

Abstract
The c.4309A>C mutation in the LRRK2 gene (LRRK2 p.N1437H) has recently been reported as the seventh pathogenic LRRK2 mutation causing monogenic Parkinson's disease (PD). So far, only two families worldwide have been identified with this mutation. By screening DNA from seven brains of PD patients, we found one individual with seemingly sporadic PD and LRRK2 p.N1437H mutation. Clinically, the patient had levodopa-responsive PD with tremor, and developed severe motor fluctuations during a disease duration of 19 years. There was severe and painful ON-dystonia, and severe depression with suicidal thoughts during OFF. In the advanced stage, cognition was slow during motor OFF, but there was no noticeable cognitive decline. There were no signs of autonomic nervous system dysfunction. Bilateral deep brain stimulation of the subthalamic nucleus had unsatisfactory results on motor symptoms. The patient committed suicide. Neuropathological examination revealed marked cell loss and moderate alpha-synuclein positive Lewy body pathology in the brainstem. There was sparse Lewy pathology in the cortex. A striking finding was very pronounced ubiquitin-positive pathology in the brainstem, temporolimbic regions and neocortex. Ubiquitin positivity was most pronounced in the white matter, and was out of proportion to the comparatively weaker alpha-synuclein immunoreactivity. Immunostaining for tau was mildly positive, revealing non-specific changes, but staining for TDP-43 and FUS was entirely negative. The distribution and shape of ubiquitin positive lesions in this patient differed from the few previously described patients with LRRK2 mutations and ubiquitin pathology, and the ubiquitinated protein substrate remains undefined.
Keywords: Autosomal Dominant Parkinsonism, LRRK2, alpha-Synuclein, Ubiquitin, Deep Brain Stimulation, Suicide
Introduction
Mutations in the leucine-rich repeat kinase 2 gene (LRRK2) are associated with Parkinson s disease (PD). Seven mutations (p.N1437H, p.R1441C/G/H, p.Y1699C, p.G2019S, and p.I2020T) are considered pathogenic with an autosomal dominant mode of inheritance [1–4]. The p.N1437H mutation has so far only been reported from two families in Norway [5,6]. The neuropathology of patients with pathogenic mutations in LRRK2 has shown a puzzling heterogeneity [3]. Most patients with LRRK2 mutations had typical PD-type Lewy body disease (LBD), but pure nigral degeneration without LBD, TAR DNA-binding protein of 43 kD (TDP-43)-positive inclusions and tau-, or ubiquitin-positive pathologies have been reported [3]. Remarkably, different types of pathology have been found in carriers of the same mutation, even within the same family [2,3,7]. Here, we describe the clinical course and provide the first neuropathology report of a newly identified Swedish PD patient with LRRK2 p.N1437H mutation.
Methods and Materials
Genetic studies
Seven brains from patients with a clinical diagnosis of PD from the collection of the Department for Neuropathology, Lund University Hospital, Sweden, were included in this study. Brain tissue was available as formalin-fixed, paraffin-embedded blocks (5 patients) and formalin-fixed sections (3 specimens from 2 patients). The initial aim was to develop a protocol for DNA extraction and genetic analysis from brains in our collection. Small samples were obtained from the tissues under sterile conditions. Genomic DNA was extracted with the QIAamp DNA FFPE Tissue Kit (QIAGEN, Hilden, Germany) according to the manufacturer s instructions, with the modifications noted below. The xylene addition and ethanol washout steps were repeated twice to optimize paraffin removal. All samples were then incubated in a buffer with proteinase K at 56°C overnight. As tissue lysis remained incomplete the following morning, additional proteinase K was added and incubated for another hour at 56°C, followed by one hour at 90°C. DNA concentration in the final elutions was between 11 and 138ng/μl. Primers were designed for sequencing of exons 2 and 3 in SNCA and exons 30, 31, 34, 35, 41, and 48 in LRRK2, which contain all known pathogenic mutations [3]. Primer sequences are available on request. PCR products were generated and purified using standard protocols. Sequencing reactions were performed in 10μl volumes containing 5μl of clean PCR product, 1:32 dilution of BigDye Terminator and 3.2 pmol of primer. After the removal of dye-terminator using the Biomek FX robot and magnetic-bead technology (CleanSEQ beads, Agencourt), sequencing products were electrophorized on an ABI 3730 capillary array and the data analyzed with SeqScape software (ABI).
