

Abstract
Aarray painting is a technique that uses microarray technology to rapidly map chromosome translocation breakpoints. previous methods to map translocation breakpoints have used fluorescence in situ hybridization (FIsH) and have consequently been labor-intensive, time-consuming and restricted to the low breakpoint resolution imposed by the use of metaphase chromosomes. array painting combines the isolation of derivative chromosomes (chromosomes with translocations) and high-resolution microarray analysis to refine the genomic location of translocation breakpoints in a single experiment. In this protocol, we describe array painting by isolation of derivative chromosomes using a MoFlo flow sorter, amplification of these derivatives using whole-genome amplification and hybridization onto commercially available oligonucleotide microarrays. although the sorting of derivative chromosomes is a specialized procedure requiring sophisticated equipment, the amplification, labeling and hybridization of Dna is straightforward, robust and can be completed within 1 week. the protocol described produces good quality data; however, array painting is equally achievable using any combination of the available alternative methodologies for chromosome isolation, amplification and hybridization.
INTRODUCTION
Chromosome breakpoint mapping
Mapping the breakpoints of chromosome translocations has been a successful approach for the identification of disease-causing genes1 and to study the mechanism of chromosome rearrangement2. Historically, the positions of translocation breakpoints have been identified by multiple rounds of fluorescence in situ hybridization (FISH) using labeled large insert clones, which are hybridized to patient metaphase chromosomes. FISH mapping is time-consuming and has limited resolution, usually mapping a chromosome break to within the sequence of a bacterial artificial chromosome (BAC) (~200 kb). Previous studies have described using flow-sorted derivative chromosome DNA as the source material for PCR walking to further refine breakpoints3. PCR primers can then be selected to amplify the breakpoint junction fragment and the product can be subsequently sequenced to provide breakpoint information at a base-pair resolution. Alternative approaches to identify breakpoints include introducing derivative chromosomes into somatic cell hybrids, identifying altered restriction fragments at breakpoints that can be cloned and sequenced4, inverse PCR5 and Vectorette PCR6. More recently, advances in microarray technology have facilitated the development of array painting7, a technique to simply and quickly map the position of translocation breakpoints in a single experiment.
Array painting
Genomic microarrays have been used since the 1990s to map genomic imbalances in comparative genomic hybridization (CGH) studies8. The same microarrays developed for use in CGH investigations can also be used in array paint experiments. In array painting, derivative chromosomes are isolated, amplified and the amplified products are differentially fluorescently labeled and co-hybridized onto a microarray. The microarray is then scanned, log2 ratios calculated from the intensities and plotted against chromosome position (Fig. 1). The position where the log2 ratios change from high to low ratios (or vice versa) defines the translocation break-point. We first described array painting in 2003, when we showed the effectiveness of the procedure to resolve two apparently balanced translocation cases on a 1-Mb whole-genome BAC array7. In addition, one case interrogated on a custom tiling BAC array was mapped to a higher resolution. In 2007, we reported ultra-high-resolution array painting using custom designed oligonucleotide microarrays9. Array painting has been successfully used in multiple reports underlining its utility for the efficient identification of chromosome breakpoints10–14.
Figure 1.

A schematic outlining the steps of an array paint experiment. (a) Derivative chromosomes are isolated from a cell line by chromosome sorting (Steps 1–22), the derivative chromosomes are then amplified using a GenomePlex Complete Whole Genome Amplification Kit, (Steps 23–38) and the derivative chromosome amplified DNA is fluorescently labeled in Cy3 or Cy5 (Steps 39–54). (b) The fluorescently labeled derivative DNA is hybridized onto a microarray using Agilent SureHyb chamber (Steps 55–80) (picture courtesy of Agilent). (c) The microarray is scanned (Steps 81–83). (d) The features on the microarray are analyzed, the log2 ratio of Cy5/Cy3 intensity is calculated for each oligonucleotide and plotted against the base-pair chromosome position (Steps 84–85). The top schematic represents the data for one parent chromosome (chr A), whereas the lower schematic represents the other parent chromosome (chr B). The transition from a high (red spots) to a low log2 ratio (green spots) indicates a chromosome breakpoint. If a feature sequence spans a chromosome breakpoint, the ratio will be close to 0 (orange spots).
Derivative chromosome isolation and amplification
A derivative (der) chromosome is a structurally rearranged chromosome, generated either by multiple aberrations within a single chromosome or by reciprocal translocation between two chromosomes, e.g., chromosome A and chromosome B translocate giving rise to der A and der B. In this protocol, we describe array painting using derivative chromosomes resulting from a reciprocal translocation (Fig. 1). This procedure could be applied to help elucidate the structure of a structurally rearranged chromosome but would be limited to identifying deletion breakpoints and would not identify inverted or duplicated genomic regions. The first stage of array painting involves the isolation of the derivative chromosomes. This can be achieved either by flow sorting as described here or by microdissection15–17.
Amplification of the derivative chromosomal DNA is required to provide sufficient DNA for a random prime labeling reaction. In this protocol, we recommend 10 ng of starting derivative DNA, which is the equivalent of many thousands of isolated derivative chromosomes; however, if this is not achievable, the protocol is likely to be successful with lower input DNA. We have previously successfully used a number of protocols for derivative amplification, including GenomiPhi (GE Healthcare)7, REPLI-g (Qiagen)18, degenerate oligonucleotide primer PCR7 and GenomePlex Complete Whole Genome Amplification (WGA) (Sigma), which is the method described in this protocol. All of these amplification processes have been routinely conducted on multiple copies of isolated derivative chromosomes; however, we reported in 2004 successful amplification of single derivative chromosomes18.
Each of the above amplification techniques have been used successfully in array paint experiments on low-resolution BAC arrays7,11–14 or higher resolution BAC tiling arrays10. We have also reported array painting on NimbleGen oligonucleotide arrays following derivative amplification with REPLI-g (Qiagen)9. In this protocol, we describe in detail array painting on Agilent oligonucleotide arrays using GenomePlex WGA to amplify the derivative DNA. WGA has the potential to introduce bias by the nonuniform amplification of the input sample. Although this may compromise the quantitative analyses in conventional array CGH experiments, array painting is largely unaffected by such bias.
Resolution of DNA microarrays
The resolution of DNA microarrays has advanced rapidly over the last decade. It is now possible to design customized high-resolution oligonucleotide arrays to specifically target breakpoint regions. For example, we have reported array painting using NimbleGen arrays with overlapping 60-mer oligonucleotides spaced every 1 bp, which refined chromosome breakpoints in four different translocations from patients with developmental disorders. Breakpoints in these cases were refined to intervals ranging from 55 to 7,223 bp (ref. 9). Although high-resolution tiling arrays have been adopted in both CGH and array painting experiments, the performance of the probes may be adversely affected by the sequence context in the region of interest; e.g., if the oligonucleotide probe is at the position of a sequence repeat, consequently some probes will not report reliably. If a probe is sited at the position of a segmental duplication that has two genomic positions, fluorescently labeled DNA from both segmental duplications would bind to the oligonucleotide regardless of their genomic position. The predicted chromosome breakpoint in an array paint experiment could be slightly inaccurate if there is a repeat at the breakpoint and probes have been designed to the repeat.
Advantages of array painting
Array painting is a very rapid approach to mapping a chromosome breakpoint. In a single experiment, a breakpoint can be refined sufficiently to permit the subsequent PCR amplification and sequencing of the junction fragment. A notable advantage of array painting is the ability to decipher complex chromosome rearrangements19, including the opportunity to detect imbalances at or close to the breakpoint. For example, we have previously mapped 12 breakpoints from 4 derivative chromosomes in a complex rearrangement, in just two array paint experiments11. In contrast to array CGH, which requires high-quality hybridizations of competing sample and reference genomic DNA, array painting is a non-competitive, simple hybridization of the selected derivative DNA and is consequently a more robust procedure. Even if a derivative cannot be resolved from its normal homolog during the flow sort procedure or if it sorts with another unrelated chromosome, it is still possible to perform a successful array paint procedure and identify a chromosome breakpoint using the contaminated derivative sort9. The log2 ratio of the Cy3 and Cy5 intensities will be close to 0 for any genomic region in common to co-sorting derivative chromosomes. The transition in log2 ratio values from a high or low ratio to 0, for a genomic region in common, will still indicate the position of a breakpoint. If a derivative chromosome co-sorts with an unrelated chromosome, the contaminating chromosome will also hybridize on the array; although it might alter normalization, this will not interfere with identifying a chromosome breakpoint for the derivative of interest. The only situation that is problematic is if by chance two derivative chromosomes from a balanced reciprocal translocation co-sorted, it would then be impossible to identify a breakpoint as all oligonucleotides representing both parent chromosomes would ratio to 0. Microdissection as a method of successful isolation of these derivatives would overcome this problem.
