

Abstract
A nodal regulator of endoplasmic reticulum stress is the transcription factor, ATF6, which is activated by ischemia and protects the heart from ischemic damage, in vivo. To explore mechanisms of ATF6-mediated protection in the heart, a whole-genome microRNA (miRNA) array analysis of RNA from the hearts of ATF6 transgenic (TG) mice was performed. The array identified 13 ATF6-regulated miRNAs, eight of which were downregulated, suggesting that they could contribute to increasing levels of their mRNAs. The down-regulated miRNAs, including miR-455, were predicted to target 45 mRNAs that we had previously shown by microarray analysis to be up-regulated by ATF6 in the heart. One of the miR-455 targets was calreticulin (Calr), which is up-regulated in the pathologic heart, where it modulates hypertrophic growth, potentially reducing the impact of the pathology. To validate the effects of miR-455, we showed that Calr protein was increased by ATF6 in mouse hearts, in vivo. In cultured cardiac myocytes, treatment with the ER stressor, tunicamycin, or with adenovirus encoding activated ATF6 decreased miR-455 and increased Calr levels, consistent with the effects of ATF6 on miR-455 and Calr, in vivo. Moreover, transfection of cultured cardiac myocytes with a synthetic precursor, premiR-455, decreased Calr levels, while transfection with an antisense, antimiR-455, increased Calr levels. The results of this study suggest that ER stress can regulate gene expression via ATF6-mediated changes in micro-RNA levels. Moreover, these findings support the hypothesis that ATF6-mediated down-regulation of miR-455 augments Calr expression, which may contribute to the protective effects of ATF6 in the heart.
Keywords: ER stress, ATF6, miRNA, calreticulin
INTRODUCTION
For the efficient synthesis and folding of most secreted and membrane proteins, conditions in the endoplasmic reticulum (ER) must be optimal; suboptimal conditions lead to improper protein folding and eventual ER stress [1]. Initially, ER stress triggers protective aspects of the conserved signaling program known as the unfolded protein response (UPR), which are oriented toward restoring the ER environment [2-4]. However, if the stress continues, and ER protein folding is not restored, apoptotic aspects of the UPR ensue [5, 6].
One of the proximal sensors of ER stress is activating transcription factor 6 (ATF6) [7], a transcription factor that binds to ER stress response elements (ERSEs) and transcriptionally induces numerous genes, a subset of which encode proteins that restore ER protein folding, thus contributing to protection [8]. We previously showed that ischemia activates ER stress in the heart [9], and activates ATF6 in cultured cardiac myocytes [10]. To examine the function of the ATF6 branch of the ER stress response, in vivo, we generated a transgenic (TG) mouse line that expresses a conditionally activated form of ATF6 in the heart [11]. In this model, ATF6 decreased infarct size and apoptosis, and improved functional recovery upon reperfusion in an ex vivo mouse model of global myocardial ischemia. To examine how ATF6 might mediate this protection, a whole-genome microarray analysis revealed the identities of 607 ATF6-regulated genes, 381 of which were upregulated, mostly as a result of ATF6-mediated increases in transcription [12]. In the present study, we determined whether ATF6 also regulates ER stress response gene expression microRNAs (miRNAs).
MiRNAs are short, 20-23 nucleotide, non-coding RNAs that act as inhibitors of gene expression by forming partial duplexes with the 3’ UTR of mRNAs [13, 14]. They act by either inhibiting mRNA translation, or by promoting the degradation of mRNAs [15]. It has been estimated that there are approximately 1,000 microRNAs encoded by the human genome, and each can have numerous mRNA targets [16]. In addition, a single mRNA can be targeted by several different miRNAs, making gene regulation by miRNAs quite complex. Thus, it is possible that ATF6-mediated down-regulation of miRNAs that target ER stress response genes may be a mechanism by which the ATF6 could regulate gene expression, post-transcriptionally. This possibility was examined in the current study, where we determined the effects of ATF6 on miRNA expression in the hearts of ATF6 transgenic mice and in cultured cardiac myocytes.
MATERIALS AND METHODS
Animals
Approximately 12 adult male C57/BL6 mice (6 non-transgenic and 6 ATF6 transgenic mice), and 100 1-4 day-old Harlan Sprague-Dawley rats were used for this study. All procedures involving animals were carried out in accordance with the San Diego State University Institutional Animal Care and Use Committee.
ATF6-MER Transgenic Mice
The generation of ATF6-MER (mutant mouse estrogen receptor) transgenic mice (TG) featuring cardiomyocyte-specific transgene expression, has been described elsewhere [11]. Non-transgenic (NTG) and ATF6-MER transgenic (TG) mice were treated with vehicle or tamoxifen, n = 3 mice per treatment group, to activate Mer-ATF6 in the ATF6 transgenic mouse hearts. Tamoxifen was suspended at 10 mg/ml in 100 ml 95% ethanol and 900 ml sunflower oil and sonicated until clarified. Mice were injected intraperitoneally once/day with 20 mg/kg tamoxifen, or with vehicle only. After 5d, RNA was extracted from mouse heart ventricles, as described previously [11].
