

Abstract
Passive immunotherapy, including adoptive T cell therapy and antibody therapy, has shown encouraging results in cancer treatment lately. However, active immunotherapy of solid cancers remains an elusive goal. It is now known that the human innate immune system recognizes pathogen-associated molecular patterns (PAMP) conserved among microbes or damage-associated molecular patterns (DAMP) released from tissue injuries to initiate adaptive immune responses during infection and tissue inflammation, respectively. In contrast, how the innate immune system recognizes endogenously arising cancer remains poorly understood at the molecular level, which poses a significant roadblock to the development of active cancer immunotherapy. We hereby review the current knowledge of how solid cancers directly and indirectly interact with cells of the human innate immune system, with a focus on the potential impact of such interactions to the resultant adaptive immune responses against cancer. We believe that understanding cancer and innate immune system interactions may allow us to better manipulate the adaptive immune system at the molecular level in order to develop effective active immunotherapy against cancer. Current and future perspectives in clinical development that exploits these molecular interactions are discussed.
Keywords: Innate immune system, apoptosis, necrosis, damage associated molecular pattern, immunotherapy, dendritic cell, tumor-associated antigen
Introduction
Despite a predominantly immunosuppressive tumor microenvironment,1, 2 spontaneous T cell and antibody responses against tumor-associated antigens (TAA) can be induced in tumor-bearing hosts.3–5 In a small fraction of patients, anti-tumor immunity may lead to spontaneous tumor regression or control of tumor expansion, with perhaps the most compelling evidence documented in patients with melanoma3 and paraneoplastic neurologic disorders.6
The ultimate goal of active cancer immunotherapy is to achieve the anti-tumor immunity that has been demonstrated in the sporadic examples of spontaneous tumor regression/containment and recent success of passive immunotherapy such as adoptive T cell therapy and antibody therapy.7–10 Recent advances in basic science have defined several ligand/receptor interactions and molecular pathways that have significant impact on subsequent adaptive immune responses in various circumstances. For example, it is now known that the human innate immune system, through its cell-surface pattern recognition receptors, recognizes PAMP conserved among microbes or DAMP released from tissue injuries to initiate adaptive immune responses during infection and tissue inflammation, respectively.11, 12 Despite this wealth of knowledge, how spontaneous anti-tumor immune responses are initiated is still poorly understood at the molecular level, which poses a major obstacle in developing effective active immunotherapy.
Direct cancer and innate immune system interactions
The major effector cells of the immune system that directly target cancer cells include natural killer cells (NK), dendritic cells (DC), macrophages, polymorphonuclear leukocytes (PMN including neutrophils, eosinophils, and basophils), mast cells, and cytotoxic T lymphocytes. NK cells, DC, PMN, mast cells, and macrophages are first-line effectors to damaged cells and cancer cells. Natural killer T cells (NKT) and γδ T cells play roles as both innate and adaptive components, through close interactions with cells of the adaptive immune system, such as CD4+ and CD8+ T lymphocytes with cytotoxic effects and memory.13 The importance of innate immune system in limiting cancer progression has been highlighted recently with the following direct molecular interactions between cancers and innate immune effector cells.
NK cells
NK cells constitute the primary innate immune cell type responsible for killing non-MHC expressing cancer cells, releasing small cytotoxic proteins such as perforin and granzyme that cause apoptosis in target cells. There are two functional types of receptors on the NK cell surface: stimulatory receptors and inhibitory receptors. Natural killer group 2D (NKG2D) molecule is perhaps the best-known stimulatory receptor.14 The ligands on tumor cells for NKG2D include MHC class-I-chain-related protein A (MICA),15 MICB,16, 17 UL16 binding protein18 in human, and minor histocompatibility molecule H60, Retinoic acid early transcript 1 protein (RAE-1 α-ε), UL16 binding protein-like transcript 1 protein in mice19–22. Fig. 1 shows the interactive diagram of such interactions in humans. The binding of the above stress-related ligands with NKG2D stimulate NK cells, leading to secretion of IFN-γ and perforin, release of inflammatory cytokines, and the induction of apoptosis in cancer cells. Other NK stimulatory receptors have also been characterized, such as NKp30,23 NKp44,24 and NKp4625 in humans, NK-cell receptor protein 1 (Nkrp1),26, 27 Ly49d/h,28, 29 NKG2C/E-CD94 in mice,14, 30 and DNAX accessory molecule31 in both humans and mice. The inhibitory receptors of NK cells consist of the human killer-cell immunoglobulin-like receptors (KIR),32, 33 the mouse Ly49a/c/g2,34–36 and NKG2A-CD94 lectin-like receptors shared by both humans and mice37. The non-classical MHC class I molecule, HLA-G, on tumors also functions as a ligand for KIR that can inhibit cytotoxicity mediated by NK cells. Ly49 family receptors specifically recognize MHC class I or MHC class-I-like molecules. The non-classical MHC class I molecule HLA-E is the ligand for human NKG2A/CD94 heterodimer receptors.38
Figure 1.
