

Abstract
In our efforts to identify novel chemical scaffolds for the development of new antiprotozoal drugs, a compound library was screened against T. gondii tachyzoites with activity discovered for N-(4-ethylbenzoyl)-2-hydroxybenzamide 1a against T. gondii as described elsewhere.1 Synthesis of a compound set was guided by T. gondii SAR with 1r found to be superior for T. gondii, also active against Thai and Sierra Leone strains of P. falciparum, and with superior ADMET properties as described elsewhere.1 Herein, synthesis methods and details of the chemical analysis of the compounds in this series are described. Further, this series of N-benzoyl-2-hydroxybenzamides was re-purposed for testing against four other protozoan parasites: T. b. rhodesiense, T. cruzi, L. donovani, and P. falciparum (K1 isolate). Structure-activity analyses led to the identification of compounds in this set with excellent anti-leishmanial activity (compound 1d). Overall, compound 1r was the best and had activity 21-fold superior to that of the standard anti-malarial drug chloroquine against the K1 P. falciparum isolate.
Introduction
Protozoan parasites cause diseases such as malaria, amoebiasis, giardiasis, toxoplasmosis, leishmaniasis and trypanosomiases, and are among the most serious public health problems in developing countries and also some of these parasites cause major health problems in the U.S., Europe and Japan.1 According to the World Health Organization (WHO), parasitic protozoan diseases plague billions of people worldwide and kill millions annually.2 Nevertheless, few medicines are available to treat those serious and sometimes lethal infections. Human African trypanosomiasis (HAT, sleeping sickness), caused by Trypanosoma brucei rhodesiense (T. b. rhodesiense) and T. b. gambiense, threatens about 50 million people living in sub-Saharan Africa and causes an estimated 25,000 deaths per year. 3 There are an estimated 10 million people suffering from American trypanosomiasis (Chagas disease), which is caused by Trypanosoma cruzi (T. cruzi).4 Visceral leishmaniasis is caused by Leishmania donovani (L. donovani) and currently threatens 350 million persons around the world.4 Plasmodium falciparum (P. falciparum) is one of the causative agents of malaria and infects about 225 million people and causes nearly 800,000 deaths, mostly in children, annually.5 Overall, in the absence of vaccines and with few chemotherapeutics available, some with reduced efficacy and adverse effects, diseases caused by protozoan parasites still contribute significantly to morbidity and mortality. Thus, discovery and development of effective, safe and affordable anti-protozoal agents remains an urgent need.
In our efforts to identify novel chemical scaffolds as potent anti-protozoal drugs, a small compound library (ASDI) was screened initially against T. gondii tachyzoites as described elsewhere.1 Moderate activity was found for N-(4-ethylbenzoyl)-2-hydroxybenzamide 1a (Figure 1) against the T. gondii parasites.1 Interestingly, some salicylimides, among other compounds, have been reported to possess anti-malarial activity ascribed to their targeting the Hemozoin Detoxification Protein (HDP), a protein known to be vital for P. falciparum.6 It is also known that certain salicylamide derivatives, such as salicilylhydroxamic acid, nitazoxanide and other thiazolides, display anti-protozoal activity.7–11 Herein, we report the details of the synthesis, modification, and biological evaluation of this set of N-benzoyl-2-hydroxybenzamides and related compounds, as potent, and in some cases selective, agents against the K1 isolate of P. falciparum and L. donovani and studied their effects on T. b. rhodiense and T. cruzi.
Figure 1.

Hit compound 1a
Chemistry
The desired N-benzoyl-2-hydroxybenzamides (1a-ab) were generated by the reaction of salicylamide (2) with various acid chlorides (3a-ab) in refluxing pyridine (Scheme 1). The crude products (mostly solids) were subsequently purified by crystallization or preparative HPLC to give the pure compounds in yields ranging from modest to very good (Table 1).
Scheme 1.

Preparation of N-benzoyl-2-hydroxybenzamides
Reagents and conditions: (a) pyridine, reflux, 4 h.
Table 1.
Variation of the Ring B in N-Benzoyl-2-hydroxybenzamides (1)
| Compound | a | b | c | d | e | f | g |
| R | 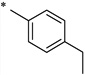 |
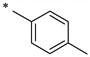 |
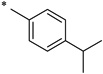 |
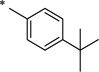 |
 |
 |
 |
| Yield (%)a | 51 | 85 | 22 | 95 | 55 | 17 | 29 |
| Compound | h | i | j | k | l | m | n |
| R | 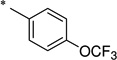 |
 |
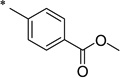
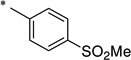 |
 |
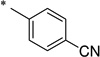
 |
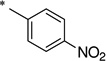
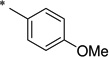 |
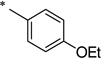 |
| Yield (%)a | 38 | 91 | 14 | 42 | 33 | 37 | 77 |
| Compound | o | p | q | r | s | t | u |
| R | 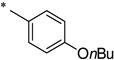 |
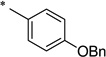 |
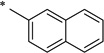
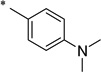 |
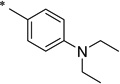 |
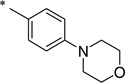 |
 |
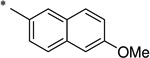 |
| Yield (%)a | 62 | 39 | 31 | 8 | 25 | 29 | 28 |
| Compound | v | w | x | y | z | aa | ab |
| R | 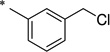 |
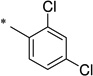 |
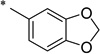 |
 |
 |
 |
 |
| Yield (%)a | 24 | 41 | 41 | 33 | 48 | 83 | 30 |
Isolated yield of product exhibiting >95% HPLC purity.
The hydroxybenzamides 1k and 1ab were further elaborated as shown in Scheme 2. Catalytic hydrogenation over palladium-on-carbon of compounds 1k and 1ab provided corresponding aniline 4a and phenol 4b derivatives in good yields (Scheme 2). Reaction of compound 4b with methyl 3-(chlorocarbonyl)propanoate provided compound 5 in modest yield (Scheme 2). The hydroxamic acid 8 was prepared in a 3-step procedure starting with protection of the hydroxyl group in 4a as its silyl ether.12 Subsequent reaction with 8-[(benzyloxy)amino]-8-oxooctanoyl chloride13 gave the intermediate 7, which was deprotected to provide compound 8 in low yield (Scheme 2).
Scheme 2.

Elaboration of Selected N-Benzoyl-2-hydroxybenzamides
Reagents and conditions: (a) Pd/C, H2, MeOH, room temp, 18 h; (b) methyl 3-(chlorocarbonyl)propanoate, pyridine, CH2Cl2, room temp, 1 h; (c) TIPSCl, Et3N, DMAP, DMF, room temp, 3.5 h, (d) 8-[(benzyloxy)amino]-8-oxooctanoyl chloride, pyridine, CH2Cl2, room temp, 2 h.
To better understand the importance of the imide linker for biological activity, designed modification of this moiety was analyzed. Syntheses are shown in Scheme 3. The N-methyl imide 10 was prepared in high yield from the corresponding 4-ethylbenzoyl chloride (3a) and N-methylsalicylamide (9)14 by following the standard procedure described above. In the same manner, the acylated salicylhydrazide 12 was prepared from 4-ethylbenzoyl chloride (3a) and salicylhydrazide (11) (Scheme 3). Reaction of the original hit compound 1a with Lawesson’s reagent in refluxing chlorobenzene provided the thioimide derivative 13 (Scheme 3).
Scheme 3.

Modifications of the Linker in N-Benzoyl-2-hydroxybenzamides
Reagents and conditions: (a) pyridine, reflux, 4 h; (b) Lawesson's reagent, chlorobenzene, 135 °C, 3 h.
Further modifications of the linker resulted in synthesis of N-[(4-ethylphenyl)carbamoyl]-2-hydroxybenzamide (16) and 1-(4-ethylphenyl)-3-(2-hydroxyphenyl)urea (19) (Scheme 4). The intermediate 15 was readily obtained from salicylamide following a known procedure.15 Standard hydrogenolysis of 15 gave the compound 16 in 69% yield (Scheme 4). The compound 19 bearing a urea linker was prepared by the reaction of isocyanate 17 with 4-ethylaniline and subsequent deprotection (Scheme 4).16
Scheme 4.

Synthesis of the Carbamoylsalicylamide 16 and the Urea 19
Reagents and conditions: (a) BnBr, K2CO3, acetone, reflux, 16 h; (b) 1) (COCl)2, CH2Cl2, room temp to 50 °C, 3 h; 2) 4-ethylaniline, MeCN, room temp, 18 h; (c) 10% Pd/C, H2, MeOH, room temp, 24 h; (d) 4-ethylaniline, CH2Cl2, room temp, 19 h; (e) BBr3, CH2Cl2, −78 °C to 0 °C.
Several compounds 21a–c bearing linkers that are chemically more stable than an imide function were analyzed. The compounds were prepared from the corresponding imide/amine precursors (20a–c) as shown in Scheme 5 by following the standard procedure described above. The oxadiazole 23 was prepared from 1a by cyclodehydration in the presence of hydroxylamine hydrochloride and sodium acetate (Scheme 5).17 Reaction of 2-(benzyloxy)aniline (24) with ethyl 3-chloro-3-oxopropanoate gave the intermediate 25, which was readily hydrolyzed to the carboxylic acid 26.13 This intermediate was converted to the final bis-amide 28 through amide bond formation13 with 4-ethylaniline and subsequent deprotection (Scheme 5).18
Scheme 5.

Preparation of Compounds with Modified Linkers
Reagents and conditions: (a) pyridine, reflux, 4 h; (b) 200 °C, 1 h; (c) NH2OH·HCl, NaOAc, absolute EtOH, 16 h; (d) pyridine, CH2Cl2, ethyl 3-chloro-3-oxopropanoate, room temp, 18 h; (e) THF, LiOH, room temp, overnight; (f) DIPEA, BOP-Cl, 4-ethylaniline, room temp, 22 h; (g) Pd(OH)2/C (25 wt %), EtOH-cyclohexene (2:1), reflux, 2.5 h.
Results and Discussion
In search of compounds with improved activity and ADMET properties,1 we modified the hit 1a at three sites: the phenol ring (A), 4-ethylphenyl ring (B), and the imide linker (Figure 1).1 ADMET studies, described in detail elsewhere.1 revealed metabolic instability to be the main concern for 1a.1 Briefly, the compound was found to be quickly metabolized in human liver microsomes (HLM), and only 0.5% of the compound remained after incubation for 60 min in the presence of NADPH.1 However, in the absence of NADPH, 53% of the compound was left unchanged after the incubation.1 The potassium ion channel coded by hERG was not inhibited by 1a at compound concentration as high as 300 µM.1 Among the recombinant cytochrome P450 (CYP450) isozymes, 3A4, 2D6, and 2C9, only the latter was inhibited to the extent of 71% at a compound concentration of 10 µM.1 The aqueous solubility in phosphate-based buffer of compound 1a was 2.76 µM at pH 4.0 and 50.8 µM at pH 7.4, respectively (for comparison, the control compound diclofenac exhibits the following solubility: 9.10 µM at pH 4.0 and 303 µM at pH 7.4).1 The tested compound presented a similar permeability as the control drugs testosterone and methotrexate.1 However, the protein binding in human plasma of 1a was high (99.9%) in comparison with the control drug, fluconazole (23.7%), and both its intrinsic clearance and its metabolism rate in HLM (282 µL/min/mg proteins) were faster than that of the control verapamil (38.8 µL/min/mg proteins).1 Nonetheless, these results encouraged us to modify the hit 1a to generate a lead compound, with the improvement of both anti-protozoal potency and metabolic stability as the primary goal.1
All of the compounds 1a-ab, 4a, 4b, 5, 8, 10, 12, 13, 16, 19, 21a–c, 23 and 28 were evaluated for in vitro activity against four pathogenic protozoa: T. b. rhodesiense (trypomastigote stage, STIB 900 strain), T. cruzi (intracellular amastigote stage, Tulahuen C4 strain), L. donovani (axenic amastigote stage, MHOM-ET-67/L82) and P. falciparum (erythrocytic stage, K1 strain, which is a chloroquine and pyrimethamine resistant strain). The toxicity of the designed compounds was assessed against a mammalian primary cell line derived from rat skeletal myoblasts (L6). The following drugs were used as the reference compounds: melarsoprol (T. b. rhodesiense), benznidazole (T. cruzi), miltefosine (L. donovani), chloroquine (P. falciparum), and podophyllotoxin (L6). The IC50 values presented below indicate the concentration of the tested compound in µg/mL that is necessary to inhibit 50% of the parasite growth. The displayed numbers are the average values collected from two replicate experiments. The selectivity index is the ratio between the IC50 for rat skeletal myoblasts (L6) and the IC50 for the respective protozoan parasite. The screening data are presented in three tables. Table 2 shows compounds bearing various para-substituents on the aromatic B ring (1a-s, 4a, 5, and 8). Table 3 presents compounds with other modification of the B ring (1t-aa, 4b); and Table 4 provides results for the structures with modifications in the linker and the A ring (10–28).