Clinical studies
Data from one patient who was found to carry a LRRK2 p.N1437H mutation was compiled. Standardized motor and cognitive tests were performed. Videos were recorded at several instances with the patient s consent (including the consent to disclose). Two relatives of the patient were interviewed by telephone. This study was approved by the responsible Ethical Review Boards.
Neuropathological studies
The patient was autopsied by an expert in forensic medicine, which is the routine and in accordance with Swedish law when suicide is the suspected cause of death. Thereafter, the brain was fixed in formaldehyde and a macroscopic neuropathological examination was performed. This was done for academic reasons to assess whether electrodes for deep brain stimulation (DBS) were appropriately placed. Neuropathological studies were performed in coronal bihemispheric sections from the frontal, frontotemporal and parieto-occipital lobes, as well as transversal sections through the brain stem and cerebellum. Furthermore, special smaller samples were taken for further characterization of neocortex, hippocampus and mesencephalon. Paraffin-embedded sections were stained with hematoxylin and eosin (H&E), Luxol fast blue (LFB), and Gallyas silver. Immunohistochemistry for ubiqutin (anti-ubiquitin, DAKO), tau (AT8 antibody, DAKO), alpha-synuclein (synuc, Zymed Laboratories), TDP-43 (Protein Tech, Cosmobio) and fused in sarcoma FUS (anti-FUS, Sigma-Aldrich) was performed on 5μm thick tissue samples. For alpha-synuclein immunohistochemistry, all sections were microwave pre-treated in 10 mM citrate buffer at pH 6.0 and 100 degrees C for 10 minutes, in order to achieve antigen retrieval. An automated immunostainer (Dako Autostainer Plus, DAKO Sweden AB) was used for the staining procedure using DAKO ChemMate Kit Peroxidase/3,3′-diaminobenzidine.The primary antibody against alpha-synuclein was diluted 1:200. Additional details of the staining protocols are available on request.
Results
Genetic studies
Sequence analysis of DNA derived from the brain of one patient showed a heterozygous mutation c.4309A>C in exon 30 of the LRRK2 gene, indicating a p.N1437H mutation (Figure 1). Identical results were obtained in two DNA samples prepared independently from this patient. No other mutations were detected in the SNCA or LRRK2 exons examined in DNA from this patient or the other patients examined.
Figure 1. Sequencing results of LRRK2 exon 30.
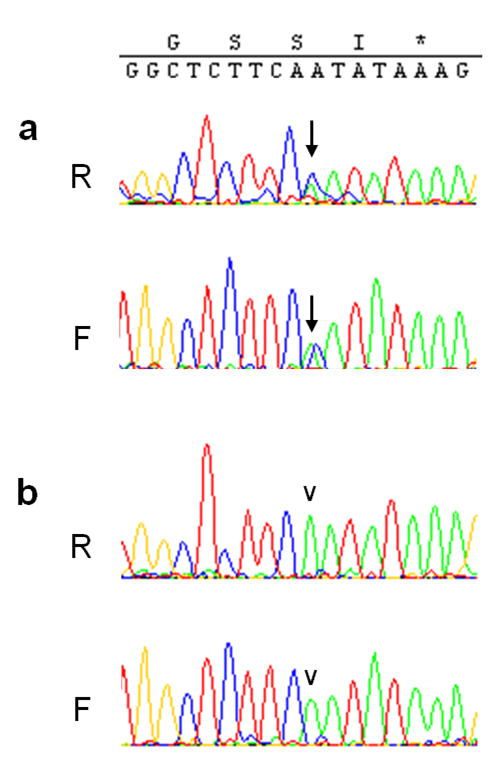
Heterozygosity for c.4309A>C (a, arrow) indicating a p.N1437H substitution, is seen in the forward (F) and reverse (R) sequencing reaction in DNA from the patient described here, but not in another patient (b, arrowhead).
Clinical studies
Ample clinical documentation was available on this patient, who was followed at Lund University Hospital during 17 years.