ATF6 Whole-Genome miRNA Array
Non-transgenic (NTG) and ATF6-MER transgenic (TG) mice [11] were treated ± tamoxifen for 5 days, n=3 mice per group, and ventricular mRNA was subjected to miRNA array analysis. About 5 mg of total mouse ventricle RNA were submitted to LC Sciences (Houston, TX) for quality control, processing and miRNA expression analysis in accordance with their specifications. All samples met quality control standards. Samples were hybridized to individual mouse miRNA chips (LC Sciences, part #MRA-1002), which were current with Sanger miRBase version 14.0 and contained roughly 700 unique mature miRNA probes. The data reported in this publication have been deposited in NCBI’s Gene Expression Omnibus [17] and are accessible through GEO Series accession number GSE33515 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33515).
miRNA Array Statistics and Data Analysis
Array statistics and data analysis for each of the 4 treatment groups were carried out essentially as described [12]. The miRNAs that exhibited significant changes in expression (p < 0.05) and were differentially expressed in vehicle vs. tamoxifen-treated TG mouse hearts by 1.5-fold, or more, were included in this study. Since tamoxifen may affect miRNA levels independently of its ability to activate ATF6-MER, miRNAs that were differentially expressed in vehicle vs. tamoxifen-treated NTG mouse hearts were excluded from this study.
miRNA Target Prediction
Potential miRNA target sites were predicted by first searching for the miRNA of interest on Sanger miRBase (http://www.mirbase.org) [18], and then identifying putative targets using TargetScan Version 5.1 (http://www.targetscan.org) [19]. Targets for miR-455 were further confirmed using miRanda (http://www.microrna.org) [20].
Primary Neonatal Rat Ventricular Myocyte Cultures
Neonatal rat ventricular myocyte cultures (NRVMCs) were prepared as previously described [12, 21, 22]. Following enzymatic dissociation with TrpE™ Express (Invitrogen Cat no. 12605), cells were plated onto plastic culture dishes and maintained in 10% FCS-containing DMEM/F-12 for ~2h. During this time, cardiac fibroblasts adhere to the culture dishes, but cardiac myocytes remain in suspension. Following this pre-plating, cells that do not plate are mostly cardiac myocytes. After the pre-plating step, NRVMCs were plated onto fibronectin-coated 6-well plates plastic culture dishes at 1×106 cells/well in 10% FCS-containing DMEM/F-12. Twenty-four hours after plating, cells were washed with medium and used for experiments. Procedures for infection of NRVMCs with adenovirus to mediate efficient gene transfer have been described previously, as has the generation and characteristics of the recombinant adenovirus that encodes a constitutively active form of ATF6, ATF6(1-373) [12].
mRNA and miRNA Quantification
For tissue samples, total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA)[11]. For cultured cell samples, both total and small RNA-enriched RNA fractions were obtained using the miRNeasy and miRNA cleanup kits (Qiagen, Valencia, CA). mRNA and miRNA levels were analyzed using the TaqMan quantitative real-time PCR (qRT-PCR) method (10 ng/assay), and quantified with an ABI 7000 Real-Time PCR System (Applied Biosystems, Foster City, CA). Taqman Primer assays for miRNAs and the reagents for reverse transcriptase and qRT-PCR reactions were obtained from Applied Biosystems. Relative expression was calculated using the comparative cycle threshold (Ct) method (2-ΔΔCt). mRNA levels were normalized to GAPDH, as previously described [11], while miRNA levels were normalized to U6 RNA. mRNA levels were measured as previously described[11] using custom primers and normalizing to GAPDH mRNA expression.
PCR Primers
The following primer pairs were used for real time quantitative PCR (RT-qPCR):
Calreticulin (rat) NM_022399:
5’- AGCAGTTCTTGGACGGAGATG
3’- TGTTGGATTCGACCCAGC
GAPDH (rat):
5- CCTGGCCAAGGTCATCCAT
3’- GTCATGAGCCCTTCCACGAT
Calr (mouse) MGI ID 88252:
5’-ACATCAGGAGCTAAAAGCA-GCC
3’-TGAAACATACGTCACCCGCA
GAPDH (mouse):
5’-CCTGGCCAAGGTCATCCAT
3’- GTCATGAGCCCTTCCACGAT
Immunoblots
NRVMC extracts were subjected to SDS-PAGE followed by immunobloting using either a Calr antibody (Abcam, catalog # AB2907) at 1:2,000, an antibody directed against a C-terminal KDEL sequence, which cross-reacts with GRP94 and 78 (Stressgen Biotechnologies, Inc., San Deigo, CA, catalog # SPA-827) at 1:1,000, or an antibody to GAPDH (Research Diagnostics, Inc., Flanders, NJ, catalog # RDI-TRK 5G4-65C) at 1:15,000.
Pre-miR-455 and Anti-miR-455
NRVMCs were subjected to overexpression or knockdown of miR-455 via transfection of premiR-455 or anti-miR455, respectively (Applied Biosystems, #PM20434 and #AM20434), using TransMessenger Transfection Reagent (Qiagen, Valencia, CA).
Statistical Analyses
Experimental results, other than those obtained in the miRNA microarray study, were subjected to Student’s T-test or to ANOVA. In the figures, unless otherwise stated in the legend, the following symbols were used to denote confidence results: ** or ## = p ≤ 0.01, * or # = p ≤ 0.05 different from all other values. The miRNA microarray results were subjected to statistical analyses as described above.