Limitations of array painting
Array painting requires the isolation of derivative chromosomes by flow sorting or microdissection, techniques that require highly trained personnel and sophisticated and expensive flow cytometry and microdissecting equipment.
Advances in new-generation sequencing technologies will offer an alternative to array painting, and it will be possible to identify translocation breakpoints by implementing paired end sequencing of the genomic DNA containing the translocation. Currently, it is still necessary to isolate derivative chromosomes before sequencing in order to ensure this approach is cost-effective. Having isolated derivative chromosomes, the costs involved to run a lane of Illumina paired end sequencing or a microarray hybridization are similar, and currently it is quicker and probably more accessible to hybridize an array. Over the next few years, this will inevitably change to favor new-generation sequencing to identify translocation breakpoints.
Potential applications
Array painting can be used to determine the composition of any isolated chromosome material and is not exclusively for mapping chromosome translocation breakpoints. The array paint procedure is comparable to the chromosome painting technique, but uses high-resolution microarrays as the target in place of metaphase chromosomes. In the same way that conventional chromosome painting can be used to determine cross species homology so can array painting. We previously reported hybridizing a white-cheeked gibbon chromosome 14 onto a human DNA microarray and determined homology to human chromosomes 2 and 17 (ref. 18). Another application would be to determine the composition of marker chromosomes. A marker chromosome is a structurally abnormal chromosome in which no part can be identified. Conventional reverse painting isolates a marker chromosome and hybridizes it onto normal metaphase chromosomes revealing its composition20. Hybridizing the marker chromosome onto a DNA microarray would show the content of the marker and refine the boundaries of the genomic regions involved to a higher resolution.
Experimental design
In this protocol, we describe array painting using flow-sorted chromosomes and oligonucleotide microarrays. The chromosomes are prepared using a chromosome stabilization buffer based on polyamines. This method generates high-molecular-weight DNA. The chromosomes are stained and the two derivative chromosomes (generated from a reciprocal translocation, Fig. 1) are selectively gated and isolated using a dual-beam flow cytometer with sorting function. Array painting is achievable using many combinations of alternative methodologies. Derivative chromosomes can be isolated by flow sorting or microdissection. It is easier to obtain multiple copies of a derivative chromosome by flow sorting, and the higher the number of starting chromosomes, the higher the quality of the amplification product. However, if flow sorting cannot isolate a derivative chromosome (e.g., if two reciprocal translocation products sorted together), microdissection would be able to isolate the derivatives from one another. There are many alternatives for derivative amplification as listed in the INTRODUCTION. We describe WGA (Sigma) using 10 ng of input DNA. The method of amplification should be selected considering the starting material and the array platform and tested to ensure their compatibility. Similarly, there are multiple array platforms available, all of which are suitable for array painting, and we describe the Agilent oligonucleotide microarray platform, which is robust and simple to perform. It is necessary to select an array of suitable probe distribution and resolution before attempting an array paint experiment.
Commercial procedures
In this protocol, we have combined commercial procedures from Sigma-Aldrich and Agilent Technologies for the DNA amplification step and the microarray hybridization steps, respectively. The manufacturer’s methodology supplied with the GenomePlex Complete WGA Kit (Sigma) was used for DNA amplification, and the Agilent protocol G4410-90010, ‘Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis—Version 6.0,’ was used for microarray hybridization.
Cell culture
Chromosomes have been successfully isolated from suspension as well as adherent cell lines. In this protocol, the suspension cell lines are generally subcultured to 50% with 50 ml of fresh growth medium and grown for 24 h before treatment with mitotic inhibiting agents. An approximate cell density of ~1–2 × 107 cells can be achieved in a single T75-cm2 flask. A mitotic index of more than 40% can be obtained for these cell types. In the case of adherent cell lines, the cells are subcultured to 50% with 30 ml of fresh growth medium and grown for 24 h before treatment with mitotic inhibiting agents. An approximate cell density of ~1 × 106 cells can be achieved in a single T150-cm2 flask. A mitotic index of more than 60% can be obtained for these types of cell line.
Mitotic arrest
The protocol we describe here is suitable for most cell lines with simple variation to the mitotic treatment time for maximal mitotic index. In general, we treat most of the lympho-blastoid cell lines with 0.1 μg ml − 1 of colcemid for a period of 4–6 h. In the case of adherent cell lines, a dosage of 0.1 μg ml − 1 and an incubation time varying from 6 to 24 h is required. The growth rate among adherent cell lines varies considerably, whereas suspension lymphoblastoid cell line growth rates are generally more similar. Adherent lines may therefore possibly require a longer incubation in colcemid. We recommend the optimization of mitotic treatment steps regarding time and dosage when working with a different cell type or animal species. It should be noted that inadequate treatment timing can result in a high number of cells at interphase; a contributing factor to the formation of debris and DNA fragments during the chromosome isolation process. An extensive exposure to colcemid can result in the formation of chromatids21, which deter accurate flow karyotype measurement and impair the purity of chromosome sorts and also cause cell death. If exposure to colcemid has caused a high rate of cell death, the concentration of colcemid should be reduced or colcemid should be replaced with another mitotic inhibiting agent such as colchicine, demecolcine, vinblastine or a commercially available cocktail mix (e.g., Procell Metaphase Arresting Solution).
Chromosome preparation and staining
During the chromosome isolation process, we take a small aliquot of hypotonic-treated cells and monitor it with Turck’s stain to assess whether the mitotic cell has been adequately swollen and whether the chromosomes are well spread within the plasma membrane (Fig. 2a). The swollen cells are then treated with polyamine isolation buffer containing Triton X-100; with adequate swelling, the chromosomes should be easily detached from one another when the cells are disrupted through vortexing or syringing. The extent of well-separated chromosomes is checked under the microscope by adding propidium iodide to a small aliquot of chromosome suspension (Fig. 2b), and chromosomes that appear to be stuck to one another forming clusters can be easily dispersed by further vortexing or syringing. The stained chromosomes are usually treated with sodium citrate and sodium sulfite for at least 1 h in the dark as an approach to improve the resolution of the flow karyotype22. The resolution of the flow karyotype can be improved for lymphoblastoid cell lines with an overnight incubation with both reagents. However, we have also found that the flow karyotype of some cell lines degrades upon using both chemical reagents; therefore, the flow karyotype of a particular cell line needs to be monitored with or without the addition of either reagent.
Figure 2.
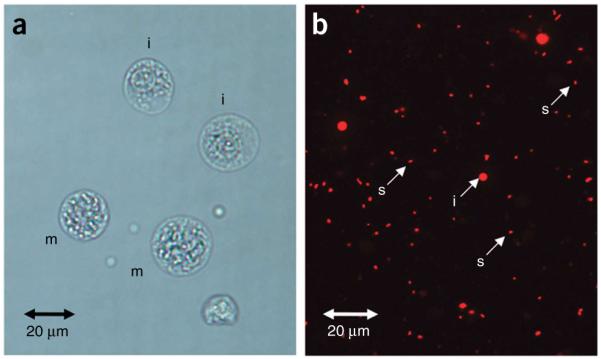
Images of chromosome preparation and staining. (a) Swollen cells in metaphase (m) and interphase (i) stained with Turck’s solution (Step 7). (b) Released single chromosomes (s) and interphase nuclei (i) after treating the swollen cells with polyamine isolation buffer containing Triton X-100, and staining with propidium iodide (Step 11).
Estimating the size of derivative chromosomes
Before starting to flow sort, it is necessary to perform G banding analysis on the samples to help generate a derivative chromosome ideogram (Fig. 3). These ideograms can be used to compare the size of the derivative chromosomes with the size of normal human chromosomes, and the position of the derivative chromosomes on a flow karyogram can then be predicted. Table 1 (ref. 23), column 2, lists the size of the parent chromosomes in Mb. By calculating the amount of each chromosome in Mb, which is present on each derivative and combining these numbers, the size of derivative chromosomes can be estimated (Fig. 3). By comparing the size of the derivative chromosome with the sizes of all the normal human chromosomes in Table 1, column 2, it is possible to predict the derivative chromosome’s position on the flow karyogram (Fig. 3). This is an important process to identify which atypical cluster on a flow karyogram is which derivative chromosome and, in addition, this process will highlight if a derivative is likely to sort with a normal chromosome (e.g., the derivative chromosome and normal chromosome are of the same size). The predicted position of a derivative chromosome on a flow karyogram may be altered slightly by its GC composition.
Figure 3.

A schematic detailing the flow sorting of the derivative chromosomes resulting from a translocation between chromosomes 3 and 20, t(3;20)(q13.2;p12.2). (a) Ideograms of chromosomes 3 (200 Mb) and 20 (62 Mb). (b) Ideograms of derived chromosome 3 (127 Mb), and of derived chromosome 20 (133 Mb). (c) Flow karyogram (100,000 events) showing the positions of all normal chromosomes in the human genome. *a chromosome 9 sorting away from the 9–12 cluster in this normal cell line due to a large heterochromatic region. (d) Flow karyogram (50,000 events) showing the actual positions of the derived chromosome 3 and derived chromosome 20 (red rings). NB: Chromosomes 1 and 2 are not in the karyogram because the voltage for both the Hoechst and Chromomycin channels are enhanced to make the derivative chromosome clusters more distinguishable.