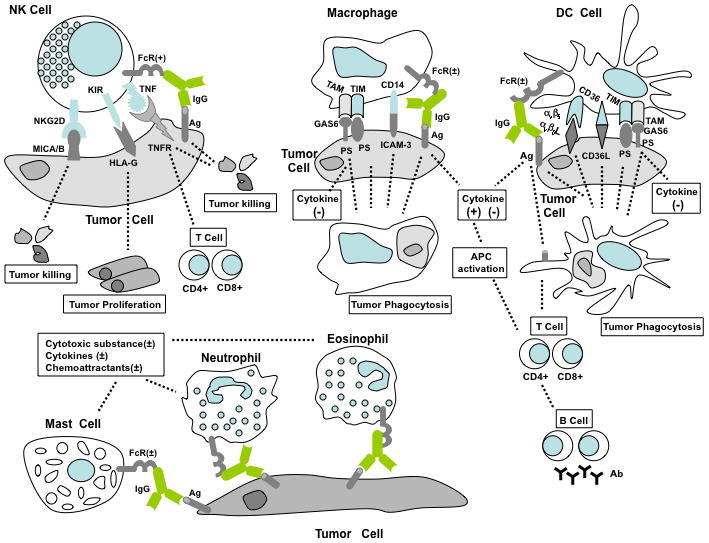
Direct cancer recognition by the innate immune system. NK, macrophages, DC, neutrophils, eosinophils, and mast cells are the cellular components of the innate immune system. NKG2D, a stimulatory receptor on NK cells, specifically recognizes MICA/B on cancers to stimulate cell killing. Inhibitory receptors such as KIR detect non-classical MHC class I molecule HLA-G on cancers to prevent NK cell cytotoxicity. Binding of TNF family ligands on NK cells to TNF family receptors on tumor cells triggers cancer apoptosis, which gives rise to subsequent CD4+ and CD8+ T cells. The function of NK cells is also mediated by activating FcR/CD16 through ADCC. Macrophages phagocytose apoptotic cancer cells via CD14, TIM, TAM and FcR receptors through the interaction with ICAM-3, PS, PS/Gas6 and immune complex, respectively. Gas6 functions as the “bridge” between PS and TAM receptors. Both activating and inhibitory Fc receptors (FcR+, activating; FcR−, inhibitory) exist on macrophages and associate with the production of cytokines and superoxide substances. DC may uptake apoptotic tumor cells through αvβ5, CD36, FcR, TIM and TAM receptors. Cytokine secretion from DC and macrophages promote antigen presenting cell activation, leading to cellular and humoral adaptive immune response. Activating FcR on DC help tumor cell antigen presentation. Activating and inhibitory FcR are also expressed on neutrophils, eosinophils, and mast cells, which directly recognize antibody-coated tumors to promote or inhibit the secretion of cytokines and chemokines from innate immune cells.
TNF family ligands are widely expressed on the NK cell surface: TNF, TNF-related apoptosis-inducing ligand (TRAIL), lymphotoxin, Fas ligand, 4-1BB ligand, lymphotoxin-like inducible protein that competes with glycoprotein D for binding herpesvirus entry mediator on T cells (LIGHT), OX40 ligand, CD40 ligand, CD30 ligand, and CD27 ligand. In parallel, the TNF family of receptors, TNF receptor, TRAIL receptor, lymphotoxin receptor, Fas, 4-1BB, HVEM/LTβ receptor, OX40, CD40, CD30, CD27 are expressed in many tumor cell lines.39–43 The complementary binding between TNF ligands and TNF receptors can efficiently induce tumor cell apoptotic death. Hence, engineered or induced expression of TNF family receptors on cancer cells represents one avenue being actively pursued for active immunotherapy. Moreover, LIGHT/HVEM (LTβR) signaling helps develop the adaptive immune response through priming and recruiting tumor-specific T cells.44–46 NK cells, activated by LIGHT, produce IFN-γ to directly promote the expansion and differentiation of T cells. Studies from mouse LIGHT tumor model suggest that intratumoral NK cells and local IFN-γ are required for priming cytotoxic T cells and tumor rejection.46
Tumors coated with antibodies against cell-surface molecules can be directly recognized by several innate immune cells through Fc receptors (FcR), the receptors for immunoglobulin. The FcR for IgG (FcγR) include two functional types of receptors, activating and inhibitory receptors. Antibody coated tumor cells can be killed by NK cells or macrophages with activating FcγR, termed ADCC or antibody-dependent cell-mediated cytotoxicity.47, 48 NK cells solely express the activating FcγR CD16 for IgG49 without inhibitory FcγR detected.