The patient's father died at age 70 years after hospitalization due to stroke. According to family history, he did not show signs of PD. The patient's mother, three siblings and one half-sibling lived to 80 to 87 years of age and did not have PD symptoms. The patient, who had remained childless, had denied any family history of PD. We found no genealogical link to the previously published Norwegian families with p.N1437H mutation (Dr. Jan Aasly, Trondheim, and Dr. Mathias Toft, Oslo, Norway, personal communication).
Like one of her sisters, the patient had decreased vision and nystagmus from birth. At 50 years of age, she noted a slowness of movements in her left arm, and started dragging her left leg. A tendency to dystonic cramps in the axial musculature developed. A year later, tremor at rest occurred in her left side. When first seen at our department at age 52, the patient kept her left arm flexed while walking. Levodopa (400mg daily) markedly improved the patient's motor symptoms and fatigue. Motor wearing-off occurred three years later, and dyskinesias were noted at the age of 57. When observed as an inpatient at age 59 years, reversible episodes of depressive moods were noted during motor OFF. Addition of selegiline (10mg/d) and subsequently amantadine hydrochloride (200mg/d) did not lead to any noticeable improvement. From age 61, the patient developed involuntary grimacing and fast movements of the upper extremities that occurred both during motor ON and OFF.
At 64 years of age, the effect of 100mg levodopa only lasted for 30–60 minutes and was inconstant and variable. At age 66, changes from motor ON to OFF appeared quickly and suddenly. The patient attempted suicide with nitrazepam tablets but was rescued. At age 69 years, a levodopa test revealed severe fatigue, depression and marked hypokinesia, tremor and rigidity after nine hours without medication. In this state, the patient was bound to a wheelchair, which she was unable to move without help, and had severe difficulties organizing and processing tasks (Hoehn & Yahr stage V). After administration of levodopa, the patient could walk up stairs supported by one person (Hoehn & Yahr stage IV). However, during motor ON, there were both hyperkinesias and painful dystonias, partly with extreme and bizarre posturing (Online Resource 1). The dystonic cramps sometimes lasted for up to one hour at a time. Bilateral DBS of the subthalamic nucleus (STN) at age 69 initially may have improved the patient's tremor and rigidity slightly, but after a few weeks, her motor functions deteriorated. During motor OFF, the patient had marked hypokinesia with freezing, a UPDRS score of 65, and severely depressed mood. Six months later the patient committed suicide by intoxication and suffocation.
Once, slight word finding difficulties were noted but were attributed to depression and the patient self-reported intermittent difficulty concentrating. Otherwise, the patient s cognition did not decline objectively. She reported vivid dreaming, but no hallucinations or nightmares when treated with between 400 and 550mg levodopa tablets daily plus 20 mg bromocriptine tablets / day (levodopa equivalent daily dose 600–750mg). Repeated assessment revealed no orthostatic hypotension, and there were no other signs of autonomous nervous system dysfunction. There was no dysphagia. Magnetic resonance imaging of the patient's brain at age 68 years revealed bilateral periventricular white matter changes but no focal changes and no atrophy (Figure 2).
Figure 2. Brain MRI imaging.

T2-weighted imaging at 68 years of age revealed marked subcortical hyperintensities bilaterally. These spared the U-fibres and were unusually widespread, taking into consideration that the patient did not have diabetes, hypertension, or any other risk factor for small vessel disease. Top row, transverse sections, bottom row, coronal sections. The subthalamic nucleus (arrow in inset) was hyperintense and distinguishable with unusual contrast from the surrounding white matter. Slice thickness was 1 or 2 mm for the slices shown.
Neuropathological studies
Macroscopic examination (Figure 3) of the patient s brain revealed severe substantia nigra depigmentation and signs of mild cortical atrophy, in spite of retained normal size. DBS electrode tracts could be followed to the subthalamic nucleus bilaterally. Microscopy of the brain sections (Figure 4 and 5) demonstrated a slightly reduced number of cortical neurons across the hemispheric mantle, but no marked cell loss. There was no discernible reactive gliosis. Conventional histochemical staining with H&E and LFB showed only minimal neurodegenerative pathology with cell atrophy, cell fragmentation with debris, minimal cell loss and mild reactive gliosis in cortical areas. However, these signs of neurodegeneration were pronounced in brainstem nuclei, including the substantia nigra pars compacta (SNpc), to a lesser extent also in the locus coeruleus (LC) and the dorsal motor nuclus of vagus (dmv), as well as, mildly, in the midline raphe nuclei and the striatum.
