not work. In fact, even bilirubin at low concentration was proposed as neurotrophic factor (46). So, under basal conditions, a sort of equilibrium could exist between the formation of BR and ROS/RNS. Conversely, we found a decrease of oxidative and nitrosative stress levels in each fraction isolated from the brain of p53(−/−) mice. The most obvious and immediate explanation could be that following BVR-A upregulation an increase of BR occurs and this latter, acting as ROS/RNS scavenger, contributes to the decrease of PC, HNE, and 3-NT. However, if true, this condition would require also an increase of the BR precursor, BV, which in turn is a product of HO activity. Since we found no changes in HO-1 protein levels, and it seems unlikely that there may be an increase of the constitutive isoform HO-2, likely the mechanism by which BVR-A exerts its antioxidant features is not only through BR but also through other mechanisms. However, with regard to BR, instead of the amount it is the rate at which this molecule is produced that could play a role. In fact, we can hypothesize that with the same amount of BV formed by the action of HO-1 and HO-2, an increase of BVR-A proteins would increase the rate at which BR is formed, thus having in a short time, more BR available to exert its antioxidant and antinitrosative effects.
Furthermore, the results about BVR-A once again account for a primary role of p53 and Nrf-2 in the induction of HO-1, because, despite that an increase of BVR-A was observed in the nucleus, no effect was observed on HO-1 protein levels.
Another target for p53 is NF-kB. Redox-dependent and -independent mechanisms were proposed to explain NF-kB activation, both of which, by using different pathways, have NF-kB as a downstream target (7). As for p53, the effect of NF-kB is cell specific although it seems to mediate opposite effects. p53 responds to a variety of intrinsic stresses with the purpose to limit cellular damage by initiating cell death, senescence, or cell cycle arrest, while NF-kB responds to a large number of extrinsic stresses and promotes cell division, which initiates the innate and adaptive immune responses (1). The novelty of our results is that we observed a robust increase of NF-kB in both cytosolic and nuclear fractions isolated from the brain of p53(−/−) mice, demonstrating a role for p53 in keeping NF-kB low even under p53 basal conditions and in the absence of other stress signals. Furthermore, these results are corroborated by those dealing with the expression levels of IKKβ. The translocation of NF-kB into the nucleus requires the dissociation from its inhibitor IkB by a phosphorylation process mediated by IKKβ. Thus, the increase of IKKβ into the cytosolic fraction isolated from the brain of p53(−/−) mice led us to speculate that the loss of p53 activates IKKβ, which, in turn, promotes NF-kB nuclear translocation. Furthermore, from another point of view, the increase of NF-kB into the nucleus could be related to the action of BVR-A that was shown to increase the ability of NF-kB to bind to DNA at least by two mechanisms involving its reductase and kinase activity: by reducing BV that is an inhibitor of NF-kB and by a direct activation of NF-kB (36). In addition, even NF-kB could contribute to the overall decrease of oxidative stress levels observed in p53(−/−) mice, since it (i) promotes the transcription of genes involved in cell survival signaling (1), and (ii) promotes glycolysis (1).
The observed significant increase of MnSOD protein levels in mitochondrial fractions in brain from p53 KO mice (Fig. 9) was very interesting. MnSOD is a p53-regulated gene (33, 53). As previously reported by others, p53 (i) inhibits MnSOD superoxide scavenging activity (70) and (ii) is linked to the regulation of MnSOD protein levels since p53 shows a dual role: at low concentration p53 increases MnSOD protein levels, whereas at high concentration p53 decreases MnSOD expression (19). On the other hand, NF-kB also was identified as the most crucial transcriptional factor regulating MnSOD induction (51), and since we found increased NF-kB protein levels as well as NF-kB nuclear translocation following p53 deletion, this finding could represent another way to explain the elevated level of MnSOD.