TABLE 1.
Sizes of chromosomes and an estimate of the number of each chromosome needed to yield required amount for amplification (10 ng).
| Chromosome | Size (Mb) | Number of chromosomes to yield 10 ng DNA |
|---|---|---|
| 1 | 247 | 19,501 |
| 2 | 243 | 19,904 |
| 3 | 200 | 23,901 |
| 4 | 191 | 25,202 |
| 5 | 181 | 26,205 |
| 6 | 171 | 27,996 |
| 7 | 159 | 30,193 |
| 8 | 146 | 33,113 |
| 9 | 140 | 35,014 |
| 10 | 135 | 35,511 |
| 11 | 134 | 35,613 |
| 12 | 132 | 36,075 |
| 13 | 114 | 44,326 |
| 14 | 106 | 46,816 |
| 15 | 100 | 48,170 |
| 16 | 89 | 51,760 |
| 17 | 79 | 56,689 |
| 18 | 76 | 59,524 |
| 19 | 64 | 72,464 |
| 20 | 62 | 76,453 |
| 21 | 47 | 100,806 |
| 22 | 50 | 92,593 |
| X | 155 | 31,606 |
| Y | 58 | 85,911 |
Chromosome sizes according to NCBI36.3.
Number of chromosomes for 10 ng of DNA from CYDAC measurements23.
Instrument requirements
Different makes of flow cytometer are available commercially. The protocol we describe here uses a MoFlo, a flow cytometer with sorting function that is equipped with two water cooled lasers, which are tuned to the required laser lines (UV and 458 nm) at a power of 300 mW. The flow sorter works on a flow chamber design based on the principle of a ‘jet-in-air’ system. We have found that in our instrument setup, high-resolution flow karyotypes are acquired through photo saturation of the fluorescence dyes by increasing the laser power to 300 mW. By this approach, the measured fluorescence is observed to be independent of the illumination intensity of the laser. The laser lines applied in the protocol are specifically tuned for the excitation of Hoechst and Chromomycin A3. For all other flow cytometer designs and laser builds, we recommend that the level of laser power be optimized carefully for maximal signal, as this is a prerequisite for obtaining a well-resolved flow karyotype and the success of this application.
Flow karyotype and chromosome analysis
We typically stain the chromosomes with Hoechst and Chromomycin A3 for bivariate flow karyotyping. To date, this is the best fluorescence dye combination to highly resolve flow karyotypes. Hoechst dye binds preferentially to AT-rich sequences, and Chromomycin dye binds preferentially to GC-rich sequences. The flow karyotype is produced based on the difference in the DNA base composition and the DNA content of each chromosome. The staining characteristics between the dyes resolve most of the human chromosomes except 9–12. The technique we describe here is applicable for most lymphoblastoid cell lines as well as primary cell culture from mouse, pig and zebrafish24. It has also been successfully used for isolating translocated chromosomes from established cancer cell lines such as breast19.
Amount of input DNA to amplification step
We describe array painting using GenomePlex WGA, which states a minimum of 10 ng starting DNA (typically tens of thousands of derivative chromosomes). Although the manufacturer’s instructions state ‘the starting amount of DNA is critical for complex starting material such as genomic DNA and a reduction of input DNA will significantly alter the gene bias of GenomePlex product,’ this protocol starts with an enriched less-complex starting material, and a lower input of DNA may still produce acceptable results.
GenomePlex WGA: fragmentation and library generation
As stated in the manufacturer’s instructions, ‘this WGA procedure utilizes a proprietary amplification technology based upon random fragmentation of genomic DNA and conversion of the resulting small fragments to PCR-amplifiable OmniPlex Library molecules flanked by universal priming sites.’ The OmniPlex Library is then PCR-amplified using universal oligonucleotide primers. When performing this procedure, the manufacturer recommends that Steps 28–30 in this protocol are conducted without interruption, as the ends of the library DNA can degrade, thus affecting subsequent steps.
Experimental controls
When preparing the GenomePlex WGA products, it is suggested to run a positive DNA (supplied in the kit) and a ‘water-only’ negative control. The purpose of any negative control is to monitor any reagent contamination and the positive control will assess the reaction independent of the user’s experimental DNA samples. To ensure successful amplification of derivative chromosome DNA, the products are run on an agarose gel as described in this protocol (Step 30). Successful amplification products should appear as a smear of products ranging in size from 100 to 1,000 bp and the negative control should have no product. Sigma states that the GenomePlex WGA Kit contains an optimized enzyme that decreases the background in the reaction and produces a negative control sample without product.
Differential random labeling and sample cleanup of derivative chromosome DNA
This procedure details the differential labeling of two derivative chromosomes derived from a translocation (termed der A and der B in Step 41 and Fig. 1). This simply means one derivative is labeled in Cy3 and the other in Cy5. In Step 50, it is stated that the sample volume needs to be ≤80.5 μl after concentrating in a Microcon reservoir membrane. It is necessary to achieve this volume to ensure the final hybridization mix will be 520 μl (see Step 55).
The success of a random prime labeling reaction is assessed in two ways. The incorporation of fluorescently conjugated nucleotides into a DNA sample is measured by calculating the Specific Activity (see Step 54) and the yield of the reaction is measured to determine the amount of DNA produced in a labeling reaction.
Environmental conditions for DNA microarray experiment
It is important to conduct microarray experiments that use Cy3 and Cy5 dCTPs in a dark, air-conditioned environment, with a temperature of ~20 to 22 °C, to maintain humidity at ~30% and to keep ozone to ~0.02 PPM. These conditions are most important when scanning microarrays, where they help to preserve the intensity of the fluorophores.
MATERIALS
REAGENTS
Cell culture medium (see REAGENT SETUP)
RMPI 1640 (Gibco, cat. no. 52400-025)
DMEM (Gibco, cat. no. 41966-029)
Fetal calf serum (Gibco, cat. no. 10108-165)
Antibiotic mixture (Sigma, cat. no. G6784)
Metaphase-arresting drug: Demecolcine 10 μg ml − 1 (Sigma, cat. no. D1925) or KaryoMAX Colcemid 10 μg ml − 1 (Gibco, cat. no. 15212-046)
Buffers (see REAGENT SETUP)
Tris (pH 7.4) (Sigma, cat. no. T3253)
EDTA (Sigma, cat. no. E-5134)
Sodium azide (Sigma, cat. no. S8032)
 NaN3 is known to be toxic. Avoid contact with skin and eyes. Wear gloves and lab coat when handling NaN3 or solutions containing it.
NaN3 is known to be toxic. Avoid contact with skin and eyes. Wear gloves and lab coat when handling NaN3 or solutions containing it.NaCl (BDH, cat. no. 3012375)
MgSO4.7H2O (Sigma, cat. no. M2773)
KCl (BDH, cat. no. 104874S)
Spermine (Sigma, cat. no. S2876)
Spermidine (Sigma, cat. no. S2501)
NaOH (Fisher Scientific, cat. no. P/5640/53)
 It is an irritant. Wear gloves, lab coat and protective eyewear when handling NaOH or solutions containing it.
It is an irritant. Wear gloves, lab coat and protective eyewear when handling NaOH or solutions containing it.Dithiothreitol (GibcoBRL, Cleland’s Reagent, cat. no. 15508-013)
EGTA (Sigma, cat. no. E3889)
Triton X-100 (Sigma, cat. no. T9284)
Propidium iodide (1 mg ml − 1) (Sigma, cat. no. P4170)
Turck’s stain (see REAGENT SETUP)
Gentian violet (Fluka, cat. no. 49144)
Glacial acetic acid (Sigma, cat. no. A9967)
 It is flammable and can cause severe burns. Do not breathe gas/fumes/vapor. Handle safely in a fume hood.