Table 2.
Effect of Variation in the para-Substituent of the B Ring on Biological Activity
| Compound | T. b .rhod. | T. cruzi | L. don. axe. | P. falc. (K1) | L6 | ||||
|---|---|---|---|---|---|---|---|---|---|
| IC50a | SIb | IC50 | SI | IC50 | SI | IC50 | SI | IC50 | |
| 1a | 81 | 0.86 | 4.8 | 15 | 7.4 | 9.3 | 5.6 | 12 | 70 |
| 1b | >100 | <0.46 | 38 | 1.2 | 3.2 | 14 | 1.4 | 33 | 46 |
| 1c | 65 | 0.29 | 2.0 | 9.5 | 1.3 | 14 | 1.50 | 12 | 19 |
| 1d | 61 | 0.038 | 6.0 | 0.38 | 0.14 | 17 | 0.029 | 80 | 2.3 |
| 1e | 87 | >1.2 | 7.7 | >13 | 7.4 | >14 | 3.9 | >26 | >100 |
| 1f | 64 | 0.60 | 51 | 0.75 | 1.6 | 23 | 1.5 | 25 | 38 |
| 1g | 70 | 0.19 | 50 | 0.26 | 0.57 | 23 | 3.8 | 3.4 | 13 |
| 1h | 65 | 1.3 | 72 | 1.2 | 5.3 | 16 | 22 | 3.8 | 84 |
| 1i | >100 | <0.55 | 100 | 0.55 | 12 | 4.7 | 14 | 3.9 | 55 |
| 1j | >100 | 1.0 | 51 | >2.0 | >100 | 1.0 | 38 | 2.7 | >100 |
| 1k | >100 | <0.53 | 56 | 0.94 | 11 | 4.9 | 22 | 2.4 | 53 |
| 1l | >50 | <0.83 | 20 | 2.1 | >50 | <0.83 | 2.9 | 14 | 42 |
| 1m | 50 | 0.78 | 31 | 1.3 | 1.7 | 23 | 1.6 | 24 | 39 |
| 1n | 60 | 0.28 | 4.1 | 4.1 | 0.45 | 37 | 1.0 | 17 | 17 |
| 1o | 55 | 0.23 | 11 | 1.2 | 0.94 | 14 | 0.26 | 50 | 13 |
| 1p | 62 | 1.0 | 4.4 | 15 | 1.5 | 42 | 4.1 | 16 | 64 |
| 1q | 81 | 0.66 | 37 | 1.4 | 18 | 3.0 | 0.49 | 110 | 54 |
| 1r | >50 | <0.16 | 2.8 | 2.9 | 0.91 | 9.1 | 0.005 | 1640 | 8.2 |
| 1s | >50 | 1.0 | >50 | 1.0 | 16 | >3.1 | 0.18 | >272 | >50 |
| 4a | 46 | 0.84 | 40 | 0.98 | 7.8 | 5.0 | 3.9 | 10 | 39 |
| 5 | >50 | 1.0 | >50 | 1.0 | 22 | >2.3 | 21 | >2.4 | >50 |
| 8 | 65 | >1.5 | 49 | >2.1 | >100 | 1.0 | 3.5 | >29 | >100 |
| standard | 0.005c | – | 0.48d | – | 0.19e | – | 0.11f | – | 0.004g |
Values indicate the inhibitory concentration of a compound or standard in µg/mL that is necessary to achieve 50% growth inhibition (IC50). Data shown in the table represent mean values from two independent determinations.
Selectivity index = (IC50 for L6)/(IC50 for the respective values for the four protozoan organisms).
Standard compounds: cmelarsoprol, dbenznidazole, emiltefosine, fchloroquine, gpodophyllotoxin.
Marked field, IC50 < 0.1 µg/mL.
Table 3.
Effect of Other Modifications of the B Ring in N-Benzoyl-2-hydroxybenzamides on Biological Activity
| Compound | T. b. rhod. | T. cruzi | L. don. axe. | P. falc. (K1) | L6 | ||||
|---|---|---|---|---|---|---|---|---|---|
| IC50a | SIb | IC50 | SI | IC50 | SI | IC50 | SI | IC50 | |
| 1a | 81 | 0.86 | 4.8 | 15 | 7.4 | 9.3 | 5.6 | 12 | 70 |
| 1t | 26 | 0.78 | 8.5 | 2.4 | 1.0 | 20 | 0.22 | 94 | 21 |
| 1u | 42 | 1.3 | 5.1 | 11 | 1.4 | 40 | 0.98 | 57 | 56 |
| 1v | 20 | 0.087 | 17 | 0.10 | 0.16 | 11 | 4.4 | 0.40 | 1.8 |
| 1w | >100 | <0.62 | 58 | 1.1 | 11 | 5.5 | 14 | 4.4 | 62 |
| 1x | >50 | <0.47 | 27 | 0.86 | >50 | <0.47 | 4.2 | 5.6 | 24 |
| 1y | 79 | 0.26 | 40 | 0.51 | 3.2 | 6.5 | 0.78 | 26 | 21 |
| 1z | >100 | <0.55 | 16 | 3.4 | 6.0 | 9.2 | 13 | 4.4 | 55 |
| 1aa | 48 | 0.33 | 36 | 0.44 | 2.0 | 7.8 | 4.7 | 3.4 | 16 |
| 4b | 62 | 0.63 | 14 | 2.8 | 1.9 | 21 | 1.1 | 36 | 39 |
| standard | 0.005c | – | 0.48d | – | 0.19e | – | 0.11f | – | 0.004g |
Values indicate the inhibitory concentration of a compound or standard in µg/mL that is necessary to achieve 50% growth inhibition (IC50). Data shown in the table represent mean values from two independent determinations.
Selectivity index = (IC50 for L6)/(IC50 for the respective protozoan parasite).
Standard compounds: cmelarsoprol, dbenznidazole, emiltefosine, fchloroquine, gpodophyllotoxin.
Marked field, 0.1 < IC50 < 1.0 µg/mL.
Table 4.
Effect of Variations in the Linker and A Ring on Biological Activity
| Compound | T. b. rhod. | T. cruzi | L. don. axe. | P. falc. (K1) | L6 | ||||
|---|---|---|---|---|---|---|---|---|---|
| IC50a | SIb | IC50 | SI | IC50 | SI | IC50 | SI | IC50 | |
| 1a | 81 | 0.86 | 4.8 | 15 | 7.4 | 9.3 | 5.6 | 12 | 70 |
| 10 | >100 | <0.094 | 29 | 0.33 | 9.1 | 1.0 | 28 | 0.33 | 9.4 |
| 12 | 17 | 0.28 | 2.2 | 2.2 | 6.2 | 0.77 | 27 | 0.18 | 4.8 |
| 13 | 61 | 0.28 | 10 | 1.7 | 5.3 | 3.3 | 4.2 | 4.1 | 17 |
| 16 | 36 | 2.0 | 74 | 0.95 | 100 | 0.70 | 26 | 2.8 | 70 |
| 19 | 18 | 0.54 | 7.0 | 1.4 | 3.7 | 2.6 | 2.1 | 4.6 | 9.6 |
| 21a | 83 | 0.31 | 8.8 | 3.0 | 1.9 | 13 | 6.9 | 3.7 | 26 |
| 21b | >50 | <0.46 | 28 | <0.81 | >50 | <0.46 | 23 | 1.0 | 23 |
| 21c | 8.2 | 1.0 | 8.9 | 0.93 | 3.0 | 2.8 | 8.5 | 0.96 | 8.2 |
| 23 | 61 | >1.6 | 39 | >2.6 | >100 | >1.0 | 27 | >3.7 | >100 |
| 28 | 35 | 0.44 | 20 | 0.76 | 13 | 1.2 | 12 | 1.3 | 16 |
| standard | 0.005c | – | 0.48d | – | 0.19e | – | 0.11f | – | 0.004g |
Values indicate the inhibitory concentration of a compound or standard in µg/mL that is necessary to achieve 50% growth inhibition (IC50). Data shown in the table represent mean values from two independent determinations.
Selectivity index = (IC50 for L6)/(IC50 for the respective protozoan parasite).
Standard compounds: cmelarsoprol, dbenznidazole, emiltefosine, fchloroquine, gpodophyllotoxin.
Marked field, 1.0 < IC50 < 10 µg/mL.
We were pleased to find that many of the synthesized analogs were more potent against the tested parasites than the hit 1a, showing that various substituents in the para-position of the B ring improved the desired anti-parasitic activity (Table 2). Compounds 1n, 1o, and 1r bearing ethoxy, n-butoxy, and N,N-diethylamino substituents, respectively, showed reasonable activity against T. cruzi, good activity against L. donovani, and very good activity against P. falciparum (K1 strain). Compound 1r exhibited the best IC50 value against P. falciparum (K1 strain) among all the tested compounds. The simple replacement of ethyl in 1a by tert-butyl in 1d led to good and very good activity against L. donovani and P. falciparum (K1 strain), respectively. The potency of compound 1d against L. donovani was comparable to that of miltefosine. Compounds bearing other alkyl substituents (ethyl 1a, isopropyl 1c) or aromatic groups (phenyl 1e, benzyl 1p) displayed similar potency (within micromolar range) against T. cruzi, L. donovani, and P. falciparum (K1 strain). Compounds with methyl (1b), fluorine (1f), methoxy (1m), and amino (4a) groups also had similar activity as compounds 1a,c,e,p, however, only against L. donovani, and P. falciparum (K1 strain). Compound 1q possessing a para-N,N-dimethylamino substitutent turned out to be not only potent, but very selective against P. falciparum (K1 strain). It was unexpected for us to see such dramatic change in the selectivity for compounds 1q,r and 4a by introducing such minor structural modifications. The morpholinyl-substituted compound 1s showed reasonable activity and selectivity against malaria parasites. To our surprise, compound 8 bearing the octanedioic acid amide hydroxyamide side chain was modestly active only against P. falciparum (K1 strain). The benzoic acid methyl ester analogue 1l gave similar results. In contrast to these results, compound 5 containing a succinamic acid methyl ester side chain was inactive against all species. On the other hand, the analog 1g containing a trifluoromethyl group is the only compound in this series that is potent and at the same time reasonably selective against L. donovani. Compound 1h bearing a trifluoromethoxy group also appeared to be selective against L. donovani, though significantly less potent (around 10-fold) than compound 1g. The methanesulfonyl group in compound 1j caused complete loss of inhibitory activity against all tested parasites. Against T. cruzi, modest inhibitory activity was displayed by several compounds already mentioned above (1a,c–e,n,p,r). Unfortunately, none of the compounds presented in Table 2 were active against T. b. rhodesiense.