However, as for other target proteins, the effect of p53 on MnSOD seems to be cell type specific since as reported by Hussain et al. in the human lymphoblast cell line TK6, the overexpression of p53 increases cellular levels of MnSOD (35). Furthermore, the same authors concluded that the increased oxidative stress observed in TK6 cells was due to increased generation of H2O2 by the elevated levels of MnSOD and possibly inadequate removal by catalase (35). Our results are in good agreement with the proposed inhibiting effect exerted by p53 on MnSOD protein levels (19). In addition, although the increase of MnSOD could raise the question of an increase of H2O2-induced oxidative stress levels, this seems not to be the case in the current study. In fact, we observed an overall significant decrease of PC, HNE, and 3-NT levels in the brain of p53(−/−) mice (Figs. 2–5), which is consistent with a protective role of MnSOD (4, 32, 69). However, the actual mechanism can be complex with other players possibly being involved. One of these could be BR produced by BVR-A due to the former's strong antioxidant properties against H2O2. Consistent with this notion, Dorè et al. demonstrated that BR at nanomolar concentrations protects neurons from the toxicity elicited by 10,000 times higher H2O2 (22).
Another intriguing aspect of our study regards the close connection between RNS, particularly nitric oxide (NO), and p53. Physiological levels of NO in the CNS are required for the maintenance of cognitive function, its role spanning from the induction and maintenance of synaptic plasticity to the control of sleep, appetite, body temperature, and neurosecretion (11). As for ROS, the activity of the p53/NO axis displays opposite effects depending on the cellular pathways involved. High levels of NO—obtained, for example, by using NO-releasing compounds—promote the intracellular accumulation of p53 (25). On the contrary, p53 is able to mediate the transcriptional transrepression of the mRNA of the iNOS by a negative feedback loop, thus reducing NO accumulation (12, 25). Moreover, high levels of NO can induce a conformational change of wt p53 resulting in impairment of its DNA-binding activity in vitro (12) as well as inhibiting its translocation to mitochondria (30).
Here we found a significant decrease in basal 3-NT levels in all but the mitochondrial fraction, following the deletion of p53. At this time, we can only speculate about a possible mechanism. Based on the evidence available, the absence of p53 (i) promotes an upregulation of MnSOD (Figs. 9 and 0, right side) (19) leading to a reduction of superoxide anion; (ii) upregulates iNOS, thus increasing NO production (Fig. 10, right side) (2); and (iii) upregulates BVR-A (Fig. 10, right side), thus increasing BR production/availability, which was demonstrated to act as an NO scavenger (5). That said, despite a possible increase of NO levels, the reduction of superoxide anion levels coupled with the increase of antinitrosative BR could be proposed as mechanism responsible for the decrease of 3-NT levels, in the brain of p53(−/−) mice.
In conclusion we demonstrated for the first time that the lack of p53 reduces significantly basal protein oxidation and lipid peroxidation in the brain of p53(−/−) mice. However, our experimental model does not allow us to completely clarify the exact mechanism that links p53 and the other proteins considered for this study, and ad hoc studies are ongoing in our laboratory. Based on the results presented in this study as well as those present in the literature, p53 activity seems to exert a dominant role in the control of many of proteins among which are Thio-1, BVR-A, MnSOD, and NF-kB, although several points remain to be addressed in future studies. In particular, while it is generally accepted that an upregulation of antioxidant systems following pro-oxidant stimuli occurs, why were we able to observe an increase of Thio-1, BVR-A, MnSOD, and NF-kB even in the absence of ROS/RNS increase? Is p53 a regulator of their expression and, if so, how? Due to the large number of processes in which p53 is involved, the implications of this work are profound. Our results provide a new basis for novel therapeutic strategies to modulate protein oxidation and lipid peroxidation having p53 as target, particularly in neurodegenerative disorders such as AD, in which oxidative stress plays a main role.
Materials and Methods
Chemicals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Nitrocellulose membranes were obtained from Bio-Rad (Hercules, CA). Anti-rabbit IgG horseradish peroxidase conjugate secondary antibody was obtained from GE Healthcare Bio-Sciences Corp. (Piscataway, NJ).
Animals
Heterozygous mice [p53(−/+)] were maintained in our laboratory to produce p53(−/−) and wt littermates. p53(−/−) mice are in the C57BL/6 background and were initially generated in the laboratory of Dr. Tyler Jacks at the Center for Cancer Research and Department of Biology, Massachusetts Institute of Technology (Cambridge, MA). The targeted disrupted p53 genes do not produce p53 protein, since 40% of their gene coding region is eliminated by the induced mutation. Male mice between 10 and 12 weeks old were used in all studies. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Kentucky and followed NIH Guidelines for the Care and Use of Laboratory Animals.