Figure 3. Macroscopic pathology.

The formalin-fixed brain weighed 1420 grams and the aspect of the whole brain was without atrophy of cerebrum or cerebellum. The basal blood vessels showed only minimal signs of atheromatosis. After sectioning, there was an impression of mild diffuse thinning of the cortical ribbon. The substantia nigra was severely depigmented bilaterally, more severely on the left side. Both DBS electrode tracts could be followed to the subthalamic nucleus. The right DBS electrode tract went through the lateral thalamic nucleus, and in the lateral thalamic segment, there was an infarct of 1.5×1mm. Macroscopic pathology revealed only slight thinning of the cortical ribbon on coronar section. No widening of sulci was noted over the convexities. Substantia nigra was depigmented (not shown). The cortical entrances of the DBS electrodes are visible in the left upper picture.
Figure 4. Brainstem Neuropathology.

a and b) Substantia nigra (SN), H&E: There was almost total loss of melanin-containing neurons of substantia nigra pars compacta (SNpc) bilaterally. Only a few clusters of pigment-containing neurons were seen in the most ventromedial parts of as well as towards its latero-dorsal end. The few remaining SN cells showed atrophy and loss of pigmentation. A few Lewy bodies were seen (LB). c) Locus coeruelus, H&E: Marked cell loss and LB pathology. d) SN, alpha-synuclein: LBs and isolated dot-like structures stained positively for alpha-synuclein. e) SN, ubiquitin: Pronounced ubiquitin pathology with dense spheroids and dot-like structures as well as irregularly shaped foamy inclusions. f) White matter (crus cerebri adjacent to SN), ubiquitin: multitudinous ubiquitin-positive structures in the form of small dots, larger speroids and intermediate cloud-like positivities. Magnification: a) x125; b–c) x400; d–f) x200; insert in e) x400, enlarged.
Figure 5. Cortical Neuropathology.

a–d) Parietal cortex, e–g) Entorhinal cortex. a and e) Alpha-synuclein: very sparse cortical alpha-synuclein pathology with single Lewy bodies (a) or Lewy neurites (e) per field. b and f) Tau: sparse tau-positive lesions of unspecific shape. c,d and g) Ubiquitin Marked ubiquitin positive pathology in the parietal cortex and very numerous lesions in the entorhinal cortex, including dense spheroids, less dense granular lesions with irregular shapes.
h) White matter subjacent to entorhinal cortex, ubiquitin: Very high burden of ubiquitin-positive lesions. Magnification: a–c) and e–h) x200; d) and inserts in g) x400.
There was an almost total loss of melanin-containing neurons of SNpc bilaterally. Only a few clusters of pigment-containing neurons were seen in its most ventromedial and laterodorsal ends. The few remaining cells showed atrophy, there was some loss of pigmentation and a few Lewy bodies were seen (LB). Apart from these isolated clusters, there were no intact melanin-containing neurons in the SNpc. There were dispersed, isolated areas with extraneuronal melanin. Areas with tissue attenuation were seen. Overall, the density of pigmented neurons was lower than in the “Severe PD” stage according to Dickson et al. [8]. There was no marked gliosis in the SNpc. In LC there were marked signs of degeneration with depigmentation of cells and loss of neurons that was severe in the central and caudal portions of the nucleus, reaching a degeneration score of 7 (maximum 9) according to Brunnström et al. [9]. In addition, there were LBs and pale bodies in some of the remaining cells. Most LBs were of classical type, but a few cortical-type LBs were also observed. There was moderate cell loss in the dorsal motor nucleus of vagus. The signs of neurodegeneration in the midline brainstem, including the raphe nuclei, as well as the striatal nuclei, were less pronounced and in less well delineated regions compared to SNpc and LC. Gallyas silver staining showed only very minimal silver positive deposits in the neocortex and basal ganglia.
