

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Familial erythrocytosis (congenital or hereditary erythrocytosis, primary familial polycythemia, erythrocytosis, familial, 1–4)
1.2 OMIM# of the disease
133100 (erythrocytosis, familial 1, ECYT1, autosomal dominant)
263400 (erythrocytosis, familial 2, ECYT2, autosomal recessive)
609820 (erythrocytosis, familial 3, ECYT3, autosomal dominant)
611783 (erythrocytosis, familial 4, ECYT4, autosomal dominant)
1.3 Name of the analysed genes or DNA/chromosome segments
Erythropoietin receptor (EPOR)/19p13.3–p13.2
von Hippel-Lindau tumor suppressor (VHL)/3p26–p25
Egl nine homolog 1 (C. elegans) (EGLN1, synonym HIF prolyl hydroxylase 2, PHD2)/1q42.1
Endothelial PAS domain protein 1 (EPAS1, synonym hypoxia-inducible factor 2 alpha, HIF2A)/2p21–p16 Other genes that can be associated with familial erythrocytosis
Hemoglobin subunit beta (HBB)/11p15.5
Hemoglobin subunit alpha 1 (HBA1)/16pter-p13.3
Hemoglobin subunit alpha 2 (HBA2)/16pter-p13.3
2,3-bisphosphoglycerate mutase (BPGM)/7q31–q34
Pyruvate kinase, liver and RBC (PKLR)/1q21 Other genes that could be associated with erythrocytosis
Hypoxia inducible factor 1, alpha subunit (basic helix-loop-helix transcription factor) (HIF1A)/14q23.2
Egl nine homolog 2 (C. elegans) (EGLN2)/19q13.2
Egl nine homolog 3 (C. elegans) (EGLN3)/14q13.1
Potentially all other genes that are involved in HIF regulation
1.4 OMIM# of the gene(s)
133171 (EPOR)
608537 (VHL)
606425 (EGLN1)
603349 (EPAS1) Other genes which can be associated with familial erythrocytosis
141900 (HBB)
141800 (HBA1)
141850 (HBA2)
613896 (BPGM)
609712 (PKLR) Other genes which could be associated with erythrocytosis
603348 (HIF1A)
606424 (EGLN2)
606426 (EGLN3)
1.5 Mutational spectrum
EPOR mutations generate a primary defect with constitutively activated receptor function while mutated VHL, EGLN1 and EPAS1 generate secondary alterations of the hypoxia-sensing pathways. Therefore, EPOR mutation-associated ECYT1 is also termed primary familial erythrocytosis whereas VHL/EGLN1/EPAS1 mutations-associated ECYT2–4 are termed secondary familial erythrocytosis.1, 2, 3
(i) ECYT1 (synonym: primary familial congenital polycythemia/PFCP): EPOR, variable exon 8 mutations at positions 5828–6003 (176 base pair region), which all result in a defect (most frequently a truncation) of the intracytoplasmic domain of the EPOR protein.1, 2, 3, 4
(ii) ECYT2: VHL, most frequently C598T/R200W, either as an isolated C598T mutation or in combination with other VHL mutations (C235T, G311T, G376T, G388C, A523G, C562G, C571G and C574T) or isolated VHL mutations other than C598T.1, 2, 3, 5, 6, 7
(iii) ECYT3: EGLN1 C950G/P317R, G1112A/R371H and A1121G/H374R.1, 2, 3
(iv) ECYT4: EPAS1 A1603G/M535V, G1605A/M535I, G1609A/G537R, G1609T/G537Y and C1617G/D539Q.1, 2, 8, 9
(v–ix) Gene mutations that increase oxygen affinity.
Hemoglobin alterations involving HBB, HBA1, HBA2, BPGM or PKLR are secondary defects, which can be associated with familial erythrocytosis; no OMIM disease code number.1, 2, 3
– High oxygen affinity hemoglobinopathies with associated erythrocytosis, particularly HBB, the most frequent mutated hemoglobin subunit, ∼100 mutations, mainly in the C-terminal region. HBA1 and HBA2 are rarely mutated hemoglobin subunits.1, 2, 3, 10, 11, 12
– 2,3-bisphosphoglycerate deficiency and pyruvate kinase hyperactivity with high adenosine triphosphate levels and consecutive increased affinity of red cells for oxygen (BPGM and PKLR).