Macrophages
Apoptotic tumor cells can be efficiently eliminated by macrophages to avoid autoimmunity. These tumor cells express the so-called “eat me” molecules at cell surface (Fig. 1) for recognition and phagocytosis by macrophages. These signals include lipid phosphatidylserine (PS), oxidized PS (ox-PS), oxidized low-density lipoprotein (oxLDL), and the multi-functional protein calreticulin (CRT).50 These molecules are translocated or redistributed to expose at the tumor cell surface during apoptosis.51, 52 CRT is also associated with the CD91 receptor on macrophages and involved in the engulfment of apoptotic cells through interaction with soluble complement protein C1q and its ligand PS. Scavenger receptors, such as SR-A, CLA-1, CD36, CD68, LOX-1 and stabilin-2, can bind ox-PS and oxLDL motifs on apoptotic tumor cells. T cell immunoglobulin mucin (TIM) proteins (TIM-1, TIM-3 and TIM-4) were recently identified as critical receptors for PS to mediate uptake of apoptotic cells.53–55 CD36 may also form complex receptors with αvβ3 integrin on macrophages; while CD14 on macrophages can serve as the receptor for ICAM-3, and trigger phagocytosis and clearance of apoptotic cells.56 Under normal circumstances in the tumor environment, the interaction between apoptotic tumor cells and macrophage phagocytes leads to immune tolerance without provoking significant pro-inflammatory cytokines. Unlike NK cells, macrophages express both activating and inhibitory FcγR simultaneously. Activating FcγR stimulate cytotoxicity to tumor cells. In contrast, FcγRIIB is the only inhibitory receptor on macrophages in mice, which is responsible for inhibitory effects on macrophage including inhibition of phagocytosis, decreased cytokine release, superoxide production, and blocking Toll-like receptor 4 (TLR4) signaling pathway.57
In the tumor milieu, macrophages are believed to be major contributors to the chronic inflammation that renders an immune suppressive environment benefiting tumor growth.2 Direct and indirect interactions of macrophages and cancer cells in the above and following sections provide molecular mechanisms underlying such effects.
DC
DC are perhaps the most potent professional antigen presenting cells, and bridge between innate and adaptive immune system. The two major groups of DC are known as the myeloid DC and the plasmacytoid DC. Functional subsets of myeloid DC in the skin, epidermal Langerhans cells and dermal interstitial DC are also characterized with distinct immune induction potentials. Activated epidermal Langerhans cells secret interleukin 15 (IL-15) and induce CD4+ and CD8+ T cell priming to elicit cellular immunity. Dermal interstitial DC stimulate B cell priming to produce humoral immunity.58, 59 Engaging DC via different receptors and subpopulations may stimulate different inflammation responses, producing multiple T cell outcomes including T helper cells of type 1 (Th1), Th2, Th17, Th21 and T regulatory cells.
With respect to direct interactions with cancer cells, DC phagocytose apoptotic cancer cells via αvβ5 integrin and CD36 receptors.60 Similar to macrophages, DC can recognize the so-called “eat me” signals on apoptotic cells through endocytotic receptors, scavenger receptors, and TIM receptors. Additionally, the apoptotic cell marker PS can be captured by TAM receptor protein tyrosine kinases (TYRO3, AXL and MER) on DC and macrophages via molecular linkers Gas6/protein S, and through αvβ3 integrin via linker MFG-E8. TAM receptors promote phagocytosis of apoptotic tumor cells and inhibit inflammation in DC and macrophages.61–63 The integrin αvβ3 complex is able to mediate engulfment of apoptotic cancer cells.64, 65 Similar to macrophages, phagocytosis of apoptotic tumor cells by DC in the absence of danger signals generally leads to immune tolerance.
DC also express both activating and inhibitory FcγR. Comparing to other fashions of antigen uptake, antibody-coated tumor cells are more efficiently internalized into DC through activating FcγR, leading to more efficient MHC class I and II–restricted antigen presentation and induction of tumor-specific effector and memory T cells.66 Therefore, inflammation and adaptive immune response could be trigged by DC-cancer cell encountering through activating FcγR signaling pathway, and this process is negatively modulated by co-expression of inhibitory FcγRIIB and TAM receptors on DC. However, it is necessary to note that uptake of antigens does not accompany induction of effector T cells. The induction of active adaptive immunity requires danger signals or maturation of DC during antigen encountering as discussed in the following sections.