RESULTS
Expression of a Conditionally Activated form of ATF6 in the Heart
ATF6 is conditionally activated in the hearts of ATF6-MER transgenic (TG) mice by tamoxifen [12]. ATF6-MER TG mouse hearts express an ATF6-MER fusion protein under the control of the α-MHC promoter. Therefore, ATF6-MER expression is restricted to cardiac myocytes starting at about the time of birth. Upon administration of tamoxifen, the transcriptional activation domain of ATF6, which, in this version of ATF6, resides between residues 39 and 94 [23], is unmasked, thus activating ATF6 by about 3-fold. Accordingly, ATF6 is activated, only in the ATF6-MER TG mice, and only upon tamoxifen treatment so that the levels of ATF6-regulated genes are changed in the ATF6-MER TG mouse hearts in a tamoxifen-dependent manner [11].
ATF6 Regulates miRNA Expression, In Vivo
To identify miRNAs whose expression is regulated by ATF6, an miRNA microarray analysis was carried out using RNA isolated from ATF6-MER TG and non-TG mouse hearts treated ± tamoxifen. The array showed that, in the mouse heart, activated ATF6 altered the levels of 13 miRs from 0.21- to 2.5-fold (Figure 1). The potential targets of the ATF6-regulated miRNAs were determined using TargetScan, miRBase and miRanda; these targets were then compared to the 607 genes previously shown to be regulated by ATF6 in the mouse heart [12]. It was found that 45 genes known to be upregulated by ATF6 were predicted targets of 8 miRNAs that were downregulated by ATF6 (Table 1, entries 1-45), consistent with possible roles for these miRNAs as modulators of ER stress response gene expression in the heart. Additionally, 9 genes known to be downregulated by ATF6 were predicted targets of 5 miRNAs that were upregulated by ATF6 (Table 1, entries 46-54), consistent with the possibility that these miRNAs might increase ER stress response gene expression in the heart.
Fig. 1. ATF6 miRNA Microarray.
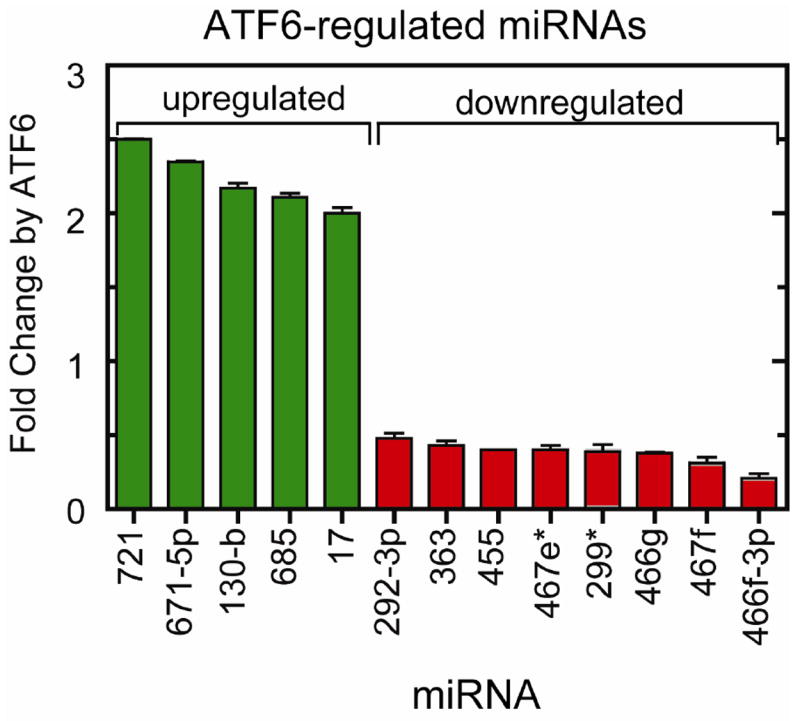
The 13 miRNAs found by miRNA microarray that were differentially expressed upon ATF6 activation in the heart are plotted as a function of the change in expression level. Those miRNAs that were upregulated are shown in green, and miRNAs that were downregulated are shown in red. The mean fold change in expression levels are shown (n = 3 mouse hearts per treatment group).
Table 1. ATF6-regulated Genes that are Putative Targets of ATF6-regulated miRNAs.
The ATF6-regulated mRNAs from a previous study [12] that are putative targets of the ATF6-regulated miRNAs from Figure 1A of this study were identified as described in the Methods, and are listed as genes 1-54 (Column 1), gene symbol (Column 2) and common name (Column 3). Also shown is the fold-change of each mRNA in response to ATF6 activation from the previous study (Column 4), as well as the name of the ATF6-regulated miRNA predicted to target the ATF6-regulated mRNA of interest (Column 5), and whether the miRNA was down-regulated (Column 6, red) or up-regulated (Column 6, green) by ATF6. ATF6-inducible mRNAs that are predicted targets of miR-455 are highlighted in yellow, and the names of known ER stress response genes are labeled in red in Column 2.