All gene alterations of the hypoxia-sensing (HIF signaling) pathway are potential candidates for secondary erythrocytosis, for example, HIF1A, EGLN2 or EGLN3; currently no known familial or sporadic cases, and no OMIM disease code number.1, 2, 3
1.6 Analytical methods
Sequencing of peripheral blood leukocytes (DNA and/or RNA/cDNA).
Restriction analysis for VHL C598T mutation: the C598T mutation abolishes an Fnu4HI restriction endonuclease recognition site.
1.7 Analytical validation
Bidirectional sequencing, double measurements.
Control samples: mutated patient sample, unmutated patient sample or unmutated cell line.
1.8 Estimated frequency of the disease
Population prevalence: <1:100 000
1.9 If applicable, prevalence in the ethnic group of investigated person
In principle, individuals from any ethnic group can develop ECYT1–4. However, a regional/ethnic association is known for VHL mutation-positive autosomal recessive ECYT2:
Chuvash polycythemia (Republic of Chuvash, Russia, mid-Volga River region, Russia).5
Ischia polycythemia (island of Ischia, Bay of Naples, Italy).6
The exact prevalence for Chuvash polycythemia and Ischia polycythemia is not known, but likely <1:100 000.
1.10 Diagnostic setting

Comment:
Diagnosis of ECYT1–4 requires detection of one of the associated gene mutations. The rank of gene testing depends on family history (recessive versus dominant inheritance) and laboratory testing (particularly erythropoietin/EPO levels); see 3.1.
Predictive testing and risk assessment in relatives for exclusion of potential heterozygous VHL mutation carrier without erythrocytosis. Owing to the autosomal recessive inheritance, predictive testing should be performed in individuals from ECYT2 families, even if the red cell parameters are normal in an individual, particularly for genetic counseling of such an individual.
Prenatal testing is not indicated in ECYT1–4 because the result of a genetic test has no consequence during pregnancy (detection of ECYT1–4 is no indication for induced abortion); the child of an affected mother can be tested after birth.

2. TEST CHARACTERISTICS
2.1 Analytical sensitivity (proportion of positive tests if the genotype is present)
>99% (false-negative rate <1%).
2.2 Analytical specificity (proportion of negative tests if the genotype is not present)
>99% (false-positve rate <1%).
2.3 Clinical sensitivity (proportion of positive tests if the disease is present)
The clinical sensitivity is undetermined but likely <50% of cases with erythocytosis and positive family history are positive for one of the EYCT 1–4-associated mutations.
2.4 Clinical specificity (proportion of negative tests if the disease is not present)
The clinical specificity is undetermined but likely >99% of cases with no erythocytosis are negative for EYCT 1–4-associated mutations.
2.5 Positive clinical predictive value (life-time risk of developing the disease if the test is positive)
100% in homozygously mutated EYCT1–4 cases.
100% in heterozygously mutated EYCT 1, 3, 4 cases.
Undetermined but likely <5% in heterozygously mutated EYCT 2 cases.
2.6 Negative clinical predictive value (probability of not developing the disease if the test is negative)
The negative clinical predictive value is undetermined because >50% of cases with erythocytosis and positive family history are negative for one of the EYCT 1–4-associated mutations.
Index case in that family had been tested:
If the index patient is carrying a mutation, the risk of mutation-negative first-degree relatives to develop the disease is 0%.
Index case in that family had not been tested:
If the index patient has not been tested for ECYT1–4 mutations, the risk of non-affected first-degree relatives to develop the disease depends on recessive or dominant transmission of the disease:
Autosomal recessive ECYT2, risk to develop the disease: 0% if only one parent is a heterozygous carrier and the other parent is not, 25% if both parents are heterozygous carriers, 50% if one parent is a heterozygous carrier and one parent is a homozygous patient and 100% if both parents are homozygous patients.