Summarizing the SAR data shown in Table 2, we observe a clear relationship between structural modifications at the para position of the B ring and anti-leishmanial and P. falciparum (K1 strain) activity. Some of the most active compounds against P. falciparum (K1 strain) are those with bulky alkyl substituents such as tert-butyl (1d). Reduction of the substituent size to ethyl (1a), methyl (1b), or even isopropyl (1c) resulted in decreased potency. This finding would suggest the presence of a large hydrophobic space in the binding pocket able to accommodate branched substituents in the area contiguous to the para position of the B ring. In comparison with the preferred tert-butyl group, a phenyl substituent (compound 1e) causes reduction in activity, therefore suggesting the preference for an alkyl group in the binding site. Anti-plasmodial potency is enhanced by introduction of bulky and strongly electron-donating N or O substituents. Compound 1r with N,N-diethylamino substitution turns out to be the most active and selective in this series against malaria parasites. Its N,N-dimethylamino analog 1q and the morpholinyl-substituted compound 1s both exhibit good potency and moderate selectivity against the same parasite. The high potency of these compounds not only reiterates the significance of hydrophobic interactions involving lipophilic partial structures of the 4-substituents but also points to potential dipolar interactions or H-bonding between the ligand and the binding pocket. The aniline 4a, which lacks the ability to engage in hydrophobic interactions via its 4-substituent, is far less active against P. falciparum (K1 strain) than compounds bearing a dialkylated nitrogen atom (1q,r,s). Potency enhancement by large, aliphatic, heteroatom-containing groups is also apparent among alkoxy-substituted compounds. High potency is observed for compounds 1n and 1o with ethoxy and n-butoxy substituents, respectively. However, the relatively small trifluoromethoxy (compound 1h) and methoxy (compound 1m) groups along with the large, aromatic benzyloxy substituent (compound 1p) are less potent in comparison with 1n,o. Enhanced potency is also observed for analogs bearing small electron-withdrawing groups such as fluoro (compound 1f) and trifluoromethyl (compound 1g). Other electron-withdrawing groups such as cyano (compound 1i), methanesulfonyl (compound 1j), and nitro (compound 1k) lead to loss of potency; as an exception, the methoxycarbonyl-substituted compound 1l maintains fair potency against P. falciparum (K1 strain). The modest activity of compound 8 bearing the octanedioic acid amide hydroxyamide moiety in comparison with the inactive compound 5 that contains a succinamic acid methyl ester side chain would suggest an alternative mode of action for the former compound against P. falciparum (K1 strain). Based on these results, we can characterize the binding site for N-benzoyl-2-hydroxybenzamides para-substituted in their B ring as a fairly large hydrophobic site with preference for branched alkyl substituents as well as strongly electron-donating groups, such as dialkylamino and alkoxy, that are able to accept H-bonds. All in all, the excellent potency of compound 1r along with its negligible cytotoxicity against the L-6 cell line (selectivity index, SI = 1640) make this compound a promising lead for the further development of anti-malarial drugs. This compound has been evaluated for its ADMET properties elsewhere1, which turned out to be more favorable than those of the hit 1a.1 Additionally, compounds 1q and 1s have reasonable selectivity index values (110 and 272, respectively) and can thus also be regarded as attractive leads.
Similar trends in the SAR were noticed for L. donovani, suggesting that this protozoan possesses a homologous molecular target for this series of compounds. The most active compounds were the tert-butyl analog 1d, the trifluoromethyl analog 1g, the ethoxy and n-butoxy compounds 1n and 1o, as well as the N,N-dimethylamino compound 1r. Decreased steric bulk of the substituents resulted in a decrease (compounds 1a,b,c,h,m), or complete loss (compound 1q) of potency. Reduced activity was also observed for compounds 1e,p containing aromatic substituents and for the morpholinyl derivative 1s. This would suggest a strict cutoff for the size of the para substituent at the L. donovani binding site. Complete loss of activity was observed for the compounds bearing large and strongly electron-withdrawing groups (1i,j,k,l), or bearing long polar chains (5 and 8). The L. donovani binding site tolerates small electron-withdrawing groups (compounds 1f,g) as well as or better than that of P. falciparum (K1 strain). This result may assist in designing further structural modifications aimed at providing potent and selective compounds against L. donovani.
In analogy to the SAR for P. falciparum (K1 strain) and L. donovani, the most active compounds against T. cruzi were analogs with branched and electron-donating substituents (1a,c,d,n and r). However, all of them were less potent against this protozoan than against P. falciparum (K1 strain) and L. donovani. On the other hand, compounds bearing electron-withdrawing groups resulted in the complete loss of anti-parasitic activity (1f,g,i–l). Surprisingly, the aromatic substituents in 1e and 1p retained moderate activity against T. cruzi, indicating possible π-π interaction in the binding pocket. None of the preceding compounds turned out to be active against T. b. rhodesiense.
In the final series of compounds, we probed the influence of other modifications of the B ring. Encouragingly, we observed an improvement in the anti-parasitic activity for some of the synthesized compounds (Table 3). The 2-naphthyl analog 1t, the 6-methoxy-2-naphthyl analog 1u, and the 2,3,4-trimethoxyphenyl analog 1y were the most active against P. falciparum (K1 strain). Compound 1t showed activity at low concentration (only 2-fold less than the standard) against malaria parasites. Also compounds 1u and 1y displayed potency within the same range as 1t against the same protozoan. We also observed modest potency of these compounds (1t,u,y) against L. donovani. Compound 1aa with 2-methyl-3-furyl as the B ring, and the symmetric analog 4a showed dual activity against malaria and leishmania parasites. However, both were less active than 1t,u,y. The piperonyl-substituted compound 1x turned out to be weakly active only against P. falciparum (K1 strain) and thus is somewhat selective against this protozoan. Compounds with 2,4-dichlorophenyl (1w) or cyclohexyl (1z) as their B rings were inactive against P. falciparum (K1 strain).
Many of the analogs presented in Table 3 showed moderate (corresponding to micromolar) activity against L. donovani, and the 3-chloromethyl analog 1v even had activity slightly better than the anti-leishmanial drug miltefosine. However, the selectivity index of compound 1v was only 11, which makes this compound less viable for further development. Analogs 1z with a cyclohexyl B ring and 1aa with a 2-methyl-3-furyl B ring turned out to be moderately active only against L. donovani. Compounds 1w and 1x were completely inactive against leishmania parasites. The fact that compounds with B rings other than benzene still retained moderate activity against L. donovani when compared to other protozoa provides valuable guidance for the design of further potential anti-leishmanial agents. Besides the hit compound 1a, the naphthyl derivatives 1t and 1u were the only compounds moderately active against T. cruzi. In accord with the previous observations, none of these compounds were active against T. b. rhodesiense parasites.
Concluding the SAR for 2-hydroxybenzamides presented in Table 3, we can state that the identity of the B ring as benzene seems to be important for the anti-P. falciparum (K1 strain) activity as presented by 1t,u,v,y. However, cyclohexane (compound 1z), and furan (compound 1aa) substitutes retain moderate anti-leishmanial activity. Free phenol functionality (compound 4b) is tolerated but alkoxy substituents (compounds 1x, y) provide a better contribution to the compounds’ anti-malarial activity. Although significantly cytotoxic, the (chloromethyl)phenyl analog 1v provided excellent potency against L. donovani, whereas 2,4-dichlorophenyl (compound 1w) gave a completely inactive analog. These conclusions are in good agreement with the initial SAR described for the para position of N-benzoyl-2-hydroxybenzamides (Table 2), and confirm the importance of the aromatic nature of the ligand’s B ring, preferably bearing electron-donating groups attached through a heteroatom.
At the same time we also prepared analogs with modified linker and A ring moieties. A series of 10 compounds was prepared and screened for anti-leishamania,-trypanosome and plasmodium (K1) activity (Table 4). We found that most of the modifications caused a decrease in anti-P. falciparum (K1 strain) activity. Only the sulfur analog 13 and urea-linked compound 19 were slightly more potent against malaria parasites than the hit compound 1a.1 On the other hand, the benzoxazinanedione 21a and indazolylbenzamide 21c were slightly less active against the same parasites than the hit 1a. Although anti-P. falciparum (K1 strain) activity was retained by compounds 13, 19, and 21a,c, their small selectivity indices suggest safety problems. The above observations support the notion that the H-bond donor-acceptor pattern in the imide linker is crucial for anti-parasitic activity. This was additionally supported by methylation of the nitrogen atom in the imide linker, which gave the inactive compound 10.
Pleasingly, the present set of structural modifications provided some compounds with improved potency against L. donovani. Good anti-leishmanial activity was shown by compounds which had already proven activity against P. falciparum (K1 strain) (13, 19, and 21a,c). In addition to these compounds, the N-methylated compound 10 and the acylated salicylhydrazide 12 showed modest potency against L. donovani. As in the case of compounds active against malaria parasites, all the compounds active against leishmania parasites suffered from lack of selectivity (SI < 5). Four compounds, the acylated salicylhydrazide 12, the urea-linked compound 19, the benzoxazinanedione 21a, and the indazolylbenzamide 21c, were only moderately active against T. cruzi. The only compound, which inhibited at moderate concentration the growth of T. b. rhodesiense was 21c. Despite its moderate activity this compound was 10-fold more potent than the parent compound 1a. Contrary to this, compounds which had chemically stable linkers, i.e., the acylated urea 16, indolylbenzamide 21b, diphenyloxadiazole 23, and bis-amide 28, turned out to be inactive across all four parasites. Summarizing the SAR for this series of compounds, we can state that the imide linker with an unsubstituted N atom is crucial for anti-malarial activity. However, we were pleased to see that compounds bearing other linker/A ring combinations, such as the urea 19, the benzoxazinanedione 21a, and the indazolylbenzamide 21c, retained moderate anti-leishmanial activity.
Conclusions
This research provides solid ground for the discovery of new and effective agents to treat protozoan infections that cause malaria and leishmaniasis. The hit compound 1a was discovered by initial HTS screening against T. gondi,1 and the preliminary ADMET results, while encouraging, revealed this agent to be metabolically unstable in HLM.1 The metabolic instability is ascribed to the imide linker, and therefore extensive modifications of this moiety as well as the B-ring were pursued.1 Several promising lead compounds active against the K1 strain of P. falciparum (K1 strain) and L. donovani were identified. Among the tested parasites P. falciparum (K1 strain) was the most sensitive to these N-benzoyl-2-hydroxybenzamides. Compound 1r is 21-fold more potent against the K1 isolate of malaria than the standard drug, chloroquine. Moreover, it exhibits an SI value of 1640, suggesting both high selectivity and low toxicity for this compound. The SAR for T. gondii reported elsewhere is similar to that described for P. falciparum K1 isolate.1 Efficacy of compound 1a was found to be much lower for other malaria isolates and 1r was also found less effective in different types of assays1 for other P. falciparum or other plasmodial isolates.
Additionally, the electron-donating diethylamino group in compound 1r stabilizes the imide linker, thus improving the chemical and metabolic stability of this analog. With its excellent anti-plasmodial potency, this highly selective agent may be used as a possible lead for the further development of anti-malarial agents. Although analog 1g is 4 times less potent than 1d, it shows better selectivity against L. donovani parasites than malaria, therefore both compounds are attractive leads for further research including assessment of activity in infected macrophages. Compound 1v, with a 3-chloromethyl substituent on the B ring, might also be regarded as an interesting compound for the development of anti-leishmanial agents. Moreover, the fact that the L. donovani binding site better tolerates small electron-withdrawing groups provides a good starting point for the design of more potent and selective agents to combat this protozoan. Compounds 1c and 12 are the most potent members of this series against T. cruzi. Because of its hydrolytic stability the latter compound can be considered an attractive scaffold for further work. T. b. rhodesiense was generally resistant to the compounds used in this study, and compound 21c was the only one showing weak activity against this parasite. Studies to determine IC90s and whether the compounds are cidal will be important in the future. While the data generated in this research may be useful in the design and synthesis of novel agents with improved potencies and safety margins to treat infections caused by L. donovani and P. falciparum (K1 strain), we call attention to the fact that it will be essential to study the potential teratogenicity of these imides, as they do bear some structural relationship to teratogen thalidomide.
Experimental Section
Antiprotozoal Assays
The in vitro activities against T. b. rhodesiense, T. cruzi, L. donovani, and P. falciparum (K1 strain) were determined as described earlier.19 The following strains and parasite forms were used: T. b. rhodesiense, STIB900, trypomastigote form; T. cruzi, Tulahuen C2C4, amastigote form in L-6 rat myoblasts; L. donovani, MHOM/ET/67/L82, axenic amastigote form; P. falciparum, K1 (chloroquine and pyrimethamine resistant) erythrocytic stage.