Cellular fraction isolation and purification
Mice were humanely euthanized, then the brain was promptly removed and homogenate, mitochondria, and nuclear fractions were immediately isolated from the freshly obtained brain by differential centrifugation methods (59) with minor modifications. Whole brain was suspended in ice-cold isolation buffer (250 mM sucrose, 10 mM HEPES, and 1 mM potassium EDTA, pH 7.2) and homogenized by six passes of a motor-driven Teflon pestle. The homogenate was centrifuged for 3 min at 1330 g at 4°C, and the resulting pellet was resuspended in isolation buffer and centrifuged at 1330 g for 3 min. The supernatants from both spins were combined and spun at 21,200 g for 10 min at 4°C. The pellet was resuspended in 15% Percoll solution (v/v in isolation buffer) and layered onto discontinuous Percoll gradients of 23% and 40% Percoll (v/v in isolation buffer). Gradients were spun at 30,700 g for 5 min at 4°C. At the 23%–40% Percoll interface, mitochondria were isolated and resuspended in respiration buffer (250 mM sucrose, 2 mM magnesium chloride, 20 mM HEPES, and 2.5 mM phosphate buffer, pH 7.2) and centrifuged at 16,700 g for 10 min at 4°C. The pellet was resuspended in respiration buffer, centrifuged at 6900 g for 10 min at 4°C, and the resulting pellet was washed in phosphate-buffered saline (PBS) at 6900 g for 10 min at 4°C. The pellet was finally resuspended in 0.5–1 ml PBS. Protein concentration was determined by the Pierce BCA method (Pierce, Rockford, IL).
Protein carbonyls
Samples (5 μl) of each fraction as well as of whole homogenate, 12% sodium dodecyl sulfate (SDS; 5 μl), and 10 μl of 10 times diluted 2,4-dinitrophenylhydrazine (DNPH) from 200 mM stock were incubated at room temperature for 20 min, followed by neutralization with 7.5 μl neutralization solution (2 M Tris in 30% glycerol). Protein (250 ng) was loaded in each well on a nitrocellulose membrane under vacuum using a slot blot apparatus. The membrane was blocked in blocking buffer (3% bovine serum albumin [BSA]) in PBS 0.01% (w/v) sodium azide and 0.2% (v/v) Tween 20 for 1 h and incubated with a 1:100 dilution of anti-DNP polyclonal antibody in PBS containing 0.01% (w/v) sodium azide and 0.2% (v/v) Tween 20 for 1 h. The membrane was washed in PBS following primary antibody incubation three times at intervals of 5 min each. The membrane was incubated after washing with an anti-rabbit IgG alkaline phosphatase secondary antibody diluted in PBS in a 1:8000 ratio for 1 h. The membrane was washed three times in PBS for 5 min each and developed with Sigma fast tablets (5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium substrate [BCIP/NBT substrate]). Blots were dried, scanned in Adobe Photoshop, and quantified in Scion Image (PC version of Macintosh-compatible NIH image). No nonspecific binding of antibody to the membrane was observed.
Protein-bound HNE and 3-NT
Samples (5 μl) of each fraction as well as of whole homogenate, 12% SDS (5 μl), and 5 μl modified Laemmli buffer containing 0.125 M Tris base (pH 6.8), 4% (v/v) SDS, and 20% (v/v) glycerol were incubated for 20 min at room temperature and were loaded (250 ng) in each well on a nitrocellulose membrane in a slot blot apparatus under vacuum. The membrane was treated as described previously and incubated with a 1:5000 dilution of antiprotein-bound HNE polyclonal antibody or 1:2000 3-NT antibody in PBS for 90 min. The membranes were further developed and quantified as described previously. A faint background staining resulting from the antibody alone was observed, but, because each sample had a control, this minor effect was controlled.