It is flammable and can cause severe burns. Do not breathe gas/fumes/vapor. Handle safely in a fume hood.DNA fluorescent dyes
1 mg ml − 1 Hoechst 33258 (Sigma, cat. no. B2883) (see REAGENT SETUP)
10 mg ml − 1 Chromomycin A3 (Sigma, cat. no. C2659) (see REAGENT SETUP)
1 M Sodium citrate (Sigma, cat. no. S4641) (see REAGENT SETUP)
500 mM Sodium sulfite (Sigma, cat. no. S8018) (see REAGENT SETUP)
3 μm beads (Sphero Rainbow Fluorescent Particles, Spherotech, cat. no. RFP 30-5)
HPLC water (VWR, cat. no. 23595.328)
TE buffer, pH7.9–8.1 at 25 °C (Promega, cat. no. V6231)
Pellet Paint (Novagen, cat. no. 70748-3)
2.5 M NaCl (BDH, cat. no. 102415K)
99.7% (vol/vol) Ethanol (BDH, cat. no. 10107)
GenomePlex Complete Whole Genome Amplification (WGA) Kit (Sigma, cat. no. WGA2)
Agarose (Invitrogen, cat. no. 15510-027)
TrackIt 50 bp DNA Ladder (Invitrogen, cat. no. 10488-043) and/or 1 kb DNA Ladder (New England Biolabs, cat. no. N3232S)
BioPrime DNA Labeling System Kit (Invitrogen, cat. no. 18094011)
10× dNTP mix (1 mM dCTP, 2 mM dATP, 2 mM dGTP, 2 mM dTTP in TE buffer) (Thermo Scientific, cat. no. CB-0315)
Cy3-labeled dCTP (Amersham, cat. no. PA53021)
Cy5-labeled dCTP (Amersham, cat. no. PA55022)
Human Cot-1 DNA (1.0 mg ml − 1) (Invitrogen, cat. no. 15279-101)
Agilent 10× Blocking Agent (Agilent, cat. no. 5188-5220) (see REAGENT SETUP)
Agilent 2× Hybridization Buffer (Agilent, cat. no. 5188-5220) (see REAGENT SETUP)
 The manufacturer states that it may be harmful if swallowed. Avoid contact with eyes, skin and clothing.
The manufacturer states that it may be harmful if swallowed. Avoid contact with eyes, skin and clothing.Agilent aCGH Wash Buffer 1 (Agilent, cat. no. 5188-5221)
Agilent aCGH Wash Buffer 2 (Agilent, cat. no. 5188-5222)
EQUIPMENT
0.22-μm syringe filter (Nalgene, cat. no. 190-2520)
20-μm mesh filter (Celltrics, Partec, cat. no. 04-004-2325)
MoFlo High Performance Cell Sorter equipped with two water-cooled lasers (Coherent, Innova 300 series)
1.5-ml microfuge tubes
Benchtop microfuge
Heat block
Thermal cycler
Microcon YM-30, 30 kDa, sample reservoirs and tubes (Millipore, cat. no. 42422)
NanoDrop 2000 Spectrophotometer (Thermo Scientific)
Hybridization Gasket Slide Kit (5) for 1 microarray per slide format (Agilent, cat. no. G2534-60003)
Hybridization Chamber Kit (Agilent, cat. no. G2534A)
Blunt forceps (supplied with hybridization chamber)
Human Genome CGH Microarray Kit 244A, consists of 5 microarrays (Agilent, cat. no. G4411B)
DNA Microarray Hybridization Oven (Agilent, cat. no. G2545A) set to 65 °C, 20 rpm
Hybridization Oven Rotator Rack (Agilent, cat. no. G2530-60029)
Three glass troughs
One slide rack
Microarray Scanner (we used Agilent G2565CA but G2565BA can be used, Agilent)
Slide holders supplied with scanner
Feature Extraction 9.5 or 10.5 software and Agilent Scan Control software—both supplied with scanner
DNA Analytics 4.0 (Agilent, cat. no. G4172AA)
Freely available ‘R’ software
Summit v3.1 (analysis software from Dako/Beckman Coulter)
REAGENT SETUP
Cell culture medium
Supplement 500 ml of RMPI 1640 or DMEM with 15% (vol/vol) fetal calf serum and antibiotics. Store solution at 4 °C for up to 1 month.
Sheath buffer
Mix the reagents to achieve the following final concentrations: 100 mM NaCl, 0.01 M Tris (pH 7.4), 0.001 M EDTA and 0.05% (wt/vol) NaN3. Autoclave and store at room temperature (RT, 18–25 °C) for up to 3 months.  This buffer contains NaN3; hence, wear gloves and lab coat when handling.
This buffer contains NaN3; hence, wear gloves and lab coat when handling.
Hypotonic solution
Mix the reagents to achieve the following final concentrations: 75 mM KCl, 10 mM MgSO4.7H2O, 0.2 mM Spermine and 0.5 mM spermidine. Make up the volume with HPLC water to 50 ml and then adjust the pH of the hypotonic solution to 8.0 using 0.25 M NaOH. Monitor the pH using pH indicator paper. Store on ice. Prepare fresh on the day of chromosome isolation.  NaOH is an irritant; hence, wear gloves, lab coat and protective eyewear when handling NaOH or solutions containing it.
NaOH is an irritant; hence, wear gloves, lab coat and protective eyewear when handling NaOH or solutions containing it.  pH to 8.0 before use (see REAGENT SETUP)
pH to 8.0 before use (see REAGENT SETUP)
Polyamine isolation buffer
Mix the reagents to achieve the following final concentrations: 15 mM Tris, 80 mM KCl, 3 mM dithiothreitol, 2 mM EDTA, 0.5 mM EGTA, 0.2 mM spermine, 0.5 mM spermidine and 0.25% (vol/vol) Triton X-100. Make up the volume to 50 ml with HPLC water. Mix well on a rotator for 30 min and then adjust the pH of the buffer to 7.5 using 0.5 M NaOH. Monitor the pH using pH indicator paper. Filter the buffer through a 0.22-μm syringe filter. Store at 4 °C. The buffer is stable for a month.  It is important to check the pH of the buffer used in the preparation of chromosome suspensions as pH can influence the stability as well as the physical structure of chromosomes.
It is important to check the pH of the buffer used in the preparation of chromosome suspensions as pH can influence the stability as well as the physical structure of chromosomes.
Turck’s stain
Mix the reagents to achieve the following final concentrations: 0.01% (wt/vol) gentian violet in 1% (vol/vol) glacial acetic acid. Store the solution at 4 °C for up to 1 year.  Glacial acetic acid is flammable and can cause severe burns; do not breathe gas/fumes/vapor. Handle safely in a fume hood.
Glacial acetic acid is flammable and can cause severe burns; do not breathe gas/fumes/vapor. Handle safely in a fume hood.
DNA fluorescent dyes
Prepare a 1 mg ml − 1 Hoechst 33258 staining solution by dissolving the reagent in sterile distilled water. Store the solution in the dark at 4 °C for up to 1 year.
Prepare 10 mg ml − 1 Chromomycin A3 staining solution by dissolving the reagent in absolute ethanol. Store the solution in the dark at − 20 °C for up to 1 year.
Sodium citrate
Prepare 1 M of the reagent in HPLC water. Store at − 20 °C for up to 1 year.
Sodium sulfite
Prepare 500 mM of the reagent in HPLC water. Store at − 20 °C for up to 1 year.
Agilent 10× blocking agent
Add 1,350 μl of nuclease-free water to the vial containing lyophilized 10× Blocking Agent. Leave at room temperature for 60 min and mix on a vortex mixer to reconstitute the sample, heating to 37 °C for 4–5 min may be needed to completely redissolve lyophilized pellet. Blocking agent can then be stored at − 20 °C for up to 2 months.
Agilent 2× Hybridization Buffer
The hybridization buffer should be carefully mixed by inverting because it separates into two phases. The buffer should then be aliquoted and stored as smaller aliquots in the dark at room temperature until the stated expiry date.  The manufacturer states that it may be harmful if swallowed. Avoid contact with eyes, skin and clothing.
The manufacturer states that it may be harmful if swallowed. Avoid contact with eyes, skin and clothing.
EQUIPMENT SETUP
Setting up flow sorting equipment
Replace the sterile sheath buffer in the sheath tank (refer to the manufacturer’s instrument setup guide) and warm up the lasers for more than an hour before flow analysis begins. Set the power of both lasers to 300 mW and keep stable using light control feedback (Coherent). The lasers are arranged such that the chromosomes pass first through the UV beam and then through the 458-nm beam. The laser beams are spatially separated at the flow chamber at a time delay set according to the manufacturer’s instruction manual. The fluorescence from Hoechst excited by the UV (330–360 nm) beam is collected using a 400-nm long pass filter combined with a 480-nm short pass filter. The fluorescence from Chromomycin A3 excited by the 458-nm beam is collected using a 490-nm long pass filter. Collect the data generated for forward scatter, Hoechst fluorescence and Chromomycin fluorescence using Hoechst fluorescence as the trigger signal. The optical light path of the flow cytometer is aligned using 3-μm fluorescent particles (Sphero Rainbow Fluorescent Particles, Spherotech) before chromosome analysis. The laser beams are aligned and adjusted to achieve a minimum peak coefficient of variance of < 1.50 and a maximum peak value for both fluorescence channels.
The data are primarily collected using a MoFlo (Beckman Coulter) equipped with two water cooled Innova 300 series lasers (Coherent). For cytometers using ‘jet-in-air’ analysis such as MoFlo, at sheath pressure of 60 psi, the lasers are delayed by 1.6 μs necessitated by the pinhole design. This time delay can be easily determined by using a pulse monitor oscilloscope. The signals from other flow cytometers are delayed and processed differently; refer to the manufacturer’s instruction manual for more detail.