Autosomal dominant ECYT1, 3 and 4, risk to develop the disease: 50% if one parent is a heterozygous patient and the other parent is not, 75% if both parents are heterozygous patients, 100% if one parent is a heterozygous patient and one parent is a homozygous patient and 100% if both parents are homozygous patients.
3. CLINICAL UTILITY
3.1 (Differential) diagnosis: the tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?
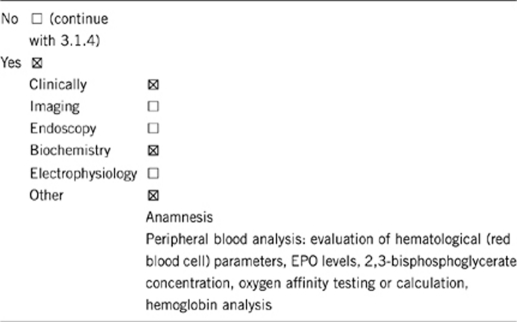
Comment:
An erythrocytosis is present when the red cell mass is greater than or equal to 125% of the predicted value for an individual's body mass and sex. The erythrocytosis is reflected by the fact that the hemoglobin and hematocrit are also usually raised. By definition it is congenital if it is present from birth. In such persons the erythrocytosis is usually detected at a young age. There can be a family history in keeping with the inherited nature of the defect. However, sporadic cases without positive family history can occur.
Familial erythrocytosis can be primary or secondary. A primary erythrocytosis occurs when there is an intrinsic defect in the erythroid cells in the bone marrow (EPOR mutation). In these cases EPO levels will be below normal. A secondary erythrocytosis is present when EPO is produced from some source and then mediates red cell production resulting in an erythrocytosis. In this situation, EPO levels will be normal (which would be inappropriate for a raised hematocrit) or elevated above the normal range. Alterations of the genes in the oxygen-sensing pathway can cause such a secondary familial erythrocytosis (VHL, EGLN1 or EPAS1 mutations). Other congenital lesions may cause a secondary erythrocytosis due to increased affinity of red cells for oxygen, such as high oxygen affinity hemoglobins, PKLR hyperactivity with high adenosine triphosphate levels and 2,3-bisphosphoglycerate deficiency.
Consider screening those with:
True erythrocytosis.
No Philadelphia chromosome-negative myeloproliferative neoplasm in terms of polycythaemia vera (PV).
No identifiable secondary cause.
Young patients.
If positive family history.
Diagnosis of familial erythrocythosis:1, 2, 3
(i) Anamnesis
– Positive family history.
– Thromboembolic complications.
(ii) Physical examination and exclusion of secondary (reactive) cause of erythrocytosis
– Cardiopulmonary status (chronic lung and/or heart diseases, including cyanotic congenital heart diseases in children and chronic hypoxia due to smoking).
– Kidney status/renal diseases with secondary endogenous increase of EPO.
– Spleen and liver status (splenomegaly and/or hepatomegaly are usually absent but can be present in neoplastic PV).
– Vertebral status (vertebral hemangiomas can be present in ECYT2).
– Exclusion of non-hematopoietic neoplasms with ectopic EPO production, particularly cerebellar hemangioblastoma, meningioma, parathyroid carcinoma and adenomas, hepatocellular carcinoma, renal cell cancer, pheochromocytoma and uterine leiomyomas.
– Exclusion of exogenous increase of EPO after administration of EPO or androgens as drugs or abuse (competitive sport).
(iii) Laboratory testing (non-phlebotomized patients)
(iii-1) Isolated increased red blood cell parameters (white blood cell counts and platelet counts are usually normal)
hemoglobin >185 g/l (men)/>165 g/l (women)
or
hemoglobin>170 g/l (men)/>150 g/l (women) if associated with a documented and sustained increase of at least 20 g/l from an individual's base line value that cannot be attributed to correction of iron deficiency
or
hemoglobin/hematocrit >99th percentile of method-specific reference range for age, sex and altitude of residence
or
elevated red blood cell mass >25% above mean normal predicted value, >125%, respectively.
(iii-2) Serum EPO level changes regarding the laboratory's reference range for normal
– EPO level decreased: consider lesions of EPO signaling pathway (ECYT1).