| 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|
| Number | ATF6-regulated mRNAs | ATF6-regulated mRNA Gene Names | mRNA Array Fold | ATF6- regulated miRNAs | miRNA Array Fold |
| 1 | SYCP2 | Synaptonemal complex protein 2 | 21.1 | miR-467f | 0.31 |
| SYCP2 | Synaptonemal complex protein 2 | 21.1 | miR-466f- 3p | 0.21 | |
| 2 | APPL1 | DCC-interacting protein 13-alpha | 9.3 | miR-467f | 0.31 |
| 3 | RTN4 | Reticulon-4 | 9.3 | miR-455 | 0.40 |
| 4 | MLLT3 | Myeloid/lymphoid or mixed-lineage leukemia translocated to chromosome 3 protein homolog | 8.7 | miR-466f-3p | 0.21 |
| 5 | THBS1 | Thrombospondin-1 Precursor | 7.2 | miR-202-3p | 0.48 |
| 6 | MXD1 | MAD protein (MAX dimerizer) | 6.5 | miR-202-3p | 0.48 |
| 7 | CHKA | Choline kinase alpha | 5.5 | miR-466f-3p | 0.21 |
| 8 | EDEM1 | ER degradation-enhancing alpha-mannosidase-like 1 | 5.4 | miR-466g | 0.38 |
| 9 | CALR | Calreticulin | 5.3 | miR-455 | 0.40 |
| 10 | MORF4L2 | Mortality factor 4-like protein 2 | 5.1 | miR-466g | 0.38 |
| 11 | XPO1 | Exportin-1 | 5.1 | miR-467f | 0.31 |
| 12 | UBFD1 | Ubiquitin domain-containing protein UBFD1 | 5.0 | miR-202-3p | 0.48 |
| UBFD1 | Ubiquitin domain-containing protein UBFD1 | 5.0 | miR-455 | 0.40 | |
| UBFD1 | Ubiquitin domain-containing protein UBFD1 | 5.0 | miR-299 | 0.39 | |
| 13 | RRAS2 | Ras-related protein R-Ras2 Precursor | 4.9 | miR-466f- 3p | 0.21 |
| RRAS2 | Ras-related protein R-Ras2 Precursor | 4.9 | miR-467f | 0.31 | |
| 14 | STRBP | Spermatid perinuclear RNA-binding protein | 4.9 | miR-202-3p | 0.48 |
| 15 | TMEM158 | Transmembrane protein 158 Precursor | 4.8 | miR-466f-3p | 0.21 |
| 16 | COL3A1 | Collagen alpha-1(III) chain Precursor | 4.6 | miR-202-3p | 0.48 |
| 17 | MAP3K3 | Mitogen-activated protein kinase kinase kinase 3 | 4.3 | miR-202-3p | 0.48 |
| 18 | SNAP23 | Synaptosomal-associated protein 23 | 4.2 | miR-202-3p | 0.48 |
| 19 | SENP2 | Sentrin-specific protease 2 | 4.2 | miR-202-3p | 0.48 |
| 20 | UGCGL1 | UDP-glucose:glycoprotein glucosyltransferase 1 Precursor | 3.7 | miR-202-3p | 0.48 |
| 21 | HN1 | Hematological and neurological expressed 1 protein | 3.5 | miR-455 | 0.40 |
| 22 | UCHL1 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 | 3.4 | miR-466g | 0.38 |
| 23 | WDR68 | WD repeat-containing protein 68 | 3.3 | miR-467f | 0.31 |
| 24 | EIF2S2 | Eukaryotic translation initiation factor 2 subunit 2 | 3.0 | miR-466g | 0.38 |
| 25 | EFNB2 | Ephrin-B2 Precursor | 3.0 | miR-467f | 0.31 |
| 26 | ENAH | Protein enabled homolog | 2.7 | miR-467e | 0.40 |
| 27 | JUNB | Transcription factor jun-B | 2.6 | miR-466f-3p | 0.21 |
| 28 | PJA2 | E3 ubiquitin-protein ligase Praja2 | 2.6 | miR-466f-3p | 0.21 |
| 29 | BMPR2 | Bone morphogenetic protein receptor type-2 Precursor | 2.6 | miR-467f | 0.31 |
| 30 | PJA2 | E3 ubiquitin-protein ligase Praja2 | 2.6 | miR-467f | 0.31 |
| 31 | SLC39A14 | Zinc transporter ZIP14 Precurso | 2.5 | miR-466g | 0.38 |
| 32 | INCENP | Inner centromere protein | 2.4 | miR-466g | 0.38 |
| 33 | SLC2A1 | Solute carrier family 2, facilitated glucose transporter member 1 | 2.4 | miR-466f-3p | 0.21 |
| SLC2A1 | Solute carrier family 2, facilitated glucose transporter member 1 | 2.4 | miR-467f | 0.31 | |
| 34 | KLHL2 | Kelch-like protein 2 | 2.4 | miR-466f-3p | 0.21 |
| KLHL2 | Kelch-like protein 2 | 2.4 | miR-467f | 0.31 | |
| 35 | ING1 | Inhibitor of growth protein 1 | 2.4 | miR-467f | 0.31 |
| 36 | ARL4C | ADP-ribosylation factor-like protein 4C | 2.3 | miR-466g | 0.38 |
| 37 | LRRC59 | Leucine-rich repeat-containing protein 59 | 2.2 | miR-202-3p | 0.48 |
| 38 | SLC6A6 | Sodium- and chloride-dependent taurine and beta-alanine transporter | 2.2 | miR-466f-3p | 0.21 |
| 39 | CUGBP2 | CUG-BP- and ETR-3-like factor 2 | 2.2 | miR-466g | 0.38 |
| 39 | CUGBP2 | CUG-BP- and ETR-3-like factor 2 | 2.2 | miR-467f | 0.31 |
| 40 | CALM1 | Calmodulin | 2.2 | miR-202-3p | 0.48 |
| 41 | CDS2 | Phosphatidate cytidylyltransferase 2 | 2.1 | miR-466f-3p | 0.21 |
| 42 | PPAP2A | Lipid phosphate phosphohydrolase 1 | 2.1 | miR-202-3p | 0.48 |
| 43 | FSTL1 | Follistatin-related protein 1 Precursor | 2.1 | miR-363 | 0.43 |
| FSTL1 | Follistatin-related protein 1 Precursor | 2.1 | miR-466g | 0.38 | |