The instrument is configured for four-way sorting on a high purity sort option of single mode per single drop envelope. A data rate ranging from 2,000 to 12,000 events per second using a 70-μm Cytonozzle tip with optimal setting of the sheath pressure to ~60 psi and the drop drive frequency to ~95 KHz is used. The drop delay of the sort set up is determined using 3 μm fluorescent particles according to the manufacturer’s recommended procedure. The sample data flow rate can be controlled by adjusting the sample differential pressure. Set a low differential pressure of 0.1–0.2 psi to regulate a data rate of ~2,000 events per second for sorting chromosomes. Set to a higher differential pressure of 0.4–0.6 psi for high-speed sorting at a rate of >10,000 events per second.  Ensure the FACS sorter room has an independent temperature control device to maintain the room temperature as fluctuation in room temperature can affect the drop delay and therefore the purity of the sort.
Ensure the FACS sorter room has an independent temperature control device to maintain the room temperature as fluctuation in room temperature can affect the drop delay and therefore the purity of the sort.
PROCEDURE
Cell culture of lymphoblastoid cell line containing chromosome translocation  ~2 to 4 weeks
~2 to 4 weeks
1| Culture a human lymphoblastoid cell line in RPMI medium supplemented with 15% fetal bovine serum (vol/vol) and 5 ml antibiotics mixture to a final concentration of 100 IU ml − 1 penicillin, 100 μg ml − 1 streptomycin and 2 mM L-glutamine in an incubator at 37 °C, 5% CO2.
2| Subculture near-confluent cells to 50% by adding the equivalent volume of fresh growth medium and dividing it into two separate T75 cm2 flasks. We recommend a volume of 50 ml per flask. The change of the phenol red dye color in the medium to yellow is an indication of confluence for suspension cell lines.
-
3| After 24 h of subculturing, add Colcemid or Demecolcine to a final concentration of 0.1 μg ml − 1 for 6 h.
 The incubation time required with the metaphase-arresting drug varies according to the rate of cell growth; the slower the cells divide, the longer the incubation time required. Optimization of this parameter is needed to achieve a high mitotic index with different cell types.
The incubation time required with the metaphase-arresting drug varies according to the rate of cell growth; the slower the cells divide, the longer the incubation time required. Optimization of this parameter is needed to achieve a high mitotic index with different cell types.
Chromosome preparation for flow sorting  3 h
3 h
4| Harvest the cell line in a 50-ml Falcon tube by centrifuging for 5 min at 289g, RT.
5| Carefully discard the supernatant without disturbing the pellet and place the tube upside down on an absorbent paper to drain off most of the medium.
6| Gently resuspend the cell pellet in 5 ml of hypotonic solution with a plastic pipette. Incubate at RT for 15 min.
-
7| Monitor the swelling of cells with Turck’s stain. Mix 5 μl of cell suspension with 5 μl of the stain on a microscope slide. View under the light microscope (Fig. 2a).
 If the cells are not swollen, leave for another 5–10 min. Continue to monitor the swelling of cells and incubate for an increased time as necessary.
If the cells are not swollen, leave for another 5–10 min. Continue to monitor the swelling of cells and incubate for an increased time as necessary. To avoid bursting of cells, view swollen cells on a microscope slide without using a coverslip. The mitotic index can be estimated by counting the proportion of swollen cells in metaphase.
To avoid bursting of cells, view swollen cells on a microscope slide without using a coverslip. The mitotic index can be estimated by counting the proportion of swollen cells in metaphase. 8| After ~15 min in hypotonic solution at RT, centrifuge the cells for 5 min at 289g, at RT. Discard the supernatant and drain the tube briefly on absorbent paper.
9| Resuspended the pellet gently in 3 ml of ice-cold polyamine isolation buffer and incubate on ice for 10 min.
10| Vortex the suspension vigorously for 10–20 s.
-
11| Monitor the chromosome suspension under the fluorescence microscope. Mix 5 μl of cell suspension with 5 μl of the propidium iodide on a microscope slide and cover with a coverslip. Check for single separated chromosomes and an absence of chromosome clusters in the suspension (Fig. 2b).

-
12| If a large number of chromosome clusters are observed (chromosomes that appear to be adhered to each other in clumps), continue to vortex for another 20 s. Monitor the chromosome suspension again under the fluorescence microscope. At this stage, many single intact chromosomes should be seen.
 If this does not improve the number of free single chromosomes, pass the suspension through a 22.5-gauge needle using a 5-ml syringe to release the chromosomes into suspension.
If this does not improve the number of free single chromosomes, pass the suspension through a 22.5-gauge needle using a 5-ml syringe to release the chromosomes into suspension. 13| Centrifuge the chromosome suspension for 2 min at 201g, RT. Filter the supernatant through a 20-μm mesh filter into a 15-ml Falcon tube. Stain the chromosome suspension at 4 °C overnight to a final concentration of 5 μg ml − 1 of Hoechst, 50 μg ml − 1 Chromomycin A3 and 10 mM MgSO4.7H2O.
14| Before running the chromosome suspension on the flow cytometer add sodium citrate and sodium sulfite to a final concentration of 10 mM and 25mM, respectively, and mix gently. Leave the suspension on ice in the dark for at least 1 h.
Derivative chromosome isolation by flow sorting  3 h
3 h
15| If possible estimate the size of each derivative chromosome using G banding23 as described in the Experimental design.
-
16| Calculate the number of derivative chromosomes required to yield 10 ng of DNA. Use Table 1 (column 2) to find the chromosome closest in size to the derivative chromosome and use Table 1, (column 3) to estimate the number of chromosomes needed to yield 10 ng of derivative DNA.
 GenomePlex WGA (see Steps 23–30) requires a minimum of 10 ng starting DNA (see Experimental design).
GenomePlex WGA (see Steps 23–30) requires a minimum of 10 ng starting DNA (see Experimental design). 17| Prepare the instrument (MoFlo) for analysis and sorting. Pre-align the optical light path and pre-determine the drop delay using 3 μm beads (see EQUIPMENT SETUP).
-
18| Obtain the flow karyotype of the cell line by analyzing the stained chromosome sample. Display the data as a bivariate flow karyogram of Hoechst versus Chromomycin fluorescence after gating out clumps and debris25 (Fig. 4a). Create a plot of linear forward scatter versus linear pulse width and draw a region gate on the main chromosome population to exclude clumps and debris (Fig. 4b).

19| Acquire a total of 100,000 events at a rate of 1,000 events per second and analyze the data using Summit v3.1.
20| Prepare two sterile UV-treated 1.5-ml microfuge tubes.
21| Create sorting gates on the derivative chromosome clusters and sort the required number of chromosomes per microfuge tube as determined in Step 16.
22| Briefly spin the microfuge tubes.
Figure 4.
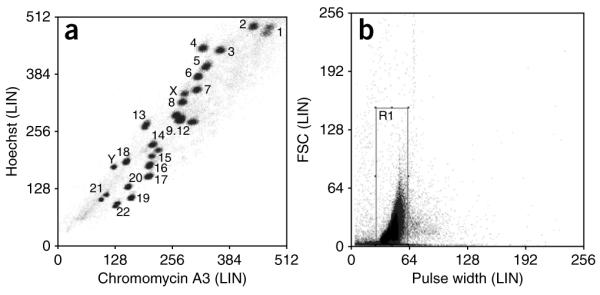
Bivariate flow karyotype of chromosomes from a normal male human lymphoblastoid cell line, GM7016A. (a) The flow karyogram is gated on R1 and displayed as a density plot of Hoechst versus Chromomycin fluorescence. (b) Box shows region gate R1, created on linear forward scatter versus linear pulse width to exclude clumps and debris.
Preparation of samples for whole-genome amplification (GenomePlex WGA)  1 h
1 h
-
23| Add 2.5 μl of Pellet Paint, 8 μl of 2.5 M NaCl and 250 μl of 99.7% (vol/vol) ethanol to each tube, mix well and precipitate at − 20 °C overnight.
 The flow-sorted chromosomes can be stored at − 20 °C until needed.
The flow-sorted chromosomes can be stored at − 20 °C until needed. 24| Pellet the flow-sorted derivative chromosomes by centrifugation for 20 min at 15,700g, RT.
25| Wash the pellets with 100 μl of 70% (vol/vol) ethanol, centrifuge again for 10 min at 15,700g, RT.
26| Remove the ethanol supernatant and allow the pellets to dry at 37 °C for 2 min.
27| Resuspend the pellets in 10 μl of TE buffer.
Fragmentation and library generation  2 h
2 h
-
28| Fragment the DNA and generate the library using the GenomePlex Complete WGA Kit (see REAGENTS) according to the manufacturer’s instructions. Use 10 ng of DNA from Step 27; also set up a positive control tube (use the DNA supplied in the kit) and a negative (water) control.