Immunostaining revealed alpha-synuclein positive LBs, Lewy neurites, dot-like structures and occasional axonal spheroids in the SNpc and LC. There was a moderate load of alpha-synuclein positive lesions in the entorhinal-hippocampal region. In the posterior part of the hippocampus, alpha-synuclein positive structures were confined to scattered long Lewy neurites within the hippocampal CA2 segment. The neocortex also contained very sparse, scattered LBs within the external layers, only single alpha-synuclein-positive lesions were seen per high power field.
Ubiquitin positivity was very pronounced both in white and grey matter in all of the regions examined. The appearance of the ubiquitin-positive lesions was unusual and variable, comprising dense, large rounded globose structures reminiscent of axonal spheroids, small, dense, rounded dots, elongated structures probably following neurites, and less dense and less well-delineated cloud-like rounded bodies. All of these lesion types were found in all examined regions. The load of ubiquitin pathology was highest in the SNpc, the neocortex, especially parietally, and the entorhinal-hippocampal cortex. Ubiquitin pathology appeared even denser in the white matter subjacent to these cortical areas, compared to the cortex itself. We did not identify Marinesco bodies. Tau staining was mild in cortical areas, with discrete dystrophic neurites and a few neurons in temporolimbic areas and isolated tau-positive lesions in the parietal cortex. There was no tau positivity in the white matter, and there were no lesions indicative of Alzheimers disease.
TDP-43 and FUS stainings were entirely negative. Clearly, ubiquitin-positive pathology was most pronounced and severe in all examined areas, followed by mild to moderate alpha-synuclein pathology.
Discussion
This is the first report of the neuropathology of a PD patient with the newly discovered LRRK2 p.N1437H mutation [5]. This mutation is considered to be disease-causing but seems to be rare. It has so far only been described in two Norwegian families [5,6,10]. It was not found in DNA from 8,611 PD patients and 6,929 control subjects [1] analyzed in a collaborative study within the GEO-PD consortium, and not in our own sample collection of 55 additional Swedish PD patients studied within this GEO-PD multicenter study [11]. The clinical course of this patient was that of typical Parkinson's disease, albeit with an earlier age at onset and early development of marked motor fluctuations and dyskinesias. Neuropathology revealed mild cortical atrophy, cell loss and gliosis in the substantia nigra, relatively sparse alpha-synuclein pathology with some Lewy bodies, and very prominent ubiquitin immunoreactivity.
The patient had seemingly sporadic PD. Her parents, who lived until ages 70 and 80 years, did not have Parkinsonism, and her siblings reached a high age without Parkinsonian signs. It remains uncertain if the LRRK2 p.N1437H mutation occurred de novo in the patient, or if one of her parents carried the mutation but remained clinically unaffected. No first-degree relatives are alive.
The age at onset of motor symptoms at 50 years in our patient is in line with 47.8 ± 8 years in the ten patients with this mutation described from Norway [5]. The mean duration from symptom onset to death was 18 ± 8 years in the Norwegian family and is consistent with the clinical course in our patient. The Norwegian patients reportedly had autonomic, psychiatric, and cognitive symptoms similar to idiopathic PD [5]. Our patient did not develop cognitive dysfunction, dysautonomia, nor signs indicating atypical Parkinsonism. However, she had recurring episodes of severe depression, performed a suicide attempt, and finally committed suicide 6.5 months after bilateral STN DBS insertion.
Mild to moderate depression may be a non-motor symptom of PD [12] and this may be related to pathology in the locus ceruleus, raphe or tegmental nuclei [13], which was marked in this patient. Severe depression and suicide are uncommon in PD patients [12]. On the other hand, suicide attempts and suicide after STN DBS operation were described after this patient was operated on in 1999 [14–16], although the causal relationship has not been finally resolved. Our patient had a long history of depressive episodes, and had risk factors for depression and suicide such as marked comorbidity with severe visual impairment, living alone and inability to work [16]. Of note, the patient had recurring suicidal thoughts and performed one suicide attempt prior to DBS insertion.
DBS had reportedly a good effect in one Norwegian patient with p.N1437H mutation [5]. Our patient displayed paradoxical worsening of bradykinesia and rigidity when DBS current was on. We have observed this phenomenon in two other patients (unpublished results) out of n=235 PD patients operated on in Lund with STN as the target for DBS. Furthermore, the patient's severe dystonia did not improve but worsened after the operation. Post mortem examination provided no obvious explanation for the lack of beneficial DBS effect.