Chemistry
All chemicals and solvents were purchased from Sigma-Aldrich and/or Fisher Scientific, and were used without further purification. Anhydrous THF and CH2Cl2 were obtained by distillation over sodium wire or CaH2, respectively. All non-aqueous reactions were carried out under argon atmosphere with exclusion of moisture from reagents, and all reaction vessels were oven-dried. 1H NMR and 13C NMR spectra were recorded on Bruker spectrometer at 400 MHz and 100 MHz, respectively, and were referenced to the residual peaks of CHCl3 at 7.26 ppm or DMSO-d6 at 2.50 ppm (1H NMR), and CDCl3 at 77.23 ppm or DMSO-d6 at 39.51 ppm (13C NMR). Chemical shifts were reported in parts per million downfield of TMS and the following abbreviations used to denote coupling patterns: s=singlet; d=doublet, t=triplet, q=quartet, br=broad, fs=fine splitting). Mass spectra were measured in the ESI mode at an ionization potential of 70 eV with an LC-MS MSD (Hewlett Packard). HRMS experiments were performed on Q-TOF-2TM (Micromass). TLC was performed on Merck 60 F254 silica gel plates. Column chromatography was performed using Merck silica gel (40–60 mesh). Preparative HPLC was carried out on an ACE 5 AQ column (150 × 20 mm), with detection at 254 and 280 nm on a Shimadzu SPD-10A VP detector; flow rate = 17 mL/min; gradient from 8% to 100% methanol in water (both solvents containing 0.05 vol% of CF3COOH). Purities of final compounds were established by analytical HPLC, which was carried out on an Agilent 1100 HPLC system with a Synergi 4 μ Hydro-RP 80A column, with detection at 254 nm on a variable wavelength detector G1314A; flow rate = 1.4 mL/min; gradient from 10% to 100% methanol in water (both solvents containing 0.05 vol% of CF3COOH) within 20 min. All the final compounds were of purity ≥95% as determined by the method described above.
General Procedure for the Synthesis of Imide Compounds
To a solution of amide (5 mmol) in pyridine (6 mL) was added an acyl chloride (5 mmol). After refluxing for 4 h, the resulting mixture was diluted with Et2O (30 mL)/H2O (20 mL), or H2O (20 mL)/1.0M HCl (5 mL). The resulting precipitate was collected by filtration, washed with water, and dried; or the resulting mixture was diluted with EtOAc, washed with H2O (20 mL) and brine (20 mL), dried over Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel or recrystallization from MeOH. Non-commercial acyl chlorides were obtained from the corresponding carboxylic acid by the following procedure: to a solution of acid (5 mmol) in anhydrous CH2Cl2 (6 mL) was added (COCl)2 (6 mmol) and 1 drop of DMF at 0 °C. The resulting mixture was allowed to warm to room temperature and stirred for 2 h. After evaporation of the solvent, the residue was used without purification in the next step. Alternatively, the acid was dissolved in SOCl2 and heated at 80 °C for 1.5 – 3 h, before cooling to room temperature and evaporating the volatiles in vacuo. The residue was used in the next step without further purification.
Hydrogenation reactions were carried out with 10% Pd/C (10–15 wt. %) and a H2 balloon in MeOH as the solvent for 18 h at room temperature.
N-(4-Ethylbenzoyl)-2-hydroxybenzamide (1a)
The general procedure was used with salicylamide (2) and 4-ethylbenzoyl chloride (3a) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (51%). 1H NMR (DMSO-d6) δ 11.70 (s, 1H), 11.60 (s, 1H), 7.72–7.88 (m, 3H), 7.28–7.42 (m, 3H), 6.88–6.98 (m, 2H), 2.62 (q, J = 8.0 Hz, 2H), 1.13 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ 164.8, 164.2, 156.4, 149.4, 134.3, 131.3, 131.1, 128.4, 127.9, 120.0, 119.1, 117.1, 28.8, 15.3; HPLC purity 99.4%; MS (ESI) m/z 270 (M + H)+; HRMS (ESI) calcd for C16H16NO3 270.1125 (M + H)+, found 270.1131.
2-Hydroxy-N-(4-methylbenzoyl)benzamide (1b)
The general procedure was used with salicylamide (2) and 4-methylbenzoyl chloride (3b) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (85%). 1H NMR (DMSO-d6) δ 11.88 (s, 1H), 11.77 (s, 1H), 7.88 (dd, J = 8.0, 2.0 Hz, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.46 (dt, J = 8.0, 2.0 Hz, 1H), 7.37 (d, J = 8.0 Hz, 2H), 6.96–7.08 (m, 2H), 2.36 (s, 3H); 13C NMR (DMSO-d6) δ 164.7, 164.1, 156.4, 143.3, 134.3, 131.1, 131.0, 129.5, 127.8, 120.0, 119.1, 117.1, 99.6, 21.1; HPLC purity 97.6%; HRMS (ESI) calcd for C15H14NO3 256.0968 (M + H)+, found 256.0978.
2-Hydroxy-N-(4-isopropylbenzoyl)benzamide (1c)
The general procedure was used with salicylamide (2) and 4-isopropylbenzoyl chloride (3c) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (22% over two steps). 1H NMR (CD3OD/CDCl3) δ 8.09 (d, J = 8.0 Hz, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.46 (t, J = 8.0 Hz, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.00 (m, 2H), 3.00 (q, J = 6.8 Hz, 1H), 1.28 (d, J = 6.8 Hz, 6H); 13C NMR (CD3OD/CDCl3) δ 167.1, 166.2, 158.0, 156.1, 136.0, 132.8, 132.3, 128.9, 128.1, 121.6, 119.3, 117.9, 35.4, 24.0; HPLC purity 97.6%; MS (ESI) m/z 284 (M + H)+.
N-(4-tert-Butylbenzoyl)-2-hydroxybenzamide (1d)
The general procedure was used with salicylamide (2) and 4-tert-butylbenzoyl chloride (3d) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (95%). 1H NMR (DMSO-d6) δ 11.94 (s, 1H), 11.91 (s, 1H), 7.84–7.92 (m, 3H), 7.57 (d, J = 8.0 Hz, 2H), 7.43 (dt, J = 8.0, 2.0 Hz, 1H), 6.94–7.05 (m, 2H), 1.31 (s, 9H); 13C NMR (DMSO-d6) δ 164.8, 164.2, 156.4, 156.0, 134.3, 131.1, 127.7, 125.8, 120.0, 119.1, 117.1, 34.9, 30.9; HPLC purity 98.0%; HRMS calcd for C18H19NO3 297.1365 (M)+, found 297.1369.
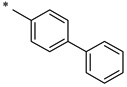
N-(2-Hydroxybenzoyl)-[1,1'-biphenyl]-4-carboxamide (1e)
The general procedure was used with salicylamide (2) and biphenyl-4-carbonyl chloride (3e) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (54% over two steps). 1H NMR (DMSO-d6) δ 11.8 (s, 1H), 11.7 (s, 1H), 8.02 (d, J = 4.0 Hz, 2H), 7.89 (t, J = 8.0 Hz, 3H), 7.77 (d, J = 8.0 Hz, 2H), 7.49 (m, 4H), 7.01 (q, J = 8.0 Hz, 2H); 13C NMR (DMSO-d6) δ 164.6, 164.3, 156.4, 144.4, 138.8, 134.2, 132.5, 131.0, 129.1, 128.4, 127.1, 127.0, 120.0, 119.1, 117.0, 99.5; HPLC purity 98.9%; MS (ESI) m/z 318 (M + H)+.
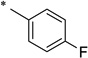
N-(4-Fluorobenzoyl)-2-hydroxybenzamide (1f)
The general procedure was used with salicylamide (2) and 4-fluorobenzoyl chloride (3f) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (17%). 1H NMR (DMSO-d6) δ 11.82 (s, 2H), 7.90–8.05 (m, 2H), 7.84 (dd, J = 7.6, 2.0 Hz, 1H), 7.36–7.48 (m, 3H), 6.94–7.02 (m, 2H); 13C NMR (DMSO-d6) δ 166.0, 164.3 (d, J = 172 Hz), 163.6, 156.4, 134.3, 131.0, 130.7 (d, J = 38 Hz), 130.3 (d, J = 12 Hz),, 119.9, 119.2, 117.1, 116.1, 115.9; HPLC purity 96.0%; HRMS (ESI) calcd for C14H11FNO3 260.0717 (M + H)+, found 260.0720.
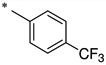
2-Hydroxy-N-(4-(trifluoromethyl)benzoyl)benzamide (1g)
The general procedure was used with salicylamide (2) and 4-(trifluoromethyl)benzoyl chloride (3g) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (29%). 1H NMR (DMSO-d6) δ 11.64 (s, 1H), 11.46 (s, 1H), 8.09 (d, J = 8.0 Hz, 2H), 7.91 (d, J = 8.0 Hz, 2H), 7.84 (dd, J = 8.0, 2.0 Hz, 1H), 7.46 (dt, J = 8.0, 2.0 Hz, 1H), 6.90–7.04 (m, 2H), 3.85 (s, 3H); 13C NMR (DMSO-d6) δ 164.8, 164.6, 156.6, 137.8, 134.4, 132.4 (q, J = 32 Hz), 131.0, 129.2 (2C), 125.8 (q, J = 4 Hz), 123.5 (q, J = 270 Hz), 119.9, 119.0, 117.1; HPLC purity 98.0%; HRMS (ESI) calcd for C15H11F3NO3 310.0685 (M + H)+, found 310.0689.
2-Hydroxy-N-(4-(trifluoromethoxy)benzoyl)benzamide (1h)
The general procedure was used with salicylamide (2) and 4-(trifluoromethoxy)benzoyl chloride (3h) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (38%). 1H NMR (DMSO-d6) δ 11.7 (br s, 2H), 8.04 (d, J = 8.0 Hz, 2H), 7.85 (dd, J = 8.0, 4.0 Hz, 1H), 7.56 (d, J = 8.0 Hz, 2H), 7.46 (t, J = 8.0 Hz, 1H), 7.01 (m, 2H); 13C NMR (DMSO-d6) δ 164.7, 164.3, 156.5, 151.2 (q, J = 1.8 Hz), 134.3, 133.0, 131.0, 130.3 (2C), 121.1 (2C), 119.9 (q, J = 258 Hz), 119.8, 119.2, 117.1; HPLC purity 99.0%; MS (ESI) m/z 326 (M + H)+.
N-(4-Cyanobenzoyl)-2-hydroxybenzamide (1i)
The general procedure was used with salicylamide (2) and 4-cyanobenzoyl chloride (3i) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (91%). 1H NMR (DMSO) δ 11.73 (s, 2H), 8.04 (app s, 4H), 7.84 (dd, J = 7.6, 2.0 Hz, 1H), 7.46 (dt, J = 7.6, 2.0 Hz, 1H), 6.95–7.05 (m, 2H), 2.36 (s, 3H); 13C NMR (DMSO) δ 164.7, 164.4, 156.6, 137.9, 134.4, 132.6, 131.0, 128.5, 119.9, 119.0, 118.4, 117.5, 114.9; HPLC purity 96.4%; HRMS (ESI) calcd for C15H9N2O3 265.0619 (M − H)+, found 265.0629.
2-Hydroxy-N-[4-(methylsulfonyl)benzoyl]benzamide (1j)
The general procedure was used with salicylamide (2) and 4-(methylsulphonyl)benzoyl chloride (3j, prepared from the corresponding carboxylic acid) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a pale pink solid (90 mg, 14%). 1H NMR (DMSO-d6) δ 11.73 (br s, 1H), 11.61 (br s, 1H), 8.14–8.09 (m, 4H), 7.84 (dd, J = 7.9, 1.7 Hz, 1H), 7.47 (ddd, J = 8.5, 7.3, 1.8 Hz, 1H), 7.03–6.98 (m, 2H), 3.31 (s, 3H); 13C NMR (DMSO-d6) δ 164.8, 164.6, 156.5, 144.0, 138.4, 134.3, 130.9, 128.8, 127.4, 119.8, 119.0, 117.0, 43.2; HPLC purity 99.7%; MS (ESI) m/z 320 (M + H)+.