| 44 | WDR1 | WD repeat-containing protein 1 | 2.0 | miR-467e | 0.40 |
| 45 | SMAD2 | Mothers against decapentaplegic homolog 2 | 2.0 | miR-455 | 0.40 |
| 46 | A2BP1 | Ataxin 2 binding protein 1 | 0.3 | miR-17 | 2.00 |
| 47 | SLC40A1 | Solute carrier family 40 (iron-regulated transporter), member 1 | 0.4 | miR-17 | 2.00 |
| 48 | HLF | Hepatic leukemia factor | 0.4 | miR-17 | 2.00 |
| 49 | CLIP4 | CAP-GLY domain containing linker protein family, member 4 | 0.5 | miR-17 | 2.00 |
| 50 | HLF | Hepatic leukemia factor | 0.4 | miR-721 | 2.50 |
| HLF | Hepatic leukemia factor | 0.4 | miR-130b | 2.17 | |
| 51 | ACSL1 | Long-chain-fatty-acid--CoA ligase 1 | 0.4 | miR-721 | 2.50 |
| ACSL1 | Long-chain-fatty-acid--CoA ligase 1 | 0.4 | miR-130b | 2.17 | |
| 52 | UCP3 | Mitochondrial uncoupling protein 3 | 0.3 | miR-721 | 2.50 |
| UCP3 | Mitochondrial uncoupling protein 3 | 0.3 | miR-130b | 2.17 | |
| 53 | TBL1XR1 | F-box-like/WD repeat-containing protein TBL1XR1 | 0.2 | miR-721 | 2.50 |
| TBL1XR1 | F-box-like/WD repeat-containing protein TBL1XR1 | 0.2 | miR-130b | 2.17 | |
| 54 | FNDC5 | Fibronectin type III domain-containing protein 5 Precursor | 0.2 | miR-671-5 | 2.35 |
ATF6 Upregulates Calr mRNA and Protein, In Vivo
One miRNA that was down-regulated by ATF6 was miR-455, which is predicted to target several ATF6-inducible mRNAs, including calreticulin (Calr) (Table 1, entry 9). Calr is localized to the ER/SR, where it functions as a major calcium-binding protein and a chaperone, that contributes to the folding of nascent proteins in this organelle [24]. Calr is also required for cardiac development [25], and is induced in animal models of hypertrophic heart disease [26-28]. Accordingly, we examined roles for miR-455 in the regulation of Calr expression.
To examine Calr expression in ATF6-MER TG mouse hearts, in vivo, the effects of tamoxifen on Calr mRNA in ATF6-MER TG and NTG mouse hearts were determined by RT-qPCR. When ATF6-MER TG and NTG mice were treated with tamoxifen, compared to vehicle-treated mice, Calr mRNA was upregulated by ~ 4.5-fold, but only in hearts from TG mice treated with tamoxifen (Fig. 2A). This change in the Calr mRNA was also reflected by coordinate increases in Calr protein levels of about 14-fold; once again, this occurred only in the TG mice treated with tamoxifen (Fig. 2 B and 2C). In a previous study, we showed Calr expression in ATF6-MER TG mice, as determined by PCR, was increased, but in that study, it did not reach statistical significance [11].
Fig 2. Effects of ATF6 on Calr Expression in Mouse Hearts.

Panel A. Non-transgenic (NTG) and (TG) mice were treated ± vehicle (Veh) or tamoxifen (Tam). Mouse heart RNA was subjected to RT-qPCR to measure Calr and GAPDH mRNA. The mean Calr/GAPDH mRNA ± S.E. is shown (n = 3 mouse hearts per treatment). *, p ≤ 0.05 different from any other values.
Panel B. Extracts prepared from NTG and TG mouse hearts, treated as described in Panel A, were subjected to SDS-PAGE, followed by immunoblotting for Calr and GAPDH.
Panel C. The immunoblots shown in Panel B were quantified by densitometry. Shown is the mean ± S.E. (n = 2 mice per treatment). **, p ≤ 0.01 different from any other values.
Effects of ER Stress and ATF6 on miR-455 and Calr Expression
To examine the effects of the prototypical ER stressor, tunicamycin (TM), as well as ATF6 on miR-455 and Calr expression, neonatal rat ventricular myocyte cultures (NRVMCs) were either treated for 20h with TM, or they were infected with a recombinant adenovirus that encodes activated ATF6, ATF6(1-373). TM and ATF6 decreased miR-455 levels to 0.25- and 0.5-fold of control, respectively, (Fig. 3A), while they increased Calr mRNA by about 20- and 40-fold, respectively (Fig. 3B). The increase in the Calr mRNA was associated with coordinate TM- and ATF6-mediated increases in Calr protein of about 2- and 4-fold, respectively (Fig. 4A and 4B). TM and ATF6 each had the expected effects on the induction of two well-known markers of ER stress, GRP94 and GRP78 (Fig. 4A, 4C, and 4D). Thus, either TM or ATF6 decreased miR-455 levels and increased Calr mRNA and protein levels.