Neuropathology revealed almost complete cell loss in the SNpc. There was a moderate burden of alpha-synuclein positive lesions in the brainstem, and very few LBs were seen in the cortex. This distribution is compatible with early Braak PD stage 5 [17] or Diffuse LB Disease [18], even though the number of cortical LBs was low. The ubiquitin pathology appeared markedly more severe than alpha-synuclein pathology in both the grey and white matter. One of the puzzling aspects of LRRK2-related parkinsonism is the wide range of different pathologies associated with mutations in this gene [3]. Thirty-eight brains from PD patients with pathogenic LRRK2 mutations have been reported so far. While the most common finding is LB disease and alpha-synuclein immunoreactive inclusions, pure nigral degeneration, TDP-43-positive inclusions, diffuse LB disease, and tau-, or non-LB alpha-synuclein-positive pathologies were described [3]. Of note, there was ubiquitin-positive neuropathology in several brains from patients with LRRK2 mutations [2,7,19,20], whereas others displayed no immunoreactivity to ubiquitin [3]. Two patients from German-Canadian Family A (LRRK2 p.Y1699C mutation) had nonspecific SN (i.e. without alpha-synuclein pathology) degeneration with ubiquitin-positive neuronal inclusions [2]. Similar inclusions were found in one out of four patients from Family D (LRRK2 p.R1441C) from Western Nebraska [21]. Ubiquitin-immunoreactive inclusions were present in the cytoplasm and the nuclei of neurons. The nuclear inclusions were similar to Marinesco bodies, while the neuronal cytoplasmic inclusions were of novel appearance [2].
One patient with clinical dementia and a pathological picture of frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions (FTLD-U), most pronounced in the temporal and limbic areas, and LRRK2 p.G2019S mutation has been described [20]. However, this person did not have parkinsonism and as the common p.G2019S mutation has incomplete penetrance, it is conceivable that this individual may have developed FTLD-U independently of LRRK2 p.G2019S carrier status. One out of eight pathologically examined members of the Sagamihara kindred with LRRK2 p.I2020T mutations and PD displayed mild Lewy pathology in the brainstem, and alpha-synuclein positive brainstem lesions also stained for tau and ubiquitin [7]. The remaining seven did not show ubiquitin reactivity, and this discrepancy remains unexplained. By contrast, our patient had ubiquitin and LB pathology as well as some unspecific tau depositions. It remains uncertain if the excessive ubiquitin pathology is a consequence of natural, perhaps accelerated aging processes and the LB disease, or if p.N1437H mutation fosters a process resulting in intense ubiquitin pathology. The cause of the patient s congenital nystagmus and vision deficit remains uncertain, as does the exact cause of the widespread white matter changes in the MRI in the absence of risk factors for cerebrovascular disease.
In idiopathic PD, alpha-synuclein immunoreactivity is generally more widespread than ubiquitin-positive pathology [22]. This has been explained by phosphorylation and subsequent ubiquitination of alpha-synuclein, for which there is some experimental evidence. This scenario is difficult to reconcile with the findings in this study, or other reports from LRRK2 mutation carriers with overwhelming ubiquitin-positive pathology out of proportion to the alpha-synuclein immunoreactivity. We hypothesize that this ubiquitin-positive pathology indicates the deposition of at least one additional, as yet unidentified protein with a potentially important role in neurodegeneration.
Supplementary Material
Video of the patient with Parkinson's disease and LRRK2 p.N1437H mutation, recorded at age 67 years, after 17 years disease duration.
Segment 1 shows the patient in motor OFF after a night (9 hours) without medication, with akinesia, slowness of movements, and tremor at rest. In this state, the patient also had difficulty speaking and rigidity; she was able to rise from her chair, albeit with difficulty, but could not walk.
The patient received a 50mg tablet levodopa-carbidopa. Twenty minutes later, the patient simultaneously developped hyperkinesia and severe dystonia in her left leg (segment 2a). Segment 2b shows how the patient walks, with great difficulty and severe balance problems, about 30 minutes after the intake of levodopa. The effect of levodopa lasted 56 minutes, and the patient abruptly switched back to motor OFF (segment 3), where she also experienced depressed mood.