– EPO level increased or in relation to erythrocytosis inappropriately normal: consider lesions of the oxygen-sensing pathway (ECYT 2–4).
(iii-3) 2,3-bisphosphoglycerate concentration decreased and/or oxygen affinity testing indicates increased affinity: consider non-ECYT diseases with erythrocytosis.
(iv) Mutation analysis
(iv-1) ECYT-associated mutations in EPOR, VHL, EGLN1 or EPAS1.
– Family history indicates homozygous/dominant transmission + EPO level decreased: EPOR mutation analysis (ECYT1).
– Family history indicates heterozygous/recessive transmission + EPO level increased/inappropriately normal (also indicative: vertebral hemangioma, patients from Russia or Italy): VHL mutation analysis (ECYT2).
– Family history indicates homozygous/dominant transmission + EPO level increased/inappropriately normal: EGLN1 and EPAS1 mutation analyses (EYCT3 and ECYT4).
(iv-2) In cases with negative results for ECYT-associated mutations analysis of rarer mutations is suggested, particularly hemoglobin chain mutations.
(iv-3) Somatic Janus kinase 2, particularly G1894T/V617F mutation, for exclusion of neoplastic PV;13, 14, 15 familial and/or juvenile PV cases are possible.16, 17
The home page of MPN&MPNr-EuroNet (COST Action BM0902) provides addresses of European laboratories, which perform mutation analysis for familial erythrocytosis: http://www.mpneuronet.eu/
3.1.2 Describe the burden of alternative diagnostic methods to the patient
Low: blood collection is required for mutation analyses but also for evaluation of red cell parameters and EPO level.
Low/medium: physical examinations (low) and additional examinations, for example, sonography/radiology (low/medium).
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Analysis of red blood cell parameters and EPO levels: low costs, low/medium sensitivity and specificity.
Physical and additional examinations: depending on the examination medium to high costs, low/medium sensitivity and specificity.
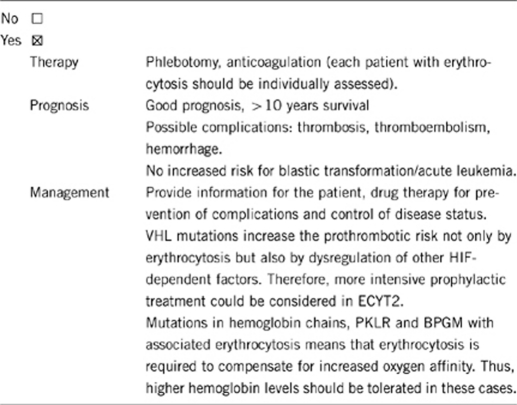
3.1.4 Will disease management be influenced by the result of a genetic test?
3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
Yes, particularly in VHL mutation-positive patients (ECYT2), who have a higher prothrombotic risk.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done
Regular follow-up examinations.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, in case of detection of ECYT1–4-associated mutations. High risk for autosomal dominant inheritance (ECYT 1, 3 and 4). Increased risk in autosomal recessive inheritance (ECYT2); in ECYT2 the homozygous VHL mutation status is associated with disease phenotype, while heterozygous mutation status is usually not. In some heterozygous ECYT2 patients, however, erythrocytosis is present and might be related to an undefined second molecular defect.1, 2, 3
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Depends on the red cell parameters of other family members but genetic testing should be performed, particularly if the index patient carries an ECYT 1–4-associated mutation.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes, detection of ECYT 1–4-associated mutations in a family member other than the index patient is associated with an increased risk of having/developing erythrocytosis.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Not applicable.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Advantage of genetic testing:1, 2, 3
ECYT 1–4-associated mutation analysis is helpful for establishment of diagnosis and subclassification of the disease.
Mutation analysis helps to determine the risk regarding homozygous and heterozygous mutation-carrying individuals.
Acknowledgments
This work was supported by EuroGentest, an EU-FP6 supported NoE, contract number 512148 (EuroGentest Unit 3: ‘Clinical genetics, community genetics and public health', Workpackage 3.2).
The authors declare no conflict of interest.