2-Hydroxy-N-(4-nitrobenzoyl)benzamide (1k)
The general procedure was used with salicylamide (2) and 4-nitrobenzoyl chloride (3k) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (42%). 1H NMR (DMSO-d6) δ 11.8 (s, 1H), 11.7 (br s, 1H), 8.36 (d, J = 8.0 Hz, 2H), 8.11 (d, J = 8.0 Hz, 2H), 7.87 (d, J = 8.0 Hz, 1H), 7.44 (t, J = 8.0 Hz, 1H), 6.99 (m, 2H); 13C NMR (DMSO-d6) δ 164.5, 164.1, 156.6, 149.6, 139.4, 134.3, 130.9, 129.1, 123.7, 119.7, 118.5, 117.0; HPLC purity 99.8%; MS (ESI) m/z 287 (M + H)+.
Methyl 4-[(2-hydroxybenzoyl)carbamoyl]benzoate (1l)
The general procedure was used with salicylamide (2) and 4-(chlorocarbonyl)benzoic acid methyl ester (3l, prepared from the corresponding carboxylic acid) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (200 mg, 33%). 1H NMR (DMSO-d6) δ 11.75 (br s, 2H), 8.11 (d, J = 8.3 Hz, 2H), 8.03 (d, J = 8.3 Hz, 2H), 7.86 (dd, J = 7.7, 1.1 Hz, 1H), 7.46, (td, J = 7.5, 1.4 Hz, 1H), 7.04–6.97 (m, 2H), 3.90 (s, 3H); 13C NMR (DMSO-d6) δ 165.5, 164.6, 164.5, 137.8, 134.3, 133.0, 131.0, 129.5, 128.1, 119.9, 119.0, 117.0, 52.5; HPLC purity 99.5%; MS (ESI) m/z 300 (M + H)+.
2-Hydroxy-N-(4-methoxybenzoyl)benzamide (1m)
The general procedure was used with salicylamide (2) and 4-methoxybenzoyl chloride (3m) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (37%). 1H NMR (DMSO-d6) δ 11.81 (s, 1H), 11.72 (s, 1H), 7.80–7.91 (m, 3H), 7.46 (dt, J = 8.4, 2.0 Hz, 1H), 7.09 (d, J = 8.4 Hz, 2H) 6.90–7.05 (m, 2H), 3.85 (s, 3H); 13C NMR (DMSO-d6) δ 164.3, 163.0, 156.4, 134.2, 131.1, 129.2, 125.9, 119.9, 119.2, 117.1, 114.2, 99.6, 55.7; HPLC purity 95.0%, HRMS (ESI) calcd for C15H14NO4 272.0917 (M + H)+, found 272.0927.
N-(4-Ethoxybenzoyl)-2-hydroxybenzamide (1n)
The general procedure was used with salicylamide (2) and 4-ethoxybenzoyl chloride (3n) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (77%). 1H NMR (DMSO-d6) δ 11.87 (s, 1H), 11.56 (s, 1H), 7.90–8.05 (m, 3H), 7.46 (dt, J = 8.4, 2.0 Hz, 1H), 6.93–7.05 (m, 4H), 4.13 (d, J = 4.0 Hz, 2H), 1.30 (t, J = 4.0 Hz, 3H); 13C NMR (DMSO-d6) δ 164.2, 162.3, 156.4, 134.2, 131.1, 129.9, 125.6, 120.0, 119.2, 117.0, 114.6, 63.7, 14.6; HPLC purity 99.6%; HRMS (ESI) calcd for C16H14NO4 284.0928 (M − H)+, found 284.0936.
N-(4-Butoxybenzoyl)-2-hydroxybenzamide (1o)
The general procedure was used with salicylamide (2) and 4-butoxybenzoyl chloride (3o) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (62%). 1H NMR (CDCl3) δ 8.12 (d, J = 8.4 Hz, 2H), 7.90 (m, 1H), 7.50 (m, 1H), 7.32 (m, 1H), 7.20 (d, J = 8.0 Hz, 1H), 6.97 (d, J = 8.4 Hz, 2H), 6.48 (br, 1H), 6.33 (br, 1H), 4.05 (d, J = 6.4 Hz, 2H), 1.81 (m, 2H), 1.52 (m, 2H), 0.99 (d, J = 7.2 Hz, 3H); 13C NMR (CDCl3) δ 167.6, 164.8, 164.2, 148.6, 132.6, 132.4, 130.7, 127.7, 126.4, 123.5, 120.8, 114.8, 68.3, 31.2, 19.3, 14.0; HPLC purity 99.8%; MS (ESI) m/z 314 (M + H)+.
N-[4-(Benzyloxy)benzoyl]-2-hydroxybenzamide (1p)
The general procedure was used with salicylamide (2) and 4-(benzyloxy)benzoyl chloride (3p) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (39%). 1H NMR (DMSO-d6) δ 11.7 (br s, 1H), 11.6 (br s, 1H), 7.87 (m, 3H), 7.41 (m, 6H), 7.17 (d, J = 8.0 Hz, 2H), 7.00 (m, 2H), 5.22 (s, 2H); 13C NMR (DMSO-d6) δ 164.2, 164.1, 162.0, 156.3, 136.5, 134.2, 131.0, 129.8, 128.5, 128.1, 127.8, 126.0, 119.9, 119.2, 117.0, 115.0, 69.6; HPLC purity 98.3%; MS (ESI) m/z 348 (M + H)+.
N-[4-(Dimethylamino)benzoyl]-2-hydroxybenzamide (1q)
The general procedure was used with salicylamide (2) and 4-(dimethylamino)benzoyl chloride (3q) as reactants. Purification of the crude material by preparative HPLC gave the title compound as yellow solid (31%). 1H NMR (DMSO-d6) δ 11.8 (s, 1H), 11.5 (s, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 8.0 Hz, 2H), 7.45 (t, J = 8.0 Hz, 1H), 7.02 (m, 2H), 6.77 (d, J = 8.0 Hz, 2H), 3.03 (s, 6H); 13C NMR (DMSO-d6) δ 164.6, 164.4, 156.7, 153.6, 134.5, 131.5, 130.0, 120.3, 120.0, 119.6, 117.5, 111.5, 40.8; HPLC purity 98.3%; MS (ESI) m/z 285 (M + H)+.
N-[4-(Diethylamino)benzoyl]-2-hydroxybenzamide (1r)
The general procedure was used with salicylamide (2) and 4-(diethylamino)benzoyl chloride (3r, prepared from the corresponding carboxylic acid) as reactants. Purification of the crude material by preparative HPLC gave the title compound as yellow solid (8% over two steps). 1H NMR (DMSO-d6) δ 11.8 (br s, 1H), 11.5 (s, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.45 (t, J = 8.0 Hz, 1H), 7.00 (m, 2H), 6.73 (d, J = 8.0 Hz, 2H), 3.41 (q, J = 8.0 Hz, 4H), 1.12 (t, J = 8.0 Hz, 6H); 13C NMR (DMSO-d6) δ 164.6, 164.4, 156.8, 151.2, 134.5, 131.5, 130.2, 120.3, 119.6, 119.0, 117.5, 110.9, 44.3, 12.7; HPLC purity 98.5%; MS (ESI) m/z 313 (M + H)+.
2-Hydroxy-N-(4-morpholinobenzoyl)benzamide (1s)
The general procedure was used with salicylamide (2) and 4-morpholinobenzoyl chloride (3s, prepared from the corresponding carboxylic acid) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (25% over two steps). 1H NMR (DMSO-d6) δ 11.8 (br s, 1H), 11.5 (s, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 8.0 Hz, 2H), 7.46 (t, J = 8.0 Hz, 1H), 7.00 (m, 4H), 3.75 (t, J = 4.0 Hz, 4H), 3.30 (t, J = 4.0 Hz, 4H); 13C NMR (DMSO-d6) δ 164.8, 164.5, 156.7, 154.4, 134.5, 131.5, 129.8, 122.8, 120.4, 119.6, 117.5, 113.8, 66.3, 47.3; HPLC purity 98%; MS (ESI) m/z 327 (M + H)+.
N-(2-Hydroxybenzoyl)-2-naphthamide (1t)
The general procedure was used with salicylamide (2) and naphthalene-2-carbonyl chloride (3t, prepared from the corresponding carboxylic acid) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (170 mg, 29% over two steps). 1H NMR (DMSO-d6) δ 11.89 (br s, 2H), 8.58 (s, 1H), 8.10 (m, 2H), 8.04 (d, J = 7.6 Hz, 1H), 7.96 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.71–7.64 (m, 2H), 7.47 (t, J = 7.5 Hz, 1H), 7.06 (d, J = 8.1 Hz, 1H), 7.01 (d, J = 7.5 Hz, 1H); 13C NMR (DMSO-d6) δ 165.1, 164.4, 156.6, 134.8, 134.2, 132.1, 131.1, 131.0, 129.2, 128.7, 128.6, 128.5, 127.7, 127.2, 123.8, 119.8, 119.1, 117.1; HPLC purity 100%; MS (ESI) m/z 292 (M + H)+.
N-(2-Hydroxybenzoyl)-6-methoxy-2-naphthamide (1u)
The general procedure was used with salicylamide (2) and 6-methoxynaphthalene-2-carbonyl chloride (3u, prepared from the corresponding carboxylic acid) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a pale yellow solid (180 mg, 28% over two steps). 1H NMR (DMSO-d6) δ 11.79 (s, 1H), 11.76 (s, 1H), 8.51 (s, 1H), 8.02 (d, J = 8.8 Hz, 1H), 7.97 (d, J = 8.8 Hz, 1H), 7.92 (dd, J = 3.4, 1.8 Hz, 1H), 7.90 (m, 1H), 7.50–7.45 (m, 2H), 7.29 (dd, J = 8.9, 2.5 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 7.01 (t, J = 7.7 Hz, 1H), 3.93 (s, 3H); 13C NMR (DMSO-d6) δ 165.0, 164.4, 159.3, 156.4, 136.7, 134.2, 131.1, 130.9, 128.6, 127.4, 127.3, 124.4, 119.9, 119.8, 119.2, 117.1, 106.0, 55.5; HPLC purity 97.2%; MS (ESI) m/z 322 (M + H)+.
N-[3-(Chloromethyl)benzoyl]-2-hydroxybenzamide (1v)
The general procedure was used with salicylamide (2) and 3-(chloromethyl)benzoyl chloride (3v) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (24%). 1H NMR (DMSO-d6) δ 11.85 (s, 2H), 7.99 (s, 1H), 7.84–7.90 (m, 2H), 7.73 (d, J = 7.6 Hz, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.46 (dt, J = 7.6, 1.6 Hz, 1H), 6.90–7.02 (m, 2H), 4.87 (s, 2H); 13C NMR (DMSO-d6) δ 164.7, 164.4, 156.5, 138.7, 134.3, 134.2, 133.2, 131.1, 129.3, 128.3, 127.5, 120.0, 119.1, 117.1, 45.5; HPLC purity 96.2%; HRMS (ESI) calcd for C15H13ClNO3 290.0578 (M + H)+, found 290.0584.
2,4-Dichloro-N-(2-hydroxybenzoyl)benzamide (1w)
The general procedure was used with salicylamide (2) and 2,4-dichlorobenzoyl chloride (3w) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (41%). 1H NMR (DMSO-d6) δ 11.60 (s, 2H), 7.68–7.82 (m, 2H), 7.60 (d, J = 7.6 Hz, 1H), 7.54 (dd, J = 7.6, 2.0 Hz, 1H), 7.47 (dt, J = 7.6, 2.0 Hz, 1H), 6.90–7.02 (m, 2H); 13C NMR (DMSO-d6) δ 166.2, 165.0, 157.3, 135.2, 134.9, 130.7, 130.6, 129.9, 129.2, 127.6, 119.8, 117.8, 117.2; HPLC purity 98.8%; HRMS (ESI) calcd for C14H8Cl2NO3 307.9887 (M − H)+, found 307.9891.