Fig. 3. Effects of Tunicamycin and ATF6 on miR-455 and Calr mRNA Levels in Cultured Cardiac Myocytes.

Panel A. Neonatal rat ventricular myocyte cultures (NRVMCs) were infected with control adenovirus (Con) or a recombinant adenovirus encoding activated ATF6(1-373) (ATF6) (n = 3 cultures per treatment). One of the control adenovirus-infected sets of cultures was treated with tunicamycin (TM) at 10 μg/ml for 20h. Cultures were then extracted and total RNA was subjected to RT-qPCR for miR-455, and U6 RNA. The mean miR-455/U6 RNA ± S.E. is shown (n = 3 cultures per treatment). *, p < 0.05 different from Con.
Panel B. NRVMCs, treated with TM or ATF6, as described in Panel A, were extracted and total RNA was subjected to RT-qPCR for Calr mRNA and GAPDH mRNA. The mean Calr/GAPDH mRNA ± S.E. is shown (n = 3 cultures per treatment). *, p < 0.05 different from Con.
Fig. 4. Effects of Tunicamycin and ATF6 on Calr, GRP94 and GRP78 Protein Levels in Cultured Cardiac Myocytes.

Panel A. NRVMCs, treated as described in Fig. 3A, were extracted and subjected to SDS-PAGE, followed by immunoblotting for GRP94 and GRP78, using a KDEL antibody, and Calr and GAPDH (n = 3 cultures per treatment).
Panel B-C. The immunoblots shown in Fig. 4A were quantified by densitometry. Shown is the mean ± S.E. (n = 3 cultures per treatment). *, p ≤ 0.05 different from Con.
Effects of Pre-miR-455 and Anti-miR-455 on Calr Expression
To examine the direct effects of miR-455 on Calr, we overexpressed either a miR-455 precursor (pre-miR-455), or an antisense to miR-455 (anti-miR-455). Compared to the pre-miR control, pre-miR-455 decreased Calr levels by about 50% (Fig. 5A and 5B), whereas, compared to anti-miR control, anti-miR-455 increased Calr levels by about 50% (Fig. 5C and D). These results are consistent with the hypothesis that by targeting the Calr transcript, miR-455 decreases Calr levels in cardiac myocytes.
Fig. 5. Effects of Pre-miR-455 and Anti-miR-455 on Calr Levels in Cultured Cardiac Myocytes.

NRVMCs were transfected with a non-targeted control precursor miRNA (Con), a precursor miRNAs predicted to generate mature miR-455 (Pre-miR-455) (Panel A), or an anti-miR-455 mRNA (Anti-miR-455) (Panel C). After 48h, Calr levels were determined by immunoblotting. Shown are the mean Calr/GAPDH levels ± S.E. (n = 3 cultures per treatment) in cultures treated with pre-miR-455 (Panel B), or with anti-miR-455 (Panel D). *, p < 0.05 different from Con.
DISCUSSION
This study shows that the ATF6 branch of the ER stress response may regulate gene expression of critical cardiac genes, such as Calr, through a mechanism that involves miRNAs.
All of the miRNAs identified in this study are intergenic, i.e. derived from their own transcriptional units in the intergenic regions of the genome [14]. This contrasts with some other miRNAs that are encoded within introns or exons of host genes with which they are cotranscribed and coexpressed [29]. Accordingly, it is most likely that ATF6 affects the levels of the miRNAs identified in this study by regulating the transcription of genes that encode those miRNAs. In this regard, a search revealed that the putative regulatory regions of some of the ATF6-upregulated miRNAs have consensus ERSEs, to which ATF6 could potentially bind and induce premiRNA gene transcription. However, no ERSEs were found in the ATF6-downregulated miRNA genes, suggesting that either ATF6 binds to non-canonical ERSEs in those genes, and in so doing, represses transcription, or that the ability of ATF6 to decrease expression of miRNAs is indirect. To the best of our knowledge, there have been no reports demonstrating ATF6 binding to non-canonical ERSEs and repressing transcription. However, there have been a few reports of indirectly ATF6 repressing gene expression. In one case, ATF6 was shown to bind to, and block the transcriptional activity of sterol regulatory element-binding protein (SREBP2) [30]. In another case, it was shown that ATF6 bound to, and blocked the ability of CREB to induce several of its target genes [31]. Therefore, ATF6 may decrease miRNA expression by indirect transcriptional repression.
Of the 13 miRNAs regulated by ATF6, we focused on miR-455 and one of its targets, Calr mRNA. Calr is a 46 kDa, ER-lumenal Ca2+-binding protein and molecular chaperone [24], which is required for proper heart development [25], and is protective when induced during cardiac pathology [28]. The results of the present study suggest that by downregulating miR-455, ATF6 contributes to the upregulation of Calr during ER stress. Very few studies have been published on miR-455; in fact, a PubMed search coupled with a search of PhenomiR [32], a knowledgebase for microRNA expression and function, revealed only 3 research papers on miR-455, none of which related to ER stress or the heart. In all 3 papers, miR-455 was upregulated in response to the maneuvers employed, but in none of the cases was it shown that a specific target of miR-455 was altered in response to that upregulation [33-35]. Accordingly, to the best of our knowledge, this is the first study to demonstrate downregulation of miR-455, and to demonstrate that the mRNA and protein levels of a predicted target of miR-455, Calr, were increased, and that this increase was partly due to miR-455 downregulation.