References
- McMullin MF. Idiopathic erythrocytosis: a disappearing entity. Hematology Am Soc Hematol Educ Program. 2009;1:629–635. doi: 10.1182/asheducation-2009.1.629. [DOI] [PubMed] [Google Scholar]
- Percy MJ, Rumi E. Genetic origins and clinical phenotype of familial and acquired erythrocytosis and thrombocytosis. Am J Hematol. 2009;84:46–54. doi: 10.1002/ajh.21313. [DOI] [PubMed] [Google Scholar]
- Gordeuk VR, Stockton DW, Prchal JT. Congenital polycythemias/erythrocytoses. Haematologica. 2005;90:109–116. [PubMed] [Google Scholar]
- Rives S, Pahl HL, Florensa L, et al. Molecular genetic analyses in familial and sporadic congenital primary erythrocytosis. Haematologica. 2007;92:674–677. doi: 10.3324/haematol.10787. [DOI] [PubMed] [Google Scholar]
- Ang SO, Chen H, Gordeuk VR, et al. Endemic polycythemia in Russia: mutation in the VHL gene. Blood Cells Mol Dis. 2002;28:57–62. doi: 10.1006/bcmd.2002.0488. [DOI] [PubMed] [Google Scholar]
- Perrotta S, Nobili B, Ferraro M, et al. Von Hippel-Lindau-dependent polycythemia is endemic on the island of Ischia: identification of a novel cluster. Blood. 2006;107:514–519. doi: 10.1182/blood-2005-06-2422. [DOI] [PubMed] [Google Scholar]
- Cario H, Schwarz K, Jorch N, et al. Mutations in the von Hippel-Lindau (VHL) tumor suppressor gene and VHL-haplotype analysis in patients with presumable congenital erythrocytosis. Haematologica. 2005;90:19–24. [PubMed] [Google Scholar]
- van Wijk R, Sutherland S, Van Wesel AC, et al. Erythrocytosis associated with a novel missense mutation in the HIF2A gene. Haematologica. 2010;95:829–832. doi: 10.3324/haematol.2009.017582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percy MJ, Furlow PW, Lucas GS, et al. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358:162–168. doi: 10.1056/NEJMoa073123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percy MJ, Butt NN, Crotty GM, et al. Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosis. Haematologica. 2009;94:1321–1322. doi: 10.3324/haematol.2009.008037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochuli M, Zurbriggen K, Schmid M, et al. A new alpha-globin variant with increased oxygen affinity in a Swiss family: Hb Frauenfeld [alpha 138(H21)Ser → Phe, TCC>TTC (alpha 2)] Hemoglobin. 2009;33:54–58. doi: 10.1080/03630260802625733. [DOI] [PubMed] [Google Scholar]
- Walker L, Eng B, McFarlane A, Waye JS. High oxygen affinity hemoglobin variant in a Canadian family: Hb Bunbury [beta94(FG1)Asp → Asn, GAC → AAC] Hemoglobin. 2007;31:101–113. doi: 10.1080/03630260601059241. [DOI] [PubMed] [Google Scholar]
- James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- Percy MJ, Jones FG, Green AR, Reilly JT, McMullin MF. The incidence of the JAK2 V617F mutation in patients with idiopathic erythrocytosis. Haematologica. 2006;91:413–414. [PubMed] [Google Scholar]
- Percy MJ, Scott LM, Erber WN, et al. The frequency of JAK2 exon 12 mutations in idiopathic erythrocytosis patients with low serum erythropoietin levels. Haematologica. 2007;92:1607–1614. doi: 10.3324/haematol.11643. [DOI] [PubMed] [Google Scholar]
- Hussein K, Bock O, Ballmaier M, et al. Familial polycythemia vera with non-germline JAK2(V617F) mutation sparing the abnormal and clonal granulopoiesis. Leukemia. 2007;21:2566–2568. doi: 10.1038/sj.leu.2404846. [DOI] [PubMed] [Google Scholar]
- Cario H, McMullin MF, Pahl HL. Clinical and hematological presentation of children and adolescents with polycythemia vera. Ann Hematol. 2009;88:713–719. doi: 10.1007/s00277-009-0758-y. [DOI] [PMC free article] [PubMed] [Google Scholar]