N-(2-Hydroxybenzoyl)benzo[d][1,3]dioxole-5-carboxamide (1x)
The general procedure was used with salicylamide (2) and benzo[d][1,3]dioxole-5-carbonyl chloride (3x, prepared from the corresponding carboxylic acid) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (234 mg, 41% over two steps). 1H NMR (DMSO-d6) δ 11.75 (br s, 1H), 11.57 (s, 1H), 7.87 (dd, J = 7.8, 1.4 Hz, 1H), 7.52 (dd, J = 8.1, 1.5 Hz, 1H), 7.47–7.41 (m, 2H), 7.08 (d, J = 8.2 Hz, 1H), 7.03–6.97 (m, 2H), 6.16 (s, 2H); 13C NMR (DMSO-d6) δ 164.2, 163.8, 156.3, 151.2, 147.9, 134.2, 131.0, 127.7, 123.3, 119.9, 119.1, 117.0, 108.3, 107.6, 102.2; HPLC purity 98.7%; MS (ESI) m/z 286 (M + H)+.
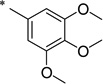
N-(2-Hydroxybenzoyl)-3,4,5-trimethoxybenzamide (1y)
The general procedure was used with salicylamide (2) and 3,4,5-trimethoxybenzoyl chloride (3y) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (33%). 1H NMR (DMSO-d6) δ 11.63 (s, 1H), 11.60 (s, 1H), 7.86 (dd, J = 7.6, 2.0 Hz, 1H), 7.23 (s, 1H), 6.96–7.10 (m, 2H); 13C NMR (DMSO-d6) δ 164.2, 164.1, 156.3, 141.4, 134.2, 131.1, 128.8, 120.0, 119.5, 117.1, 105.3, 60.2, 56.2; HPLC purity 96.9%; HRMS (ESI) calcd for C17H18NO6 332.1129 (M + H)+, found 332.1135.
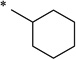
N-(Cyclohexanecarbonyl)-2-hydroxybenzamide (1z)
The general procedure was used with salicylamide (2) and cyclohexanecarbonyl chloride (3z) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (354 mg, 48%). 1H NMR (DMSO-d6) δ 11.44 (s, 1H), 10.80 (s, 1H), 7.81 (dd, J = 7.9, 1.7 Hz, 1H), 7.44 (ddd, J = 8.3, 7.2, 1.7 Hz, 1H), 6.98 (dd, J = 8.2, 0.9 Hz, 1H), 6.94 (d, J = 7.1, 1.1 Hz, 1H), 2.87 (tt, J = 11.0, 3.3 Hz, 1H), 1.89–1.86 (m, 2H), 1.75–1.72 (m, 2H), 1.66-1.63 (m, 1H), 1.39–1.13 (m, 5H); 13C NMR (DMSO-d6) δ 176.2, 164.8, 156.7, 134.2, 130.7, 119.7, 118.6, 117.0, 44.6, 28.5, 25.4, 25.1; HPLC purity 100%; MS (ESI) m/z 248 (M + H)+.
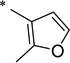
N-(2-Hydroxybenzoyl)-2-methylfuran-3-carboxamide (1aa)
The general procedure was used with salicylamide (2) and 2-methylfuran-3-carbonyl chloride (3aa) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (83%). 1H NMR (DMSO-d6) δ 11.65 (s, 1H), 11.24 (s, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.68, (s, 1H), 7.45 (t, J = 7.6 Hz, 1H), 6.82–7.05 (m, 2H), 6.77 (s, 1H), 2.50 (s, 3H); 13C NMR (DMSO-d6) δ 164.1, 161.0, 158.8, 156.2, 141.6, 134.2, 131.0, 119.9, 119.2, 117.0, 115.9, 108.8, 13.5; HPLC purity 98.7%; HRMS (ESI) calcd for C13H12NO4 246.0761 (M + H)+, found 246.0765.
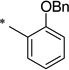
2-(Benzyloxy)-N-[(2-hydroxyphenyl)carbonyl]benzamide (1ab)
The general procedure was used with salicylamide (2) and 2-(benzyloxy)benzoyl chloride (3ab) as reactants. The reaction was carried out on 4 mmolar scale and provided crude material as a brown oil (423 mg, 30%), which was pure enough to be used in the next step without further purification. Purification of a portion of the crude material by preparative HPLC gave the title compound as a white solid (60 mg). 1H NMR (DMSO-d6) δ 11.68 (br s, 1H), 11.39 (br s, 1H), 7.79 (dd, J = 7.9, 1.6 Hz, 1H), 7.73 (dd, J = 7.7, 1.7 Hz, 1H), 7.52 (ddd, J = 8.5, 7.5, 1.8 Hz, 1H), 7.44 (ddd, J = 8.3, 7.3, 1.8 Hz, 1H), 7.38–7.35 (m, 2H), 7.25–7.21 (m, 4H), 7.09 (td, J = 7.6, 0.5 Hz, 1H), 6.96 (dd, J = 8.3, 0.5 Hz, 1H), 6.93 (td, J = 8.0, 0.8 Hz, 1H), 5.29 (s, 2H); 13C NMR (DMSO-d6) δ 164.8, 164.4, 156.8, 155.8, 136.3, 134.4, 133.4, 130.9, 130.8, 128.4 (2C), 127.9, 127.3 (2C), 123.8, 121.1, 119.8, 118.4, 117.0, 113.6, 69.9; HPLC purity 99.2%; MS (ESI) m/z (M + H)+.
N-(4-Aminobenzoyl)-2-hydroxybenzamide (4a)
Compound 4a was prepared by standard hydrogenolysis of compound 1i as a pale yellow solid (2.468 g, 96%). 1H NMR (DMSO-d6) δ 11.77 (br s, 1H), 11.44 (s, 1H), 7.90 (d, J = 7.4 Hz, 1H), 7.64 (d, J = 8.2 Hz, 2H), 7.45 (t, J = 7.3 Hz, 1H), 7.03 (d, J = 8.1 Hz, 1H), 6.99 (t, J = 7.4 Hz, 1H), 6.62 (d, J = 8.2 Hz, 2H), 6.05 (br, 2H); 13C NMR (DMSO-d6) δ 164.1, 163.9, 156.3, 153.5, 134.0, 131.0, 129.7, 119.9, 119.5, 119.1, 117.0, 112.8; HPLC purity 97.7%; MS (ESI) m/z 257 (M + H)+.
2-Hydroxy-N-(2-hydroxybenzoyl)benzamide (4b)
Compound 4b was prepared by standard hydrogenolysis of compound 1ab as a white solid (76%). 1H NMR (DMSO-d6) δ 11.54 (br s, 3H), 7.82 (dd, J = 7.8, 1.5 Hz, 2H), 7.45 (td, J = 8.5, 1.6 Hz, 2H), 7.02–6.96 (m, 4H); 13C NMR (DMSO-d6) δ 164.6, 156.9, 134.3, 130.7, 119.7, 119.0, 117.1; HPLC purity 98.5%; MS (ESI) m/z 258 (M + H)+.
Methyl 4-[[4-[(2-Hydroxybenzoyl)carbamoyl]phenyl]amino]-4-oxobutanoate (5)
To a suspension of 4a (244 mg, 0.95 mmol) and pyridine (0.085 mL, 1.05 mmol) in CH2Cl2 (2 mL) was added a solution of methyl 3-(chlorocarbonyl)propanoate (150 mg, 1.0 mmol) in CH2Cl2 (2 mL) at 0 °C. The mixture was stirred in the cold bath for 30 min and then at room temperature for 1 h before quenching with HCl (0.1M) to give pale green precipitate, which was filtered off, washed with water, and dried. The crude material was recrystallized from MeOH and purified further by HPLC to give the title compound as a white solid (90 mg, 26%). 1H NMR (DMSO-d6) δ 11.72 (s, 1H), 11.61 (s, 1H), 10.38 (s, 1H), 7.88 (d, J = 8.5 Hz, 3H), 7.75 (d, J = 8.7 Hz, 2H), 7.46 (td, J = 7.5, 1.8 Hz, 1H), 7.03 (d, J = 8.2 Hz, 1H), 7.00 (t, J = 8.2 Hz, 1H), 3.60 (s, 3H), 2.69–2.60 (m, 4H); 13C NMR (DMSO-d6) δ 172.8, 170.5, 164.2, 156.4, 143.3, 134.2, 131.0, 128.9, 127.6, 119.9, 119.1, 118.4, 117.0, 51.4, 31.0, 28.3; HPLC purity 97.7%; MS (ESI) m/z 371 (M + H)+.
N-Hydroxy-N'-[4-[[(2-hydroxyphenyl)carbonyl]carbamoyl]phenyl]octanediamide (8)
Compound 8 was prepared by standard hydrogenolysis of 7 as a pale yellow solid (15 mg, 7% over two steps). Compound 7 was obtained from 6 by following the same methodology as employed for the preparation of 5 from 4a, with the exception of using 8-[(benzyloxy)amino]-8-oxooctanoyl chloride as the acylating agent and extending the reaction time to 2 h. 1H NMR (DMSO-d6) δ 11.73 (br s, 1H), 11.62 (br s, 1H), 10.33 (br s, 1H), 10.25 (br s, 1H), 8.65 (br s, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 8.7 Hz, 2H), 7.77 (d, J = 8.8 Hz, 2H), 7.46 (td, J = 7.5, 1.6 Hz, 1H), 7.04–6.98 (m, 2H), 2.35(t, J = 7.5 Hz, 2H), 1.94 (t, J = 7.3 Hz, 2H), 1.59 (t, J = 7.5 Hz, 2H), 1.49 (t, J = 7.0 Hz, 2H), 1.29 (br, 4H); 13C NMR (DMSO-d6) δ 171.9, 169.1, 164.2, 156.3, 143.5, 134.2, 131.0, 128.8, 127.6, 119.9, 119.1, 118.5, 117.0, 36.4, 32.2, 28.4, 25.0, 24.8; HPLC purity 96.0%; MS (ESI) m/z 428 (M + H)+.
N-(4-Ethylbenzoyl)-2-hydroxy-N-methylbenzamide (10)
The general procedure was used with 2-hydroxy-N-methylbenzamide (9)14 and 4-ethylbenzoyl chloride (3a) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (84% over two steps). 1H NMR (CDCl3) δ 8.08 (d, J = 8.0 Hz, 2H), 7.75 (d, J = 7.6 Hz, 1H), 7.44 (t, J = 8.0 Hz, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.27 (t, J = 7.6 Hz, 1H), 7.18 (t, J = 8.0 Hz, 1H), 6.58 (br s, 1H), 2.79 (d, J = 4.8 Hz, 3H), 2.73 (q, J = 7.6 Hz, 2H), 1.25 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3) δ 166.4, 165.1, 151.1, 148.2, 131.6, 130.4, 129.9, 128.9, 128.4, 126.4, 126.2, 123.2, 29.1, 26.7, 15.2; HPLC purity 98.7%; MS (ESI) m/z 284 (M + H)+.
N'-(4-Ethylbenzoyl)-2-hydroxybenzhydrazide (12)
The general procedure was used with salicylhydrazide (11) and 4-ethylbenzoyl chloride (3a) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (36%). 1H NMR (DMSO-d6) δ 12.0 (br s, 1H), 10.7 (br s, 1H), 10.6 (s, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 8.0 Hz, 2H), 7.46 (t, J = 8.0 Hz, 1H), 7.36 (d, J = 8.0 Hz, 2H), 6.97 (m, 2H), 2.68 (q, J = 8.0 Hz, 2H), 1.21 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ 168.2, 165.9, 159.8, 148.6, 134.6, 130.2, 128.7, 128.3, 128.1, 119.5, 117.8, 115.0, 28.5, 15.7; HPLC purity 97.5%; MS (ESI) m/z 285 (M + H)+.
4-Ethyl-N-(2-hydroxythiobenzoyl)benzamide (13)
To a suspension of compound 1a (231 mg, 0.86 mmol) in chlorobenzene (3 mL) was added Lawesson’s reagent (522 mg, 1.29 mmol) under argon. The resulting mixture was refluxed for 3 h to produce a clear dark-red solution. After evaporation of the bulk of solvent, the crude product was directly purified by column chromatography on silica gel using hexane-EtOAc (2:1) as the eluent to give 13 as a yellow solid (16.5 mg, 7%). 1H NMR (CDCl3) δ 8.10 (d, J = 8.0 Hz, 2H), 7.86 (dd, J = 8.0, 1.6 Hz, 1H), 7.64 (br, 1H), 7.48 (t, J = 8.0 Hz, 1H), 7.41 (m, 4H), 7.16 (d, J = 8.0 Hz, 1H), 2.75 (q, J = 8.0 Hz, 2H), 1.28 (t, J = 8.0 Hz, 3H); 13C NMR (CDCl3) δ 200.6, 166.0, 151.4, 145.6, 134.6, 131.6, 130.7, 130.6, 128.4, 126.4, 126.1, 123.1, 29.1, 15.2; HPLC purity 97.8%; MS (ESI) m/z 286 (M + H)+.