The patient reported muscle pain and subjectively experienced difficulty breathing when the levodopa effect started and waned. During the entire duration of the levodopa effect observed during this test, there were no episodes where the patient was entirely without hypokinesia and without hyperkinesia and/or painful dystonia, suggesting there was no ”therapeutic window” with optimal levodopa concentration.
Acknowledgments
Andreas Puschmann, Håkan Widner, Jan Reimer, and Christer Nilsson received funding from The Swedish Parkinson Academy, Stiftelsen Olle Engkvist Byggmästare, AFA Insurance, The Swedish Parkinson Foundation, Apotekare Hedbergs Foundation, Elsa Schmitz Foundation, and Lund University Hospital Research Funds. Owen A. Ross, Carles Vilariño-Güell, Sarah J. Lincoln, Jennifer M. Kachergus, Stephanie A. Cobb, Zbigniew K. Wszolek, Matthew J. Farrer are supported by NIH/NINDS Morris K. Udall Center for Excellence in PD Research at Mayo Clinic (P50 NS072187) grant. Zbigniew K. Wszolek is partially supported by the NIH/NINDS 1RC2NS070276, NS057567, P50NS072187, Mayo Clinic Florida (MCF) Research Committee CR program (MCF #90052030), Dystonia Medical Research Foundation, and the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch (MCF #90052031/PAU #90052).
We thank the patient s family for their kind cooperation. Gunnar Gunnarsson, Dept. for Neurosurgery, Lund University Hospital, provided the photographs of Figure 3. Karin Nilsson, Dept. for Geriatric Psychiatry, Lund University, undertook genealogical investigations. Jan Reimer, Dept. for Neurology, Lund, performed and videorecorded the levodopa test. We thank Dr. J. Aasly, Trondheim, and Dr. M. Toft, Oslo, for comparing genealogical information with the Norwegian LRRK2 p.N1437H families they described.
Footnotes
Conflict of Interest:
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ross OA, Soto-Ortolaza AI, Aasly JO, Abahuni N, Annesi G, Bacon JA, et al. LRRK2 exonic variants and susceptibility to Parkinson s disease. Lancet Neurol. 2011 Oct;10(10):898–908. doi: 10.1016/S1474-4422(11)70175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004 Nov 18;44(4):601–7. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Wider C, Dickson DW, Wszolek ZK. Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neurodegener Dis. 2010;7(1–3):175–9. doi: 10.1159/000289232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004 Nov 18;44(4):595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 5.Aasly JO, Vilarino-Guell C, Dächsel JC, Webber PJ, West AB, Haugarvoll K, et al. Novel pathogenic LRRK2 p.Asn1437His substitution in familial Parkinson's disease. Mov Disord. 2010 Oct 15;25(13):2156–63. doi: 10.1002/mds.23265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johansen KK, White LR, Farrer MJ, Aasly JO. Subclinical signs in LRRK2 mutation carriers. Parkinsonism Relat Disord. 2011 Aug;17(7):528–32. doi: 10.1016/j.parkreldis.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Hasegawa K, Stoessl AJ, Yokoyama T, Kowa H, Wszolek ZK, Yagishita S. Familial parkinsonism: study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord. 2009 May;15(4):300–6. doi: 10.1016/j.parkreldis.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, et al. Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol. 2009 Dec;8(12):1150–7. doi: 10.1016/S1474-4422(09)70238-8. [DOI] [PubMed] [Google Scholar]
- 9.Brunnström H, Friberg N, Lindberg E, Englund E. Differential degeneration of the locus coeruleus in dementia subtypes. Clin Neuropathol. 2011 May-Jun;30(3):104–10. doi: 10.5414/npp30104. [DOI] [PubMed] [Google Scholar]
- 10.Corti O, Lesage S, Brice A. What Genetics Tells us About the Causes and Mechanisms of Parkinson's Disease. Physiol Rev. 2011 Oct;91(4):1161–218. doi: 10.1152/physrev.00022.2010. [DOI] [PubMed] [Google Scholar]
- 11.Puschmann A. Heredity in Parkinson's disease. From rare mutations to common genetic risk factors. Lund, Sweden: Lund University Faculty of Medicine Doctoral Dissertation Series; 2011. p. 95. [Google Scholar]