 The fragmentation and library generation steps should be continued without interruption.
The fragmentation and library generation steps should be continued without interruption.
Amplification of WGA library products  6 h
6 h
29| Amplify the WGA library products using the WGA kit according to the manufacturer’s instructions.
-
30| Run 4–8 μl (5–10%) of the samples, positive and negative controls on a 1.5% (wt/vol) agarose gel26 (70 V for 30 min) (Fig. 5).
 The DNA size should range from 100 to 1,000 bp, with the mean size of ~400 bp.
The DNA size should range from 100 to 1,000 bp, with the mean size of ~400 bp. According to the manufacturer’s instructions, the samples can be stored at −20 °C until ready to purify and analyze. The stability of WGA DNA is equivalent to genomic DNA stored under the same conditions.
According to the manufacturer’s instructions, the samples can be stored at −20 °C until ready to purify and analyze. The stability of WGA DNA is equivalent to genomic DNA stored under the same conditions.
Figure 5.
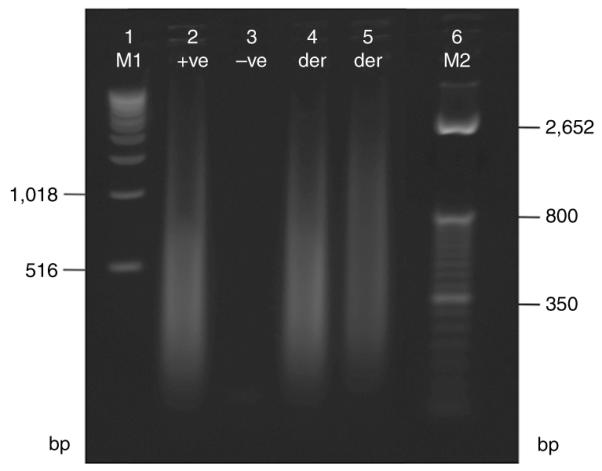
Agarose gel electrophoresis (1.5%) of amplified derivative chromosome products. A positive control from the GenomePlex Complete Whole Genome Amplification Kit is in lane 2 ( + ve), and a water negative control is in lane 3 ( − ve). Lanes 4 and 5 are two different derivative (der) chromosomes following amplification. Note the majority of the product is around 400 bp. M1 = 1 kb Ladder, M2 = TrackIt 50 bp Ladder.
Sample purification  1 h
1 h
31| Add 435 μl of TE to the amplified samples.
32| Insert a Microcon YM-30 sample reservoir into a 1.5-ml centrifuge tube for each sample.
-
33| Pipette the diluted, amplified samples into the sample reservoir and close the lid.
 Do not touch the membrane with the pipette tip.
Do not touch the membrane with the pipette tip. 34| Centrifuge the samples for 11 min at 8,000g, RT.
35| Add 500 μl of TE buffer and centrifuge the samples for 11 min at 8,000g, RT.
36| Place the sample reservoir upside down in a new 1.5-ml microfuge tube, centrifuge samples for 3 min at 1,000g, RT, to transfer the now purified, concentrated samples.
37| Discard the sample reservoirs.
-
38| Measure 1 μl of the amplified samples on a NanoDrop spectrophotometer to assess the DNA yield.
 The manufacturer states that the amplification could be ~500-fold. The minimum amount needed for DNA probe labeling is 300 ng (see Step 39).
The manufacturer states that the amplification could be ~500-fold. The minimum amount needed for DNA probe labeling is 300 ng (see Step 39).
Random labeling of derivative chromosome DNA  1 h (plus overnight incubation)
1 h (plus overnight incubation)
39| Differentially label amplified derivative chromosome DNA as previously reported27, with the slight modification as detailed here. Mix 300 ng of each amplified derivative chromosome DNA from the previous step with 60 μl of 2.5× random primers supplied in the BioPrime DNA Labeling System Kit and add water (also supplied in the kit) to a final volume of 130.5 μl.
40| Denature the samples in a heat block for 10 min at 100 °C and immediately cool on ice.
-
41| Add the following reagents to the sample tubes on ice:
Component der A
Amount per reaction (μl)der B
Amount per reaction (μl)10× dNTP mix 15 15 Cy3-labeled dCTP 0 1.5 Cy5-labeled dCTP 1.5 0 Klenow fragment (40 U μl − 1) 3 3  Do not use the 10× dNTP mixture supplied in the BioPrime DNA Labeling System Kit (see REAGENTS).
Do not use the 10× dNTP mixture supplied in the BioPrime DNA Labeling System Kit (see REAGENTS). 42| Incubate the labeling reactions in the dark at 37 °C overnight (16 h).
43| Stop the reactions by adding 15 μl of stop buffer (supplied in the kit), mix and spin.
Removal of unincorporated nucleotides from labeled reactions  1 h
1 h
44| Clean up the samples according to the manufacturer’s instructions (manufacturer’s protocol G4410-90010 ‘Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis-version 6.0,’ section 4) . Add 315 μl of TE buffer to each of the labeled derivative chromosome DNA samples.
45| Insert two Microcon YM-30 sample reservoirs into 1.5 ml centrifuge tubes.
-
46| Pipette the two diluted amplified samples into separate sample reservoirs, close the lid.
 Do not touch the membrane with the pipette tip.
Do not touch the membrane with the pipette tip. -
47| Centrifuge the samples for 10 min at 8,000g, RT. Discard the flow-through.
 The samples should be retained on the filter and the filter should be moist. Unincorporated nucleotides should make the flow-through slightly colored.
The samples should be retained on the filter and the filter should be moist. Unincorporated nucleotides should make the flow-through slightly colored.
48| Add 480 μl of TE buffer and centrifuge the samples for 10 min at 8,000g, RT. Discard the flow-through (flow-through should be less colored than in Step 47).
49| Place the sample reservoir upside down in a new 1.5-ml microfuge tube. Centrifuge samples for 1 min at 8,000g, RT, to collect the purified, concentrated samples.
50| Measure and record the volume of each eluate. If the sample volume exceeds 80.5 μl, return the sample to its sample reservoir and centrifuge for 1 min at 8,000g, RT. Discard flow-through.
51| Place the sample reservoir upside down in a new 1.5-ml microfuge tube. Centrifuge the sample for 1 min at 8,000g, RT, to transfer the purified, concentrated sample to the microfuge tube.
52| Measure and record the volume of each eluate; bring the sample volume to 80.5 μl with TE buffer. (If eluate volume is still more than 80.5 μl, repeat Steps 50 and 51 until volume is ≤ 80.5 μl.) Discard the sample reservoirs.
53| Measure 1.5 μl of the labeled derivative chromosome samples on a NanoDrop spectrophotometer to assess the yield and specific activity (SA). Using the Microarray program, the NanoDrop will record the dye incorporation as pmol μl − 1 and yield as ng μl − 1.
-
54| Calculate the specific activity using the calculation below
where specific activity is the pmol dye per μg genomic DNA. SA and yield of labeled derivative chromosome DNA should be approximately 20–30 pmol μg − 1 DNA and between 20 and 30 μg total DNA in 80.5 μl sample volume.
SA and yield of labeled derivative chromosome DNA should be approximately 20–30 pmol μg − 1 DNA and between 20 and 30 μg total DNA in 80.5 μl sample volume. Labeled derivative chromosome DNA can be stored overnight or over a weekend in the dark at − 20 °C. Generally proceed to hybridization as soon as possible.
Labeled derivative chromosome DNA can be stored overnight or over a weekend in the dark at − 20 °C. Generally proceed to hybridization as soon as possible.
Sample preparation for hybridization to an Agilent 244k Array  1 h
1 h
-
55| Hybridize the samples onto an Agilent 244k Array according to the manufacturer’s instructions (G4410-90010 ‘Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis-version 6.0,’ section 5).
Add the following reagents to the sample tubes:Component Amount (μl) Cy3-labeled der A and Cy5-labeled der B 158 Cot-1 DNA (1 mg ml − 1) 50 Agilent 10× Blocking Agent 52 Agilent 2× hybridization buffer 260 Total volume 520  Aliquot hybridization buffer. Gently mix before use (see REAGENT SETUP).
Aliquot hybridization buffer. Gently mix before use (see REAGENT SETUP). 56| Mix the samples by pipetting up and down. Spin to collect the sample.
57| Denature the samples for 3 min at 95 °C in a heat block.
58| Immediately transfer the sample tubes for 30 min to a 37 °C heat block in the dark.
59| Spin the samples in a microfuge for 1 min at maximum speed to collect the sample.
Agilent 244k microarray hybridization  30 min (plus 40 h hybridization)
30 min (plus 40 h hybridization)
60| Assemble a gasket slide into an Agilent SureHyb chamber base with the gasket label facing up.