N-[(4-Ethylphenyl)carbamoyl]-2-hydroxybenzamide (16)
The intermediate 14 was prepared on 20 mmolar scale to give the crude product as a white solid. The crude material was recrystallized from acetone to provide the product as a white crystals (3.0 g, 66%), which was subsequently converted into compound 15.15 The latter was obtained as a white solid (540 mg, 72%) containing an unidentified impurity (17% by HPLC). NMR data for the product 15: 1H NMR (DMSO-d6) δ 10.59 (s, 1H), 10.52 (s, 1H), 7.77 (dd, J = 7.5, 1.6 Hz, 1H), 7.60–7.53 (m, 3H), 7.46 (d, J = 8.3 Hz, 2H), 7.42–7.31 (m, 4H), 7.22–7.11 (m, 3H), 5.29 (s, 2H), 2.57 (q, J = 7.5 Hz, 2H), (t, J = 7.6 Hz, 3H); 13C NMR (DMSO-d6) δ 167.5, 156.2, 150.3, 139.2, 136.1, 135.2, 133.8, 130.3, 128.5, 128.2, 128.1, 127.6, 122.0, 121.0, 119.8, 113.7, 70.3, 27.5, 15.6; MS (ESI) m/z 375 (M + H)+. Standard hydrogenolysis of the intermediate 15 afforded the final compound 16 as a clean product (254 mg of a white powdery solid, 69%). 1H NMR (DMSO-d6) δ 11.83 (br s, 1H), 10.69 (s, 1H), 10.57 (s, 1H), 7.96 (dd, J = 7.9, 1.4 Hz, 1H), 7.50 (dd, J = 8.0, 1.4 Hz, 1H), 7.48 (d, J = 8.5 Hz, 2H), 7.18 (d, J = 8.3 Hz, 2H), 7.06 (d, J = 8.2 Hz, 1H), 7.01 (t, J = 7.6 Hz, 1H), 2.57 (q, J = 7.5 Hz, 2H), 1.16 (t, J = 7.6 Hz, 3H); 13C NMR (DMSO-d6) δ 166.7, 156.8, 150.4, 139.3, 135.2, 135.0, 130.8, 128.2, 120.0, 119.9, 117.2, 117.0, 27.6, 15.7; HPLC purity 99.8%; MS (ESI) m/z 285 (M + H)+.
1-(4-Ethylphenyl)-3-(2-hydroxyphenyl)urea (19)
To a solution of 17 (0.27 mL, 2.0 mmol) in CH2Cl2 (4 mL) was added dropwise 4-ethylaniline (0.25 mL, 2.0 mmol). The resulting mixture was stirred for 19 h at room temperature, and the resulting precipitate was then filtered off and washed with CH2Cl2 (25 mL) to give the desired crude 18 as a pale yellow solid (507 mg, 94%). 1H NMR (DMSO-d6) δ 9.20 (s, 1H), 8.17 (s, 1H), 8.12 (dd, J = 7.8, 1.7 Hz, 1H), 7.35 (d, J = 8.4 Hz, 2H), 7.1 (d, J = 8.4 Hz, 2H), 7.01 (dd, J = 7.9, 1.4 Hz, 1H), 6.93 (td, J = 7.7, 1.8 Hz, 1H), 6.88 (td, J = 7.7, 1.5 Hz, 1H), 3.87 (s, 3H), 2.54 (q, J = 7.6 Hz, 2H), 1.15 (t, J = 7.6 Hz, 3H); 13C NMR (DMSO-d6) δ 152.4, 147.6, 137.5, 137.1, 128.8, 128.0, 121.6, 120.5, 118.2, 118.1, 110.7, 55.8, 27.5, 15.8; HPLC purity 99.7%; MS (ESI) m/z 271 (M + H)+. To a solution of 18 (270 mg, 1.0 mmol) in CH2Cl2 (25 mL) was added dropwise BBr3 (4.0 mL of a 1.0M solution in CH2Cl2, 4.0 mmol) at −78 °C. The resulting mixture was allowed to warm to 0 °C within 2 h. After stirring for further 2 h at 0 °C, the reaction mixture was quenched with MeOH (25 mL) and washed with sat. NaHCO3 (2 × 50 mL), and H2O (2 × 50 mL). The organic phase was dried over Na2SO4, filtered, and concentrated to give the crude product which was dissolved in MeOH to yield 19 as pale yellow crystals (175 mg, 68%). 1H NMR (DMSO-d6) δ 9.89 (s, 1H), 9.19 (s, 1H), 8.11 (s, 1H), 8.03 (dd, J = 7.6, 1.3 Hz, 1H), 7.35 (d, J = 8.3 Hz, 2H), 7.10 (d, J = 8.3 Hz, 2H), 6.83 (dd, J = 7.6, 1.4 Hz, 1H), 6.78 (td, J = 7.2, 1.5 Hz, 1H), 6.73 (td, J = 7.3, 1.4 Hz, 1H), 2.54 (q, J = 7.6 Hz, 2H), 1.15 (t, J = 7.5 Hz, 3H); 13C NMR (DMSO-d6) δ 152.6, 145.6 137.6, 137.0, 128.0, 127.9, 121.6, 119.1, 118.5, 118.0, 114.4, 27.5, 15.8; HPLC purity 100%; MS (ESI) m/z 257 (M + H)+.
3-(4-Ethylbenzoyl)-2H-benz[e][1,3]oxazine-2,4(3H)-dione (21a)
The general procedure was used with 2H-benz[e][1,3]oxazine-2,4(3H)-dione (20a) and 4-ethylbenzoyl chloride (3a) as reactants. Purification of the crude material by preparative HPLC gave the title compound as a white solid (29%). 1H NMR (DMSO-d6) δ 8.17 (d, J = 8.4 Hz, 2H), 8.00 (d, J = 6.8 Hz, 1H), 7.91 (t, J = 7.6 Hz, 1H), 7.52 (m, 2H), 7.44 (d, J = 8.0 Hz, 2H), 2.72 (q, J = 7.6 Hz, 2H), 1.21 (t, J = 7.6 Hz, 3H); 13C NMR (DMSO-d6) δ 167.5, 160.4, 153.3, 153.0, 145.8, 137.0, 131.3, 128.9, 128.4, 127.1, 125.7, 116.8, 114.3, 28.4, 15.0; HPLC purity 97.4%; MS (ESI) m/z 296 (M + H)+.
N-(1-Acetyl-1H-indol-3-yl)-4-ethylbenzamide (21b)
To a suspension of 1-(1H-indol-3-yl)ethanone (796 mg, 5 mmol) in 95% ethanol (6 mL) was added NH2OH·HCl (417 mg, 6 mmol) and NaOAc (492 mg, 6 mmol). After refluxing for 2 h, the mixture was cooled, diluted with EtOAc (50 mL), washed with H2O (20 mL) and brine (20 mL), dried over Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel using hexane–EtOAc (2:1) as the eluent to give (E)-1-(1H-indol-3-yl)ethanone oxime as a white solid (797 mg, 92%).20 The oxime was dissolved in ethanol (6 mL) followed by addition of conc. H2SO4 (several drops). The mixture was refluxed for 4 h, concentrated in vacuo, and then diluted with H2O (40 mL) and washed with CHCl3 (3 × 20 mL). The aqueous layer was basified with 2N NaOH to pH 9 and extracted with EtOAc (3 × 40 mL). The combined organic layers were washed with brine (20 mL), dried over Na2SO4, and concentrated in vacuo. The crude product was purified by column chromatography on silica gel using CHCl3/MeOH (10:1) as the eluent to give 1-(3-amino-1H-indol-1-yl)ethanone (20b)20 as a dark-green solid (310 mg, 51%). Then the final compound 21b was obtained as a dark-green solid (380 mg, 25% over three steps) following the general procedure. 1H NMR (DMSO-d6) δ 11.7 (s, 1H), 8.33 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 4.0 Hz, 1H), 8.02 (d, J = 8.0 Hz, 2H), 7.45 (m, 3H), 7.20 (m, 2H), 2.72 (q, J = 8.0 Hz, 2H), 2.52 (s, 3H), 1.23 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ 163.6, 161.1, 150.3, 137.5, 130.9, 129.7, 128.8, 127.1, 124.5, 123.4, 123.1, 121.2, 112.3, 111.1, 28.7, 15.7, 14.5; HPLC purity 98.8%; MS (ESI) m/z 307 (M + H)+.
4-Ethyl-N-(1H-indazol-3-yl)benzamide (21c)
To a solution of 2-fluorobenzonitrile (0.54 mL, 5 mmol) in n-BuOH (6 mL) was added NH2NH2·H2O (0.78 mL, 25 mmol). After refluxing for 4 h, the mixture was cooled, diluted with EtOAc (50 mL), washed with H2O (20 mL) and brine (20 mL), dried over Na2SO4, and concentrated in vacuo. The resulting crude 1H-indazol-3-amine (20c)21 was converted without purification following the general procedure to compound 21c, which was obtained as a white solid (465 mg, 35% over two steps). 1H NMR (CDCl3) δ 9.76 (s, 1H), 8.73 (s, 1H), 8.12 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 2H), 7.39 (m, 2H), 7.33 (d, J = 8.0 Hz, 2H), 7.19 (m, 1H), 2.73 (q, J = 7.6 Hz, 2H), 1.28 (t, J = 7.6 Hz, 3H); 13C NMR (CDCl3) δ 165.5, 149.1, 141.8, 141.6, 131.2, 128.3, 127.7, 123.4, 120.8, 116.2, 109.6, 28.9, 15.3; HPLC purity 98.1%; MS (ESI) m/z 266 (M + H)+.
2-[3-(4-Ethylphenyl)-[1,2,4]oxadiazol-5-yl]phenol (23)
Neat compound 1a (22 mg, 0.08 mmol) was heated to its melting point (~ 200 °C) for 1 h. The resulting product 22 was cooled to room temperature and suspended in absolute ethanol (2 mL). Solid NaOAc (9 mg, 0.12 mmol) and NH2OH·HCl (8.4 mg, 0.12 mmol) were added. The mixture was stirred for 16 h, diluted with EtOAc (50 mL), washed with H2O (20 mL) and brine (20 mL), dried over Na2SO4, and concentrated in vacuo.17 The crude product was purified directly by column chromatography on silica gel using hexane–EtOAc (10:1) as the eluent to give 7 as a white solid (15 mg, 69% over two steps). 1H NMR (CDCl3) δ 10.5 (s, 1H), 8.06 (d, J = 8.0 Hz, 2H), 8.03 (dd, J = 8.0, 4.0 Hz, 1H), 7.56 (dd, J = 8.0, 4.0 Hz, 1H), 7.38 (d, J = 8.0 Hz, 2H), 7.17 (d, J = 8.0 Hz, 1H), 7.05 (d, J = 8.0 Hz, 1H), 2.76 (q, J = 8.0 Hz, 2H), 1.31 (t, J = 8.0 Hz, 3H); 13C NMR (CDCl3) δ 174.1, 167.1, 158.1, 148.4, 135.1, 128.5, 127.8, 127.6, 123.1, 120.1, 117.7, 108.2, 28.9, 15.3; HPLC purity 99.8%; MS (ESI) m/z 267 (M + H)+.