- 12.Reichmann H, Schneider C, Lohle M. Non-motor features of Parkinson's disease: depression and dementia. Parkinsonism Relat Disord. 2009 Dec;15(Suppl 3):S87–92. doi: 10.1016/S1353-8020(09)70789-8. [DOI] [PubMed] [Google Scholar]
- 13.Dickson DW, Fujishiro H, Orr C, DelleDonne A, Josephs KA, Frigerio R, et al. Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat Disord. 2009 Dec;15(Suppl 3):S1–5. doi: 10.1016/S1353-8020(09)70769-2. [DOI] [PubMed] [Google Scholar]
- 14.Doshi PK, Chhaya N, Bhatt MH. Depression leading to attempted suicide after bilateral subthalamic nucleus stimulation for Parkinson's disease. Mov Disord. 2002 Sep;17(5):1084–5. doi: 10.1002/mds.10198. [DOI] [PubMed] [Google Scholar]
- 15.Funkiewiez A, Ardouin C, Caputo E, Krack P, Fraix V, Klinger H, et al. Long term effects of bilateral subthalamic nucleus stimulation on cognitive function, mood, and behaviour in Parkinson's disease. J Neurol Neurosurg Psychiatry. 2004 Jun;75(6):834–9. doi: 10.1136/jnnp.2002.009803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voon V, Krack P, Lang AE, Lozano AM, Dujardin K, Schupbach M, et al. A multicentre study on suicide outcomes following subthalamic stimulation for Parkinson's disease. Brain. 2008 Oct;131(Pt 10):2720–8. doi: 10.1093/brain/awn214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003 Mar-Apr;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 18.Kosaka K, Tsuchiya K, Yoshimura M. Lewy body disease with and without dementia: a clinicopathological study of 35 cases. Clin Neuropathol. 1988 Nov-Dec;7(6):299–305. [PubMed] [Google Scholar]
- 19.Wszolek ZK, Vieregge P, Uitti RJ, Gasser T, Yasuhara O, McGeer P, et al. German-Canadian family (family A) with parkinsonism, amyotrophy, and dementia - Longitudinal observations. Parkinsonism Relat Disord. 1997 Nov;3(3):125–39. doi: 10.1016/s1353-8020(97)00013-8. [DOI] [PubMed] [Google Scholar]
- 20.Dächsel JC, Ross OA, Mata IF, Kachergus J, Toft M, Cannon A, et al. Lrrk2 G2019S substitution in frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions. Acta Neuropathol. 2007 May;113(5):601–6. doi: 10.1007/s00401-006-0178-1. [DOI] [PubMed] [Google Scholar]
- 21.Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology. 2004 May 11;62(9):1619–22. doi: 10.1212/01.wnl.0000125015.06989.db. [DOI] [PubMed] [Google Scholar]
- 22.Ferrer I, Martinez A, Blanco R, Dalfo E, Carmona M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: preclinical Parkinson disease. J Neural Transm. 2010 Sep 23; doi: 10.1007/s00702-010-0482-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video of the patient with Parkinson's disease and LRRK2 p.N1437H mutation, recorded at age 67 years, after 17 years disease duration.
Segment 1 shows the patient in motor OFF after a night (9 hours) without medication, with akinesia, slowness of movements, and tremor at rest. In this state, the patient also had difficulty speaking and rigidity; she was able to rise from her chair, albeit with difficulty, but could not walk.
The patient received a 50mg tablet levodopa-carbidopa. Twenty minutes later, the patient simultaneously developped hyperkinesia and severe dystonia in her left leg (segment 2a). Segment 2b shows how the patient walks, with great difficulty and severe balance problems, about 30 minutes after the intake of levodopa. The effect of levodopa lasted 56 minutes, and the patient abruptly switched back to motor OFF (segment 3), where she also experienced depressed mood.
The patient reported muscle pain and subjectively experienced difficulty breathing when the levodopa effect started and waned. During the entire duration of the levodopa effect observed during this test, there were no episodes where the patient was entirely without hypokinesia and without hyperkinesia and/or painful dystonia, suggesting there was no ”therapeutic window” with optimal levodopa concentration.