61| Add 490 μl of the 520 μl reaction mix along the length of the hybridization area of the gasket slide taking care not to get too close to the gasket edge.
-
62| Place the microarray slide on top of the gasket slide with the ‘active side’ facing down.
 Each microarray is printed on the side of the glass slide containing the ‘Agilent’-labeled barcode or the ‘active side.’ The numeric barcode is on the ‘inactive side,’ which has no array. Remember the word Agilent goes onto the hybridization mix, so write on the bench ‘Agilent side facing down’.
Each microarray is printed on the side of the glass slide containing the ‘Agilent’-labeled barcode or the ‘active side.’ The numeric barcode is on the ‘inactive side,’ which has no array. Remember the word Agilent goes onto the hybridization mix, so write on the bench ‘Agilent side facing down’. 63| Add the SureHyb chamber cover onto the glass slides and assemble. Tighten the clamp onto the chamber.
64| Using a rotating motion, allow the liquid to wet the slides. Vertically rotate the assembled chamber to wet the slides and assess the mobility of the large bubbles. Ensure there are no small bubbles stuck at the side of the slide. If small bubbles can be seen, gently tap the chamber on the bench to dislodge them.
65| Place the assembled chamber in a rotating rack set to 20 rpm in a hybridization oven. Use an empty chamber as a balance. Hybridize for 40 h at 65 °C.
Setting up Wash Buffers and the equipment  30 min
30 min
-
66| Wash the Agilent 244k arrays according to the manufacturer’s instructions (manufacturer’s protocol G4410-90010 ‘Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis-version 6.0,’ section 5). The wash steps are listed below.
 When washing microarrays, do not exceed four slides per trough.
When washing microarrays, do not exceed four slides per trough. -
67| Fill two clean glass troughs with Wash Buffer 1 (one to dismantle slides in and one for the first wash) and one trough with Wash Buffer 2 (for second wash).
 Do not use detergent to wash glassware used in the wash procedure. Detergent may leave fluorescent residue.
Do not use detergent to wash glassware used in the wash procedure. Detergent may leave fluorescent residue. 68| Put a slide rack and magnetic flea (1.5 inches in length) into one trough containing Wash Buffer 1 and place on a magnetic stirrer at RT.
69| Put a glass slide rack into the trough containing Wash Buffer 2 and heat in a microwave for ~30 s on a high setting to warm to 37 °C. Once the correct temperature is achieved, add a magnetic flea and place in an incubator to maintain this temperature until required.
Microarray washing  30 min
30 min
70| Turn on the Agilent Scanner 20 min before washing the slides to allow the lasers to warm up.
-
71| Remove a SureHyb chamber containing a microarray from the 65 °C incubator and disassemble the chamber.
 Take care as the metal chambers are hot to hold.
Take care as the metal chambers are hot to hold. If, when assembling the hybridization mix, microarray slide and gasket slide, one was aware that a small amount of sample spilled outside of the gasket, this may have caused the gasket slide and array slide to have adhered tightly or even the glass slides to have adhered to the metal chamber. If in doubt, disassemble the entire chamber (having removed the screw) submerged under Wash Buffer 1.
If, when assembling the hybridization mix, microarray slide and gasket slide, one was aware that a small amount of sample spilled outside of the gasket, this may have caused the gasket slide and array slide to have adhered tightly or even the glass slides to have adhered to the metal chamber. If in doubt, disassemble the entire chamber (having removed the screw) submerged under Wash Buffer 1. 72| Remove the glass slide sandwich from the metal chamber by holding the edges (maintain the orientation) and immediately transfer and submerge in Wash Buffer 1.
-
73| Keep the sandwich completely submerged in Wash Buffer 1. Remove the microarray slide away from the gasket slide using blunt forceps.
 Do not allow the microarray slide surface to dry out at any stage during the wash process.
Do not allow the microarray slide surface to dry out at any stage during the wash process. 74| Quickly transfer the microarray slide to the slide rack in the second trough containing Wash Buffer 1 on the magnetic stirrer. Repeat Steps 72–74 for additional microarrays.
75| Turn on the magnetic stirrer to achieve good but not vigorous mixing (maintain a small vortex in the middle of buffer). Wash for 5 min.
76| 15 s before the first wash has finished, remove the trough containing Wash Buffer 2 from the incubator.
77| At the end of wash 1, turn off the magnetic stirrer, remove the trough from the magnetic stirrer, place the pre-warmed trough of Wash Buffer 2 onto the stirrer and transfer the slides to it.
78| Turn on the magnetic stirrer and wash for 1 min.
79| After 1 min, turn off the stirrer and remove the trough containing the microarrays.
-
80| Slowly (taking 10 s per slide) remove the slides from Wash Buffer 2 holding the edges of each slide vertically. Place each slide into a slide holder supplied with the scanner. With the open slide holder door facing toward you, insert the glass slide with the active side labeled with the Agilent barcode facing up. When the door is closed, the numeric barcode side of the slide is fully visible at the back of the holder.
 The microarray slides are hydrophobic, so slow removal from Wash Buffer 2 ensures the slides are dry and ready to be scanned.
The microarray slides are hydrophobic, so slow removal from Wash Buffer 2 ensures the slides are dry and ready to be scanned. Scan microarray slides immediately to minimize the effect of environmental oxidants on signal intensities.
Scan microarray slides immediately to minimize the effect of environmental oxidants on signal intensities.
Microarray scanning  30 min
30 min
81| Using Agilent Scan Control scanning software (ver 8.3 for scanner C), select the following scan settings:
Scan region Scan area 61 × 21.6 mm Scan resolution (μm) 5 Dye channel Red and Green Green PMT (%) 100 Red PMT (%) 100 82| Set the following settings for automatic file naming:
Prefix1 Instrumental Serial Number Prefix2 Array Barcode -
83| Scan microarrays using an Agilent Scanner B/C and Scan Control software, or an alternative scanner to generate a tiff image of the microarray (e.g., GenePix 4000B Scanner).
 When an image is viewed in Feature Extraction, the maximum red and green values as seen in the set image color display range on auto scale should be >2,000.
When an image is viewed in Feature Extraction, the maximum red and green values as seen in the set image color display range on auto scale should be >2,000.
Data extraction using agilent feature extraction software and analysis  ~2 h
~2 h
84| Extract the fluorescence intensity for both the Cy3 (green) and Cy5 (red) channels using Feature Extraction following the manufacturer’s instructions.
85| Import Feature Extraction text files into DNA Analytics 4.0 or use another software, e.g., the standard functions in the freely available software ‘R’ that will plot normalized log2 ratios against chromosome position for each oligonucleotide on the array.
![]()
Steps 1–3, cell culture: ~2–4 weeks
Steps 4–14, chromosome preparation for flow sorting: 3 h
Steps 15–22, flow sorting derivative chromosomes: 3 h
Steps 23–27, preparation of samples for GenomePlex WGA: 1 h
Step 28, fragmentation and library generation: 2 h
Steps 29–30, amplification of WGA library products: 6 h
Steps 31–38, sample purification: 1 h
Steps 39–43, random labeling of derivative chromosome DNA: 1 h (plus overnight incubation)
Steps 44–54, removal of unincorporated nucleotides from labeled reactions: 1 h
Steps 55–59, sample preparation for hybridization to an Agilent 244k array: 1 h
Steps 60–65, Agilent 244k microarray hybridization: 30 min (plus 40 h incubation)
Steps 66–69, setting up Wash Buffers and equipment: 30 min
Steps 70–80, microarray washing: 30 min
Steps 81–83, microarray scanning: ~10 min per 244k microarray slide
Steps 84–85, data extraction using Agilent Feature Extraction Software and analysis: ~2 h
![]()
Troubleshooting advice can be found in Table 2.
Table 2.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 11 | Low chromosome yield | Low mitotic index | Optimize incubation time with metaphase- arresting drug |
| Undissociated chromosome clusters | Extend vortexing time or syringe the suspension to release the chromosomes into suspension |
||
| 18 | High percentage of debris | Check pH of hypotonic solution and polyamine isolation buffer |
Replace reagents (Hypotonic and polyamine isolation buffer) |
| Chromosome fragmentation due to physical force |
Reduce vortexing time | ||
| Poor data resolution | Insufficient staining time | Extend fluorescence dyes staining time | |
| Effect from sodium sulfite and sodium citrate |
Omit either reagent | ||
| 30 | Low yield after GenomePlex WGA | Derivative chromosome DNA is degraded or was considerably < 10 ng |
Increase the amount of input DNA |
| Fragmentation time was not optimal | A 4-min fragmentation time is crucial | ||
| Negative control produced a product | Reagents contaminated | Replace reagents | |
| 47 | Flow through is brightly colored and the membrane has retained only a small amount of color |
Cleanup column membrane failed | Retrieve the sample by putting flow through back into a new column and clean up as described in the protocol |
| 54 | Low yield and low SA after labeling | Klenow inactive | Test reagents |
| 83 | No fluorescent signal on microarray | Check the slide was in the correct orientation during hybridization |
Repeat experiment |
| Low signal | Poor atmospheric conditions | Monitor the ozone levels, temperature and humidity in the laboratory where the experiment was conducted and, in particular, in the proximity of the scanner |
ANTICIPATED RESULTS
The scanned microarray image after an array paint hybridization on a genome-wide array should have a minority of spots showing bright red, green or orange fluorescence.