Western blot analysis
For Western blot analyses, protein levels were analyzed based on their cellular localization into the whole cell. Thus, HO-1 and Thio-1 in membrane fraction; BVR-A, NF-kB, Ikkβ, and Nrf-2 in cytosolic fraction; BVR-A, NF-kB, and Nrf-2 into the nuclear fraction. Briefly, 50 μg of protein was denaturated in sample buffer for 5 min at 100°C, and proteins were separated on 12% precast Criterion gels (Bio-Rad) by electrophoresis at 100 mA for 2 h in 3-(N-morpholino)propane sulfonic acid (MOPS) buffer (Bio-Rad) into Bio-Rad apparatus. The proteins from the gels were then transferred to nitrocellulose membrane using the Transblot-Blot SD Semi-Dry Transfer Cell at 20 mA for 2 h. Subsequently, the membranes were blocked at 4°C for 1 h with fresh blocking buffer made of 3% BSA in PBS containing 0.01% (w/v) sodium azide and 0.2% (v/v) Tween 20 (PBST). The membranes were incubated at room temperature in PBST for 2 h with the following primary antibodies, as separate experiments: polyclonal anti-rabbit Thioredoxin 1 (Cell Signaling Technology, Danvers, MA; dilution 1:1000), polyclonal anti-rabbit HO-1 (Assay design–Stressgen, Ann Harbor, MI; dilution 1:1000), polyclonal anti-rabbit BVR-A (Sigma-Aldrich; dilution 1:1000), polyclonal anti-rabbit NF-kB (p65 subunit) (Assay design–Stressgen; dilution 1:1000), polyclonal anti-rabbit IKKβ (Assay design–Stressgen; dilution 1:1000), polyclonal anti-rabbit Nrf-2 (Abcam, Cambridge, MA; dilution 1:1000), and polyclonal anti-rabbit β-actin (Sigma-Aldrich; dilution 1:2000). The membranes were then washed three times for 5 min with PBST followed by incubation with anti-mouse alkaline phosphatase or horseradish peroxidase conjugate secondary antibody (1:3000) in PBST for 2 h at room temperature. Membranes were then washed three times in PBST for 5 min and developed using BCIP/NBT color developing reagent for alkaline phosphatase secondary antibody or ECL plus WB detection reagents for horseradish peroxidase conjugate secondary antibody. Blots were dried and scanned in TIF format using Adobe Photoshop on a Canoscan 8800F (Canon) or STORM UV transilluminator (λex=470 nm, λem=618 nm; Molecular Dynamics, Sunnyvale, CA) for chemiluminescence. The images were quantified with Image Quant TL 1D version 7.0 software (GE Healthcare). The optical density of bands was calculated as volume (optical density×area) adjusted for the background.
2D gel electrophoresis and identification of MnSOD
2D gel electrophoresis, gel staining, image analysis, trypsin digestion, mass spectrometry, and database interrogation for the identification of MnSOD in mitochondria were performed as previously described by Di Domenico et al. (20).
Statistical analysis
Data are expressed as mean±SD of n=6 independent samples. All statistical analyses were performed using a two-tailed Student's t-test. p<0.05 was considered significantly different from control.
Abbreviations Used
- 3-NT
3-nitrotyrosine
- AD
Alzheimer disease
- ARE
antioxidant-response element
- BAX
Bcl-2 associated X protein
- BCIP/NBT
5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium
- BR
bilirubin-IX-alpha
- BSA
bovine serum albumin
- BV
biliverdin-IX-alpha
- BVR-A
biliverdin reductase-A
- CNS
central nervous system
- CuZnSOD
copper- and zinc-containing SOD
- DNPH
2,4-dinitrophenylhydrazine
- ECSOD
extracellular SOD
- H2O2
hydrogen peroxide
- HNE
4-hydroxy-2-nonenal
- HO-1
heme oxygenase-1
- Hsp
heat-shock protein
- IKKβ
IkB kinase type β
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- MCI
mild cognitive impairment
- MnSOD
manganese superoxide dismutase
- NF-kB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NO
nitric oxide
- NQO1
quinine oxidoreductase
- Nrf-2
nuclear factor-erythroid 2 (NF-E2) related factor 2
- p53(−/−)
p53 knockout
- PBS
phosphate-buffered saline
- PCs
protein carbonyls
- POX
proline oxidase
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- Thio-1
thioredoxin-1
- wt
wild type
Acknowledgments
This work was supported by NIH grants [AG-05119] to D.A.B. and [CA 139843] to D.S.C. The authors thank the Redox Chemistry and Biology Core of the Markey Cancer Center for the use of facilities and resources.
Author Disclosure Statement
There are no conflicts of interest for any author.