In addition to Calr, there are several other putative targets of miR-455 in the list of ATF6-regulated genes in the heart. For example, reticulon-4 (RTN4) (Table 1, entry 3), which has not been studied in the heart, has been shown to protect cultured cells from ER stress-induced apoptosis [36], and protects mice from neurodegeneration in models of protein unfolding diseases, such as amyotrophic lateral sclerosis [37]. Another putative target of miR-455 is mothers against decapentaplegic homolog 2 (SMAD2) (Table 1, entry 45), which has been studied in the heart, where it has been shown to increase the secretion of the TGF-β family member, GDF15, which is antihypertrophic [28, 38]. The effects of these genes are consistent with a previous study of ours demonstrating that ATF6 is protective in the heart [11], and antihypertrophic in cultured cardiac myocytes [12].
All together, the ATF6-regulated miRNAs described in this study were predicted to target numerous transcripts, including 6 encoded by known ER stress response genes (Table 1, entries 3, 5, 8, 9, 40 and 43). It is worth noting that all of the ER stress response gene mRNAs listed in Table 1 are upregulated by ATF6, and that they are putative targets of miRNAs that are downregulated by ATF6, consistent with roles for all of these miRNAs in decreasing ER stress response gene product levels.
CONCLUSIONS
By regulating a unique gene program, the ATF6 branch of the ER stress response protects the heart from ischemic damage. This study showed that in addition to its ability to regulate the transcription of potentially protective genes, ATF6 also regulates miRNA expression in the mouse heart. Moreover, some of the ATF6-regulated miRNAs identified in this study exerted effects on gene expression that were consistent with the previously characterized effects of ATF6 on specific mRNAs in the heart. One of the miRNAs that was downregulated by ATF6 was miR-455, which was shown to augment expression of the cardioprotective gene, Calr, which may contribute to the protective effects of ATF6 in the heart.
Highlights.
The ER stress-activated transcription factor, ATF6, is cardioprotective.
ATF6 increases expression of calreticulin in the heart.
We found 13 miRNAs to be regulated by ATF6 in the heart, in vivo.
MicroR-455, which targets the calreticulin mRNA, was downregulated by ATF6.
Thus, ATF6 may induce calreticulin, partly, by decreasing miR-455.
Acknowledgments
This work was supported by grants from the National Institutes of Health, HL-075573, HL-085577, HL104535 and RO3 EB011698. PJB was a Fellow of the Rees-Stealy Research Foundation and the San Diego State University Heart Institute, a scholar of the San Diego Chapter of the Achievement Rewards for College Scientists (ARCS) Foundation and a recipient of an American Heart Association States Affiliate Pre-doctoral Fellowship, Award # 0815210F.
ABBREVIATIONS
- ATF6
activation of transcription factor 6
- Calr
calreticulin
- miRNA
microRNA
- UPR
unfolded protein response
- MER
mutant mouse estrogen receptor
- MER-ATF6
fusion protein with the MER fused to the C-terminal of ATF6
- TG
transgenic
- NTG
non-transgenic
- NRVMC
neonatal rat ventricular myocyte cultures
- qRT-PCR
quantitative real time PCR
- ERSE
ER stress response element
Footnotes
DISCLOSURES
None declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Glembotski CC. Endoplasmic reticulum stress in the heart. Circ Res. 2007;101:975–84. doi: 10.1161/CIRCRESAHA.107.161273. [DOI] [PubMed] [Google Scholar]
- 2.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–33. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 3.Austin RC. The unfolded protein response in health and disease. Antioxid Redox Signal. 2009;11:2279–87. doi: 10.1089/ars.2009.2686. [DOI] [PubMed] [Google Scholar]
- 4.Glembotski CC. The role of the unfolded protein response in the heart. J Mol Cell Cardiol. 2008;44:453–9. doi: 10.1016/j.yjmcc.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–30. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 6.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–64. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–9. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 9.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–82. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 10.Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J Biol Chem. 2009;284:29735–45. doi: 10.1074/jbc.M109.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martindale JJ, Fernandez R, Thuerauf D, Whittaker R, Gude N, Sussman MA, et al. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ Res. 2006;98:1186–93. doi: 10.1161/01.RES.0000220643.65941.8d. [DOI] [PubMed] [Google Scholar]
- 12.Belmont PJ, Tadimalla A, Chen WJ, Martindale JJ, Thuerauf DJ, Marcinko M, et al. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J Biol Chem. 2008;283:14012–21. doi: 10.1074/jbc.M709776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Rooij E. The art of microRNA research. Circ Res. 2011;108:219–34. doi: 10.1161/CIRCRESAHA.110.227496. [DOI] [PubMed] [Google Scholar]