Signal intensities are extracted for all array features and log2 ratios for Cy5/Cy3 intensities are calculated as described in Step 85. Log2 ratios are then plotted against chromosome base-pair position for each oligonucleotide on the array. As the Cy5 intensities are divided by the Cy3 intensities, the log2 ratios for the oligonucleotide probes corresponding to the derivative chromosome labeled in Cy5 will be high, whereas the log2 ratios for the oligonucleotide probes corresponding to the derivative chromosome labeled in Cy3 will be low. Along the length of a chromosome involved in the rearrangement, there will be a transition between either a high to a low ratio, or a low to a high ratio. The transition identifies the chromosome breakpoint. The genomic density of features on the microarray will determine the accuracy of the breakpoint determination. If there is a data point that has an intermediate log2 ratio, neither high nor low, then it is likely that the chromosome breakpoint falls within the sequence of that array feature (Fig. 1).
The dynamic range of log2 ratios in an array paint experiment conducted on the Agilent platform in our hands is usually at least − 4 to + 4 as shown in Figure 6, which shows the array paint results for a typical Agilent 244k hybridization following this protocol. Both chromosome breakpoints are immediately obvious where there is a dramatic shift from high to low log2 ratio values (Fig. 6a,c). Zooming in to the transition sites (Fig. 6b,d) it is possible to determine the genomic positions for the two chromosome breakpoints containing regions. However, owing to an uneven probe distribution on catalog array designs, the size of these regions will vary. In this instance, the chromosome A region is 7.9 kb and the chromosome B region is 27 kb. An average spacing for features on this array is 10 kb.
Figure 6.

Array paint of derivative chromosomes A and B on a Agilent 244k whole genome array. (a) Log2 ratios plotted against chromosome A position of ~10 Mb region using the freely available software R. Transition from a high to a low log2 ratio indicates the chromosome A breakpoint. (b) Zoom in of chromosome A breakpoint region. Flanking clones are ~7.9 kb apart on a 244k feature array. (c) Log2 ratios plotted against chromosome B position of an ~10-Mb region using the freely available software R. Transition from a low to a high log2 ratio indicates the chromosome B breakpoint. (d) Zoom in of chromosome B breakpoint region. Flanking clones are ~27 kb apart on a 244k feature array.
ACKNOWLEDGMENTS
The labeling procedure described in Steps 39–43 was originally developed by Heike Fiegler28 and slightly modified for this protocol. We acknowledge Diana Rajan and Leong Siew Hong for proofreading and checking this protocol. We thank John Crolla (Wessex Regional Genetics Laboratory) for supplying the t(3;20) cell line used in Figure 3. This work was supported by the Wellcome Trust [Grant no. WT077008].
References
- 1.Millar JK, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- 2.Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMullan TW, et al. A candidate gene for congenital bilateral isolated ptosis identified by molecular analysis of a de novo balanced translocation. Hum. Genet. 2002;110:244–250. doi: 10.1007/s00439-002-0679-5. [DOI] [PubMed] [Google Scholar]
- 4.van Bakel I, Holt S, Craig I, Boyd Y. Sequence analysis of the breakpoint regions of an X;5 translocation in a female with Duchenne muscular dystrophy. Am. J. Hum. Genet. 1995;57:329–336. [PMC free article] [PubMed] [Google Scholar]
- 5.Spitz F, et al. A t(2;8) balanced translocation with breakpoints near the human HOXD complex causes mesomelic dysplasia and vertebral defects. Genomics. 2002;79:493–498. doi: 10.1006/geno.2002.6735. [DOI] [PubMed] [Google Scholar]
- 6.Mills KI, et al. Amplification and sequencing of genomic breakpoints located within the M-bcr region by Vectorette-mediated polymerase chain reaction. Leukemia. 1992;6:481–483. [PubMed] [Google Scholar]
- 7.Fiegler H, et al. Array painting: a method for the rapid analysis of aberrant chromosomes using DNA microarrays. J. Med. Genet. 2003;40:664–670. doi: 10.1136/jmg.40.9.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinkel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 9.Gribble SM, et al. Ultra-high resolution array painting facilitates breakpoint sequencing. J. Med. Genet. 2007;44:51–58. doi: 10.1136/jmg.2006.044909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baptista J, et al. Breakpoint mapping and array CGH in translocations: comparison of a phenotypically normal and an abnormal cohort. Am. J. Hum. Genet. 2008;82:927–936. doi: 10.1016/j.ajhg.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fauth C, et al. Micro-array analyses decipher exceptional complex familial chromosomal rearrangement. Hum. Genet. 2006;119:145–153. doi: 10.1007/s00439-005-0103-z. [DOI] [PubMed] [Google Scholar]
- 12.Foster RE, et al. Characterization of a 3;6 translocation associated with renal cell carcinoma. Genes Chromosomes Cancer. 2007;46:311–317. doi: 10.1002/gcc.20403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gribble SM, et al. The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J. Med. Genet. 2005;42:8–16. doi: 10.1136/jmg.2004.024141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez-Perales S, et al. Cloning of a new familial t(3;8) translocation associated with conventional renal cell carcinoma reveals a 5 kb microdeletion and no gene involved in the rearrangement. Hum. Mol. Genet. 2004;13:983–990. doi: 10.1093/hmg/ddh111. [DOI] [PubMed] [Google Scholar]
- 15.Vermeesch JR, et al. Tetrasomy 12pter-12p13.31 in a girl with partial Pallister–Killian syndrome phenotype. Eur. J. Med. Genet. 2005;48:319–327. doi: 10.1016/j.ejmg.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 16.Backx L, et al. Array painting using microdissected chromosomes to map chromosomal breakpoints. Cytogenet Genome Res. 2007;116:158–166. doi: 10.1159/000098181. [DOI] [PubMed] [Google Scholar]
- 17.Veltman IM, et al. Chromosomal breakpoint mapping by arrayCGH using flow-sorted chromosomes. Biotechniques. 2003;35:1066–1070. doi: 10.2144/03355dd03. [DOI] [PubMed] [Google Scholar]
- 18.Gribble SM, et al. Applications of combined DNA microarray and chromosome sorting technologies. Chromosome Res. 2004;12:35–43. doi: 10.1023/b:chro.0000009325.69828.83. [DOI] [PubMed] [Google Scholar]
- 19.Howarth KD, et al. Array painting reveals a high frequency of balanced translocations in breast cancer cell lines that break in cancer-relevant genes. Oncogene. 2008;27:3345–3359. doi: 10.1038/sj.onc.1210993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carter NP, et al. Reverse chromosome painting: a method for the rapid analysis of aberrant chromosomes in clinical cytogenetics. J. Med. Genet. 1992;29:299–307. doi: 10.1136/jmg.29.5.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Telenius H, et al. Chromatid contamination can impair the purity of flow-sorted metaphase chromosomes. Cytometry. 1993;14:97–101. doi: 10.1002/cyto.990140116. [DOI] [PubMed] [Google Scholar]
- 22.van den Engh G, Trask B, Lansdorp P, Gray J. Improved resolution of flow cytometric measurements of Hoechst- and chromomycin-A3-stained human chromosomes after addition of citrate and sulfite. Cytometry. 1988;9:266–270. doi: 10.1002/cyto.990090313. [DOI] [PubMed] [Google Scholar]
- 23.Mayall BH, et al. The DNA-based human karyotype. Cytometry. 1984;5:376–385. doi: 10.1002/cyto.990050414. [DOI] [PubMed] [Google Scholar]
- 24.Freeman JL, et al. Definition of the zebrafish genome using flow cytometry and cytogenetic mapping. BMC Genomics. 2007;8:195. doi: 10.1186/1471-2164-8-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng BL, Yang F, Carter NP. Flow analysis and sorting of microchromosomes ( < 3 Mb) Cytometry A. 2007;71:410–413. doi: 10.1002/cyto.a.20394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sambrook J, Russell DW, editors. Molecular Cloning, a Laboratory Manual. Vol. 1. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2001. Gel electrophoresis; pp. 5.1–5.13. chapter 54. [Google Scholar]
- 27.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fiegler H, Redon R, Carter NP. Construction and use of spotted large-insert clone DNA microarrays for the detection of genomic copy number changes. Nat. Protoc. 2007;2:577–587. doi: 10.1038/nprot.2007.53. [DOI] [PMC free article] [PubMed] [Google Scholar]