References
- 1.Ak P. Levine AJ. p53 and NF-kappaB: different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010;24:3643–3652. doi: 10.1096/fj.10-160549. [DOI] [PubMed] [Google Scholar]
- 2.Ambs S. Ogunfusika MO. Merriam WG. Bennett WP. Billiar TR. Harris CC. Up-regulation of inducible nitric oxide synthase expression in cancer-prone p53 knockout mice. Proc Natl Acad Sci U S A. 1998;95:8823–8828. doi: 10.1073/pnas.95.15.8823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arner ES. Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur J Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 4.Bakthavatchalu V. Dey S. Xu Y. Noel T. Jungsuwadee P. Holley AK. Dhar SK. Batinic-Haberle I. St. Clair DK. Manganese superoxide dismutase is a mitochondrial fidelity protein that protects Polgamma against UV-induced inactivation. Oncogene. 2011. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 5.Barone E. Trombino S. Cassano R. Sgambato A. De Paola B. Di Stasio E. Picci N. Preziosi P. Mancuso C. Characterization of the S-denitrosylating activity of bilirubin. J Cell Mol Med. 2009;13:2365–2375. doi: 10.1111/j.1582-4934.2008.00680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bedogni B. Pani G. Colavitti R. Riccio A. Borrello S. Murphy M. Smith R. Eboli ML. Galeotti T. Redox regulation of cAMP-responsive element-binding protein and induction of manganese superoxide dismutase in nerve growth factor-dependent cell survival. J Biol Chem. 2003;278:16510–16519. doi: 10.1074/jbc.M301089200. [DOI] [PubMed] [Google Scholar]
- 7.Bowie A. O'Neill LA. Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- 8.Brand KA. Hermfisse U. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J. 1997;11:388–395. doi: 10.1096/fasebj.11.5.9141507. [DOI] [PubMed] [Google Scholar]
- 9.Butterfield DA. Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 10.Calabrese V. Cornelius C. Mancuso C. Barone E. Calafato S. Bates T. Rizzarelli E. Kostova AT. Vitagenes, dietary antioxidants and neuroprotection in neurodegenerative diseases. Front Biosci. 2009;14:376–397. doi: 10.2741/3250. [DOI] [PubMed] [Google Scholar]
- 11.Calabrese V. Mancuso C. Calvani M. Rizzarelli E. Butterfield DA. Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 12.Calmels S. Hainaut P. Ohshima H. Nitric oxide induces conformational and functional modifications of wild-type p53 tumor suppressor protein. Cancer Res. 1997;57:3365–3369. [PubMed] [Google Scholar]
- 13.Cenini G. Sultana R. Memo M. Butterfield DA. Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer disease. Free Radic Biol Med. 2008;45:81–85. doi: 10.1016/j.freeradbiomed.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cenini G. Sultana R. Memo M. Butterfield DA. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer's disease. J Cell Mol Med. 2008;12:987–994. doi: 10.1111/j.1582-4934.2008.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatoo W. Abdouh M. Bernier G. p53 Pro-oxidant activity in the central nervous system: implication in aging and neurodegenerative diseases. Antioxid Redox Signal. 2011;15:1729–1737. doi: 10.1089/ars.2010.3610. [DOI] [PubMed] [Google Scholar]
- 16.Chatoo W. Abdouh M. David J. Champagne MP. Ferreira J. Rodier F. Bernier G. The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J Neurosci. 2009;29:529–542. doi: 10.1523/JNEUROSCI.5303-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai C. Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16:528–536. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhar SK. Xu Y. Chen Y. St. Clair DK. Specificity protein 1-dependent p53-mediated suppression of human manganese superoxide dismutase gene expression. J Biol Chem. 2006;281:21698–21709. doi: 10.1074/jbc.M601083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dhar SK. Xu Y. St. Clair DK. Nuclear factor kappaB- and specificity protein 1-dependent p53-mediated bi-directional regulation of the human manganese superoxide dismutase gene. J Biol Chem. 2010;285:9835–9846. doi: 10.1074/jbc.M109.060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Domenico F. Casalena G. Sultana R. Cai J. Pierce WM. Perluigi M. Cini C. Baracca A. Solaini G. Lenaz G. Jia J. Dziennis S. Murphy SJ. Alkayed NJ. Butterfield DA. Involvement of Stat3 in mouse brain development and sexual dimorphism: a proteomics approach. Brain Res. 2010;1362:1–12. doi: 10.1016/j.brainres.2010.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Domenico F. Cenini G. Sultana R. Perluigi M. Uberti D. Memo M. Butterfield DA. Glutathionylation of the pro-apoptotic protein p53 in Alzheimer's disease brain: implications for AD pathogenesis. Neurochem Res. 2009;34:727–733. doi: 10.1007/s11064-009-9924-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dore S. Takahashi M. Ferris CD. Zakhary R. Hester LD. Guastella D. Snyder SH. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci U S A. 1999;96:2445–2450. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ewing JF. Weber CM. Maines MD. Biliverdin reductase is heat resistant and coexpressed with constitutive and heat shock forms of heme oxygenase in brain. J Neurochem. 1993;61:1015–1023. doi: 10.1111/j.1471-4159.1993.tb03615.x. [DOI] [PubMed] [Google Scholar]
- 24.Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–253. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- 25.Forrester K. Ambs S. Lupold SE. Kapust RB. Spillare EA. Weinberg WC. Felley-Bosco E. Wang XW. Geller DA. Tzeng E. Billiar TR. Harris CC. Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc Natl Acad Sci U S A. 1996;93:2442–2447. doi: 10.1073/pnas.93.6.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franklin EM. Browne S. Horan AM. Inomata K. Hammam MA. Kinoshita H. Lamparter T. Golfis G. Mantle TJ. The use of synthetic linear tetrapyrroles to probe the verdin sites of human biliverdin-IX alpha reductase and human biliverdin-IX beta reductase. FEBS J. 2009;276:4405–4413. doi: 10.1111/j.1742-4658.2009.07148.x. [DOI] [PubMed] [Google Scholar]
- 27.Gupta R. Rao Gogineni V. Nalla AK. Chetty C. Klopfenstein JD. Tsung AJ. Mohanam S. Rao JS. Oncogenic role of p53 is suppressed by si-RNA bicistronic construct of uPA, uPAR and cathepsin-B in meningiomas both in vitro and in vivo. Int J Oncol. 2011;38:973–983. doi: 10.3892/ijo.2011.934. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Hadj Amor IY. Smaoui K. Chaabene I. Mabrouk I. Djemal L. Elleuch H. Allouche M. Mokdad-Gargouri R. Gargouri A. Human p53 induces cell death and downregulates thioredoxin expression in Saccharomyces cerevisiae. FEMS Yeast Res. 2008;8:1254–1262. doi: 10.1111/j.1567-1364.2008.00445.x. [DOI] [PubMed] [Google Scholar]
- 29.Hafsi H. Hainaut P. Redox control and interplay between p53 isoforms: roles in the regulation of basal p53 levels, cell fate, and senescence. Antioxid Redox Signal. 2011;15:1655–1667. doi: 10.1089/ars.2010.3771. [DOI] [PubMed] [Google Scholar]
- 30.Hernlund E. Kutuk O. Basaga H. Linder S. Panaretakis T. Shoshan M. Cisplatin-induced nitrosylation of p53 prevents its mitochondrial translocation. Free Radic Biol Med. 2009;46:1607–1613. doi: 10.1016/j.freeradbiomed.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 31.Hirota K. Nakamura H. Masutani H. Yodoi J. Thioredoxin superfamily and thioredoxin-inducing agents. Ann N Y Acad Sci. 2002;957:189–199. doi: 10.1111/j.1749-6632.2002.tb02916.x. [DOI] [PubMed] [Google Scholar]
- 32.Holley AK. Bakthavatchalu V. Velez-Roman JM. St. Clair DK. Manganese superoxide dismutase: guardian of the powerhouse. Int J Mol Sci. 2011;12:7114–7162. doi: 10.3390/ijms12107114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holley AK. Dhar SK. St. Clair DK. Manganese superoxide dismutase versus p53: the mitochondrial center. Ann N Y Acad Sci. 2010;1201:72–78. doi: 10.1111/j.1749-6632.2010.05612.x. [DOI] [PubMed] [Google Scholar]