- 14.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 15.van Rooij E, Marshall WS, Olson EN. Toward microRNA-based therapeutics for heart disease: the sense in antisense. Circ Res. 2008;103:919–28. doi: 10.1161/CIRCRESAHA.108.183426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation. 2010;121:1022–32. doi: 10.1161/CIRCULATIONAHA.109.889048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–4. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 20.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010;11:R90. doi: 10.1186/gb-2010-11-8-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simpson P, Savion S. Differentiation of rat myocytes in single cell cultures with and without proliferating nonmyocardial cells. Cross-striations, ultrastructure, and chronotropic response to isoproterenol. Circ Res. 1982;50:101–16. doi: 10.1161/01.res.50.1.101. [DOI] [PubMed] [Google Scholar]
- 22.Sei CA, Irons CE, Sprenkle AB, McDonough PM, Brown JH, Glembotski CC. The alpha-adrenergic stimulation of atrial natriuretic factor expression in cardiac myocytes requires calcium influx, protein kinase C, and calmodulin-regulated pathways. J Biol Chem. 1991;266:15910–6. [PubMed] [Google Scholar]
- 23.Thuerauf DJ, Morrison LE, Hoover H, Glembotski CC. Coordination of ATF6-mediated transcription and ATF6 degradation by a domain that is shared with the viral transcription factor, VP16. J Biol Chem. 2002;277:20734–9. doi: 10.1074/jbc.M201749200. [DOI] [PubMed] [Google Scholar]
- 24.Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J. 2009;417:651–66. doi: 10.1042/BJ20081847. [DOI] [PubMed] [Google Scholar]
- 25.Mesaeli N, Nakamura K, Zvaritch E, Dickie P, Dziak E, Krause KH, et al. Calreticulin is essential for cardiac development. J Cell Biol. 1999;144:857–68. doi: 10.1083/jcb.144.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sato Y, Ferguson DG, Sako H, Dorn GW, 2nd, Kadambi VJ, Yatani A, et al. Cardiac-specific overexpression of mouse cardiac calsequestrin is associated with depressed cardiovascular function and hypertrophy in transgenic mice. J Biol Chem. 1998;273:28470–7. doi: 10.1074/jbc.273.43.28470. [DOI] [PubMed] [Google Scholar]
- 27.Tsutsui H, Ishibashi Y, Imanaka-Yoshida K, Yamamoto S, Yoshida T, Sugimachi M, et al. Alterations in sarcoplasmic reticulum calcium-storing proteins in pressure-overload cardiac hypertrophy. Am J Physiol. 1997;272:H168–75. doi: 10.1152/ajpheart.1997.272.1.H168. [DOI] [PubMed] [Google Scholar]
- 28.Papp S, Dziak E, Kabir G, Backx P, Clement S, Opas M. Evidence for calreticulin attenuation of cardiac hypertrophy induced by pressure overload and soluble agonists. Am J Pathol. 2010;176:1113–21. doi: 10.2353/ajpath.2010.090392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu N, Olson EN. MicroRNA regulatory networks in cardiovascular development. Dev Cell. 2010;18:510–25. doi: 10.1016/j.devcel.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng L, Lu M, Mori K, Luo S, Lee AS, Zhu Y, et al. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004;23:950–8. doi: 10.1038/sj.emboj.7600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seo HY, Kim MK, Min AK, Kim HS, Ryu SY, Kim NK, et al. Endoplasmic reticulum stress-induced activation of activating transcription factor 6 decreases cAMP-stimulated hepatic gluconeogenesis via inhibition of CREB. Endocrinology. 2010;151:561–8. doi: 10.1210/en.2009-0641. [DOI] [PubMed] [Google Scholar]
- 32.Ruepp A, Kowarsch A, Schmidl D, Buggenthin F, Brauner B, Dunger I, et al. PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010;11:R6. doi: 10.1186/gb-2010-11-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ujifuku K, Mitsutake N, Takakura S, Matsuse M, Saenko V, Suzuki K, et al. miR-195, miR-455-3p and miR-10a(*) are implicated in acquired temozolomide resistance in glioblastoma multiforme cells. Cancer Lett. 2010;296:241–8. doi: 10.1016/j.canlet.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 34.Walden TB, Timmons JA, Keller P, Nedergaard J, Cannon B. Distinct expression of muscle-specific microRNAs (myomirs) in brown adipocytes. J Cell Physiol. 2009;218:444–9. doi: 10.1002/jcp.21621. [DOI] [PubMed] [Google Scholar]
- 35.Panguluri SK, Bhatnagar S, Kumar A, McCarthy JJ, Srivastava AK, Cooper NG, et al. Genomic profiling of messenger RNAs and microRNAs reveals potential mechanisms of TWEAK-induced skeletal muscle wasting in mice. PLoS One. 2010;5:e8760. doi: 10.1371/journal.pone.0008760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuang E, Wan Q, Li X, Xu H, Zou T, Qi Y. ER stress triggers apoptosis induced by Nogo-B/ASY overexpression. Exp Cell Res. 2006;312:1983–8. doi: 10.1016/j.yexcr.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 37.Yang YS, Harel NY, Strittmatter SM. Reticulon-4A (Nogo-A) redistributes protein disulfide isomerase to protect mice from SOD1-dependent amyotrophic lateral sclerosis. J Neurosci. 2009;29:13850–9. doi: 10.1523/JNEUROSCI.2312-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, et al. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res. 2006;98:342–50. doi: 10.1161/01.RES.0000202804.84885.d0. [DOI] [PubMed] [Google Scholar]