N-(4-Ethylphenyl)-N'-(2-hydroxyphenyl)propanediamide (28)
Compound 25 was obtained from 24 by following the same methodology as employed for the preparation of 5 from 4a, with the exception of using ethyl 3-chloro-3-oxopropanoate as the acylating agent and extending the reaction time to 18 h. The crude material was subsequently purified by column chromatography on silica gel with hexane/EtOAc (gradient from 0 to 30% EtOAc) as the eluent to provide pure compound 25 as a pale yellow oil (497 mg, 75%). 1H NMR (CDCl3) δ 9.53 (br s, 1H), 8.38 (dd, J = 7.9, 1.5 Hz, 1H), 7.50 (d, J = 7.0 Hz, 2H), 7.42–7.32 (m, 3H), 7.04 (td, J = 7.7, 1.5 Hz, 1H), 6.99–6.95 (m, 2H), 5.16 (s, 2H), 4.17 (q, J = 7.1 Hz, 2H), 3.47 (s, 2H), 1.26 (t, J = 7.1 Hz, 3H); 13C NMR (CDCl3) δ 169.1, 162.8, 147.5, 136.4, 128.5 (2C), 128.1, 127.7, 127.4 (2C), 124.1, 121.3, 120.3, 111.7, 70.7, 61.6, 42.5, 14.0; HPLC purity 94%; MS (ESI) m/z 314 (M + H)+.
Hydrolysis13 of the ester 25 gave the desired acid 26 as orange crystals (260 mg, 91% on a 1.0 mmolar scale). The crude product was pure enough to be used in the next step without further purification. 1H NMR (DMSO-d6) δ 9.63 (br s, 1H), 8.00 (dd, J = 8.0, 1.4 Hz, 1H), 7.51 (d, J = 7.0 Hz, 2H), 7.40–7.29 (m, 3H), 7.09 (dd, J = 8.2, 1.2 Hz, 1H), 7.03 (td, J = 7.8, 1.5 Hz, 1H), 6.90 (td, J = 7.9, 1.3 Hz, 1H), 5.22 (s, 2H), 3.50 (s, 2H); 13C NMR (DMSO-d6) δ 170.1, 164.6, 148.2, 137.0, 128.4 (2C), 127.8, 127.6, 127.3 (2C), 124.4, 121.7, 120.6, 112.9, 69.6, 43.1; HPLC purity 97.8%; MS (ESI) m/z 284 (M + H)+. Compound 27 was obtained as a yellow oil (300 mg, 85%) by following the literature procedure.13 The crude product was of adequate purity to be used in the next step without further purification. 1H NMR (DMSO-d6) δ 10.21 (br s, 1H), 9.99 (br s, 1H), 8.12 (d, J = 7.5 Hz, 1H), 7.58 (d, J = 6.9 Hz, 2H), 7.52 (d, J = 8.3 Hz, 2H), 7.38–7.30 (m, 3H), 7.18–7.13 (m, 3H), 7.04 (t, J = 7.4 Hz, 1H), 6.92 (t, J = 7.6 Hz, 1H), 5.22 (s, 2H), 3.59 (s, 2H), 2.57 (q, J = 7.5 Hz, 2H), 1.16 (t, J = 7.5 Hz, 3H); 13C NMR (DMSO-d6) δ 166.4, 165.0, 147.8, 139.2, 136.9, 136.4, 128.4 (2C), 128.0 (2C), 127.8, 127.7, 127.4 (2C), 124.2, 120.9, 120.8, 119.4 (2C), 112.7, 69.7, 44.6, 27.7, 15.8; HPLC purity >95%; MS (ESI) m/z 411 (M + Na)+.
The final compound 28 was obtained as a yellow solid by following a literature procedure.18 Purification by preparative HPLC provided the desired compound as a white solid (110 mg, 55% on a 0.67 mmolar scale). 1H NMR (DMSO-d6) δ 9.68 (br s, 1H), 9.43 (br s, 1H), 9.20 (br s, 1H), 9.48 (dd, J = 8.0, 1.0 Hz, 1H), 7.03 (d, J = 8.3 Hz, 2H), 6.69 (d, J = 8.3 Hz, 2H), 6.46 (td, J = 8.0, 1.3 Hz, 1H), 6.41 (dd, J = 8.0, 1.3 Hz, 1H), 6.30 (td, J = 8.0, 1.3 Hz, 1H), 3.10 (s, 2H), 2.09 (q, J = 7.6 Hz, 2H), 0.69 (t, J = 7.5 Hz, 3H); 13C NMR (DMSO-d6) δ 166.0, 165.3, 147.2, 139.1, 136.4, 128.0 (2C), 126.4, 124.3, 121.1, 119.4 (2C), 119.0, 115.2, 45.0, 27.6, 15.7; HPLC purity 97.7%; MS (ESI) m/z 299 (M + H)+.
Acknowledgment
The authors are grateful for the financial support provided by NIAID grant U01 AI082180, The Cornwell-Mann Family Foundation, The Pritzker Family Foundation, The Rooney Alden and Mussillami Families. Special acknowledgment is addressed to Dr. W. Tückmantel and Drs. K. El Bissati, D. Jacobus, J. McCall, C. Eid, M-F Cesbron-Delauw, J Rogers, and N. Grihalde for useful discussion and comments.
Abbreviations
- ADMET
absorption, distribution, metabolism, excretion, toxicity
- CYP450
cytochrome P450
- DMSO-d6
deuterated dimetyhyl sulfoxide
- hERG
the human Ether-à-go-go-Related Gene
- HLM
human liver microsomes
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- HTS
high-throughput screening
- L. donovani
Leishmania donovani
- L6
toxicity of the tested compounds assessed against a mammalian primary cell line derived from rat skeletal myoblasts
- NADPH
nicotinamide adenine dinucleotide phosphate
- SAR
structure-activity relationships
- SI
selectivity index
- T. b. rhodesiense
Trypanosoma brucei rhodesiense
- T. cruzi
Trypanosoma cruzi
- T. gondii
Toxoplasma gondii
- TLC
thin layer chromatography
- TMS
tetramethylsilane
- P. falciparum
Plasmodium falciparum
References
- 1.Fomovska A, Huang Q, Bissati KE, Ernest J. Mui, Witola WH, Cheng G, Zhou Y, Sommerville C, Roberts CW, Bettis S, Prigge ST, Afanador GA, Hickman MR, Lee PJ, Leed SE, Auschwitz JM, Pieroni M, Stec J, Muench SP, Rice DW, Kozikowski AP, McLeod R. Novel N-benzoyl-2-hydroxybenzamide disrupts unique parasite secretory pathway. Antimicrob. Agents Chemother. 2012 doi: 10.1128/AAC.06450-11. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Working to overcome the global impact of neglected tropical diseases: First WHO report on neglected tropical diseases. Geneva, Switzerland: World Health Organization; 2010. [Google Scholar]
- 3.Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet. 2010;375:148–159. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- 4.Stuart K, Brun R, Croft S, Fairlamb A, Gürtler RE, McKerrow J, Reed S, Tarleton R. Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Invest. 2008;118:1301–1310. doi: 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. Malaria Fact sheet No. 94. 2011
- 6.Rathore D, Jani D, Nagarkatti R. US20070148185 (A1) Novel therapeutic target for protozoal diseases. 2007
- 7.Adagu IS, Nolder D, Warhurst DC, Rossignol JF. In vitro activity of nitazoxanide and related compounds against isolates of Giardia intestinalis, Entamoeba histolytica and Trichomonas vaginalis. J. Antimicrob. Chemother. 2002;49:103–111. doi: 10.1093/jac/49.1.103. [DOI] [PubMed] [Google Scholar]
- 8.Cortes HC, Mueller N, Esposito M, Leitao A, Naguleswaran A, Hemphill A. In vitro efficacy of nitro- and bromo-thiazolyl-salicylamide compounds (thiazolides) against Besnoitia besnoiti infection in Vero cells. Parasitology. 2007;134:975–985. doi: 10.1017/S0031182007002417. [DOI] [PubMed] [Google Scholar]
- 9.Esposito M, Moores S, Naguleswaran A, Muller J, Hemphill A. Induction of tachyzoite egress from cells infected with the protozoan Neospora caninum by nitro- and bromo-thiazolides, a class of broad-spectrum anti-parasitic drugs. Int. J. Parasitol. 2007;37:1143–1152. doi: 10.1016/j.ijpara.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 10.Fonseca-Salamanca F, Martinez-Grueiro MM, Martinez-Fernandez AR. Nematocidal activity of nitazoxanide in laboratory models. Parasitol. Res. 2003;91:321–324. doi: 10.1007/s00436-003-0974-7. [DOI] [PubMed] [Google Scholar]
- 11.Roberts CW, Roberts F, Henriquez FL, Akiyoshi D, Samuel BU, Richards TA, Milhous W, Kyle D, McIntosh L, Hill GC, Chaudhuri M, Tzipori S, McLeod R. Evidence for mitochondrial-derived alternative oxidase in the apicomplexan parasite Cryptosporidium parvum: a potential anti-microbial agent target. Int. J. Parasitol. 2004;34:297–308. doi: 10.1016/j.ijpara.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Cai ZR, Jabri SY, Jin H, Kim CU, Lansdown RA, Metobo SE, Mish MR, Pastor RM. WO 2006/125048 A2 Integrase inhibitor compounds. 2006
- 13.Wittich S, Scherf H, Xie C, Brosch G, Loidl P, Gerhäuser C, Jung M. Structure-activity relationships on phenylalanine-containing inhibitors of histone deacetylase: in vitro enzyme inhibition, induction of differentiation, and inhibition of proliferation in Friend leukemic cells. J. Med. Chem. 2002;45:3296–3309. doi: 10.1021/jm0208119. [DOI] [PubMed] [Google Scholar]
- 14.Hossain N. WO 2005/037814 (A1) Novel tricyclic spiroderivatives as modulators of chemokine receptor activity. 2005
- 15.Qiao C, Gupte A, Boshoff HI, Wilson DJ, Bennett EM, Somu RV, Barry CE, 3rd, Aldrich CC. 5'-O-[(N-acyl)sulfamoyl]adenosines as antitubercular agents that inhibit MbtA: an adenylation enzyme required for siderophore biosynthesis of the mycobactins. J. Med. Chem. 2007;50:6080–6094. doi: 10.1021/jm070905o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adler T, Bonjoch J, Clayden J, Font-Bardia M, Pickworth M, Solans X, Sole D, Vallverdu L. Slow interconversion of enantiomeric conformers or atropisomers of anilide and urea derivatives of 2-substituted anilines. Org. Biomol. Chem. 2005;3:3173–3183. doi: 10.1039/b507202f. [DOI] [PubMed] [Google Scholar]
- 17.Kline T, Fromhold M, McKennon TE, Cai S, Treiberg J, Ihle N, Sherman D, Schwan W, Hickey MJ, Warrener P, Witte PR, Brody LL, Goltry L, Barker LM, Anderson SU, Tanaka SK, Shawar RM, Nguyen LY, Langhorne M, Bigelow A, Embuscado L, Naeemi E. Antimicrobial effects of novel siderophores linked to beta-lactam antibiotics. Bioorg. Med. Chem. 2000;8:73–93. doi: 10.1016/s0968-0896(99)00261-8. [DOI] [PubMed] [Google Scholar]
- 18.Hanessian S, Liak TJ, Vanasse B. Facile cleavage of benzyl ethers by catalytic transfer hydrogenation. Synthesis. 1981:396–397. [Google Scholar]
- 19.Orhan I, Sener B, Kaiser M, Brun R, Tasdemir D. Inhibitory activity of marine sponge-derived natural products against parasitic protozoa. Mar. Drugs. 2010;8:47–58. doi: 10.3390/md8010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qu J, Kumar N, Alamgir M, Black DS. A versatile synthetic route to 11H-indolo[3,2-c]isoquinolines. Tetrahedron Lett. 2009;50:5628–5630. [Google Scholar]
- 21.Vasudevan A, Souers AJ, Freeman JC, Verzal MK, Gao J, Mulhern MM, Wodka D, Lynch JK, Engstrom KM, Wagaw SH, Brodjian S, Dayton B, Falls DH, Bush E, Brune M, Shapiro RD, Marsh KC, Hernandez LE, Collins CA, Kym PR. Aminopiperidine indazoles as orally efficacious melanin concentrating hormone receptor-1 antagonists. Bioorg. Med. Chem. Lett. 2005;15:5293–5297. doi: 10.1016/j.bmcl.2005.08.049. [DOI] [PubMed] [Google Scholar]