- 34.Holley AK. St. Clair DK. Preventing Dr. Jekyll from becoming Mr. Hyde: is manganese superoxide dismutase the key to prevent radiation-induced neoplastic transformation? Cancer Biol Ther. 2009;8:1972–1973. doi: 10.4161/cbt.8.20.9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hussain SP. Amstad P. He P. Robles A. Lupold S. Kaneko I. Ichimiya M. Sengupta S. Mechanic L. Okamura S. Hofseth LJ. Moake M. Nagashima M. Forrester KS. Harris CC. p53-induced up-regulation of MnSOD and GPx but not catalase increases oxidative stress and apoptosis. Cancer Res. 2004;64:2350–2356. doi: 10.1158/0008-5472.can-2287-2. [DOI] [PubMed] [Google Scholar]
- 36.Kapitulnik J. Maines MD. Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol Sci. 2009;30:129–137. doi: 10.1016/j.tips.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 37.Kim YC. Masutani H. Yamaguchi Y. Itoh K. Yamamoto M. Yodoi J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. J Biol Chem. 2001;276:18399–18406. doi: 10.1074/jbc.M100103200. [DOI] [PubMed] [Google Scholar]
- 38.Kraus JL. Conti F. Madonna S. Tchoghandjian A. Beclin C. Alternative responses of primary tumor cells and glioblastoma cell lines to N,N-bis-(8-hydroxyquinoline-5-yl methyl)-benzyl substituted amines: cell death versus P53-independent senescence. Int J Oncol. 2010;37:1463–1470. doi: 10.3892/ijo_00000798. [DOI] [PubMed] [Google Scholar]
- 39.Lanni C. Uberti D. Racchi M. Govoni S. Memo M. Unfolded p53: a potential biomarker for Alzheimer's disease. J Alzheimers Dis. 2007;12:93–99. doi: 10.3233/jad-2007-12109. [DOI] [PubMed] [Google Scholar]
- 40.Lebedeva MA. Eaton JS. Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim Biophys Acta. 2009;1787:328–334. doi: 10.1016/j.bbabio.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lim YP. Lim TT. Chan YL. Song AC. Yeo BH. Vojtesek B. Coomber D. Rajagopal G. Lane D. The p53 knowledgebase: an integrated information resource for p53 research. Oncogene. 2007;26:1517–1521. doi: 10.1038/sj.onc.1209952. [DOI] [PubMed] [Google Scholar]
- 42.Liu B. Chen Y. St. Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008;44:1529–1535. doi: 10.1016/j.freeradbiomed.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lozano G. Mouse models of p53 functions. Cold Spring Harb Perspect Biol. 2010;2:a001115. doi: 10.1101/cshperspect.a001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maines MD. The heme oxygenase system and its functions in the brain. Cell Mol Biol (Noisy-le-grand) 2000;46:573–585. [PubMed] [Google Scholar]
- 45.Mancuso C. Barone E. The heme oxygenase/biliverdin reductase pathway in drug research and development. Curr Drug Metab. 2009;10:579–594. doi: 10.2174/138920009789375405. [DOI] [PubMed] [Google Scholar]
- 46.Mancuso C. Capone C. Ranieri SC. Fusco S. Calabrese V. Eboli ML. Preziosi P. Galeotti T. Pani G. Bilirubin as an endogenous modulator of neurotrophin redox signaling. J Neurosci Res. 2008;86:2235–2249. doi: 10.1002/jnr.21665. [DOI] [PubMed] [Google Scholar]
- 47.Mark RJ. Lovell MA. Markesbery WR. Uchida K. Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- 48.Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 49.Matheu A. Maraver A. Klatt P. Flores I. Garcia-Cao I. Borras C. Flores JM. Vina J. Blasco MA. Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 50.Meiller A. Alvarez S. Drane P. Lallemand C. Blanchard B. Tovey M. May E. p53-dependent stimulation of redox-related genes in the lymphoid organs of gamma-irradiated—mice identification of haeme-oxygenase 1 as a direct p53 target gene. Nucleic Acids Res. 2007;35:6924–6934. doi: 10.1093/nar/gkm824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miao L. St. Clair DK. Regulation of superoxide dismutase genes: implications in disease. Free Radic Biol Med. 2009;47:344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nam SY. Sabapathy K. p53 promotes cellular survival in a context-dependent manner by directly inducing the expression of haeme-oxygenase-1. Oncogene. 2011;30:4476–4486. doi: 10.1038/onc.2011.150. [DOI] [PubMed] [Google Scholar]
- 53.Pani G. Bedogni B. Anzevino R. Colavitti R. Palazzotti B. Borrello S. Galeotti T. Deregulated manganese superoxide dismutase expression and resistance to oxidative injury in p53-deficient cells. Cancer Res. 2000;60:4654–4660. [PubMed] [Google Scholar]
- 54.Polyak K. Xia Y. Zweier JL. Kinzler KW. Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 55.Rivera A. Maxwell SA. The p53-induced gene-6 (proline oxidase) mediates apoptosis through a calcineurin-dependent pathway. J Biol Chem. 2005;280:29346–29354. doi: 10.1074/jbc.M504852200. [DOI] [PubMed] [Google Scholar]
- 56.Sablina AA. Budanov AV. Ilyinskaya GV. Agapova LS. Kravchenko JE. Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schulze-Osthoff K. Beyaert R. Vandevoorde V. Haegeman G. Fiers W. Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. EMBO J. 1993;12:3095–3104. doi: 10.1002/j.1460-2075.1993.tb05978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simons AL. Mattson DM. Dornfeld K. Spitz DR. Glucose deprivation-induced metabolic oxidative stress and cancer therapy. J Cancer Res Ther. 2009;5(Suppl 1):S2–S6. doi: 10.4103/0973-1482.55133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sims NR. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem. 1990;55:698–707. doi: 10.1111/j.1471-4159.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- 60.Stambolsky P. Weisz L. Shats I. Klein Y. Goldfinger N. Oren M. Rotter V. Regulation of AIF expression by p53. Cell Death Differ. 2006;13:2140–2149. doi: 10.1038/sj.cdd.4401965. [DOI] [PubMed] [Google Scholar]
- 61.Stocker R. Yamamoto Y. McDonagh AF. Glazer AN. Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 62.Sultana R. Perluigi M. Butterfield DA. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer's disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signal. 2006;8:2021–2037. doi: 10.1089/ars.2006.8.2021. [DOI] [PubMed] [Google Scholar]
- 63.Sun J. Hoshino H. Takaku K. Nakajima O. Muto A. Suzuki H. Tashiro S. Takahashi S. Shibahara S. Alam J. Taketo MM. Yamamoto M. Igarashi K. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002;21:5216–5224. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomko RJ., Jr. Bansal P. Lazo JS. Airing out an antioxidant role for the tumor suppressor p53. Mol Interv. 2006;6:23–25, 2. doi: 10.1124/mi.6.1.5. [DOI] [PubMed] [Google Scholar]
- 65.Trinei M. Giorgio M. Cicalese A. Barozzi S. Ventura A. Migliaccio E. Milia E. Padura IM. Raker VA. Maccarana M. Petronilli V. Minucci S. Bernardi P. Lanfrancone L. Pelicci PG. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene. 2002;21:3872–3878. doi: 10.1038/sj.onc.1205513. [DOI] [PubMed] [Google Scholar]
- 66.Velez JM. Miriyala S. Nithipongvanitch R. Noel T. Plabplueng CD. Oberley T. Jungsuwadee P. Van Remmen H. Vore M. St. Clair DK. p53 Regulates oxidative stress-mediated retrograde signaling: a novel mechanism for chemotherapy-induced cardiac injury. PLoS One. 2011;6:e18005. doi: 10.1371/journal.pone.0018005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vousden KH. Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 68.Weisiger RA. Fridovich I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J Biol Chem. 1973;248:4793–4796. [PubMed] [Google Scholar]
- 69.Wenzel P. Schuhmacher S. Kienhofer J. Muller J. Hortmann M. Oelze M. Schulz E. Treiber N. Kawamoto T. Scharffetter-Kochanek K. Munzel T. Burkle A. Bachschmid MM. Daiber A. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008;80:280–289. doi: 10.1093/cvr/cvn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao Y. Chaiswing L. Velez JM. Batinic-Haberle I. Colburn NH. Oberley TD. St. Clair DK. p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005;65:3745–3750. doi: 10.1158/0008-5472.CAN-04-3835. [DOI] [PubMed] [Google Scholar]