PMN and mast cells
Tumor-associated PMN and mast cells can have a significant role in tumorigenesis and metastasis.67 However, fewer studies have been focused on the direct molecular recognition between tumor cells and PMN. The known examples are activating and inhibiting FcγR on PMN and mast cells to interact with antibody coated antigens on tumor cells. Activating FcγR induces neutrophils to release cytokines and chemoattractants which influence recruitment and activation of DC and macrophages in tumor environment.48, 68, 69 Activation of inhibitory FcγRIIB on neutrophils decreases products of reactive oxygen species, which are cytotoxic against tumors. While in mast cells, stimulating FcγRIIB can decrease Ig-E mediated release of granular molecules, IL-4 cytokine and histamine which trigger inflammatory response in tumor environment.57 One study has shown that increased direct contact between tumor cells and PMN plus macrophages in mice is responsible for resisting lethal doses of cancer cells.70, 71 However, the molecular mechanism for such efficacy remains unclear.
Clinical development based on direct cancer and innate immune system interactions
A few NK-cell-based cancer therapies are now being tested in clinical trials, most of which utilize direct cytotoxic activity of NK cells against cancer, such as activation of NK cell-surface stimulatory receptors or blocking surface inhibitory receptors. Based on preclinical studies showing tumor regression induced through genetic overexpression of NKG2D, several drugs that selectively up-regulate NKG2D ligands on tumor cells are introduced to complement chemotherapy such as DNA damage-inducing cisplatin and 5-FU,72 the histone deacetylase inhibitor sodium valproate73. Low-dose proteasome inhibitor bortezomib has also been applied in human breast cancer74 and hepatocellular carcinoma75 to increase NK activating ligands and subsequent tumor lysis. TRAIL on NK cells can efficiently trigger cancer cell apoptosis even after chemotherapy, which induces resistance to intrinsic apoptotic process in cancer. Thus, modulating TRAIL pathway on NK cells is also a new approach combining NK cell-based therapy with chemotherapy.76, 77 In addition to activation of NK-surface stimulatory receptors, therapeutic monoclonal antibodies such as the anti-KIR monoclonal antibody blocking inhibitory signaling in NK cells have been tested in clinical trials on acute myeloid leukemia and multiple myeloma patients.78
Several clinically useful monoclonal antibodies have now been approved for lymphoma and leukemia, with some functioning in part through ADCC, such as B-lymphocyte antigen CD20-targeted humanized monoclonal antibodies Rituximab, Tositumomab, and Veltuzumab.79, 80
Cancer and innate immune system interactions through DAMP and their partner receptors
In addition to the direct cancer/innate immune system interactions, a large number of molecules released due to cancer cell death, may function as DAMP and interact with innate immune cells (Table 1). Such cancer-derived DAMP include both intracellular molecules and extracellular matrix (ECM) molecules released from apoptotic and necrotic tumor cells. Intracellular molecules that can function as DAMP include heat shock proteins (HSP), high-mobility group box-1 protein (HMGB1), adenosine triphosphate (ATP), mitochondrial formyl peptides, mitochondrial DNA, and uric acid. Special attention is given to NY-ESO-1 and possibly others, which are initially identified as TAA but lately have been recognized with similar properties as DAMP. ECM danger molecules include hyaluronan and heparan sulfate fragments, S100 family proteins, fibronectin, surfactant protein A, biglycan, versican and so on. TLR on innate immune cells represent the major pattern recognition receptors sensing DAMP-related danger signals.11 Other receptors such as cytoplasm NOD-like receptors and RIG-I-like receptors also play significant roles in responding to DAMP derived from cancers.81
Table 1.
Cancer-derived DAMP and their receptors on the innate immune cells.
| DAMP | Receptors | Target Cells | Immunological Outcomes | Ref |
|---|---|---|---|---|
| HSP family members | CD91, TLR2/4, CD14, CD40, Scavenger receptor LOX-1 | DC Macrophages | Antigen presentation, cross- presentation; DC activation | 84, 88, 89 |
| HMGB1/DNA/RNA | RAGE, TLR9 | Plasmacytoid DC | Cytokine production: IFN-α, TNF-α | 92 |
| HMGB1/IL-1β | IL-1R/IL-1RAcP | Macrophages Neutrophils | Chemokine/Cytokine production: MIP-2, TNF-α | 93 |
| HMGB1/LPS | TLR4 | Monocytes | Cytokine production: TNF-α, IL-6, IL-1β, IL-10 | 94, 95 |
| HMGB1/nucleosome | TLR2 | DC Macrophages | Cytokine production: TNF-α, IL-6, IL-1β, IL-10; Upregulation of co-stimulatory molecules: MHC class II, CD86, CD83 | 96 |
| HMGB1/CXCL12 | CXCR4, TLR4 | DC Macrophages | Migration of DC and macrophages | 97 |
| HMGB1 | TREM-1 | Monocytes Neutrophils | Chemokine/Cytokine production: TNF-α, IL-6, IL-8 | 98 |
| HMGB1 | CD24/Siglec-10 | DC | Cytokine reduction:IL-6, CCL2, TNF-α; Inhibition of NF-kβ | 99 |
| Biglycan | TLR2/4 | Macrophages | Chemokine/Cytokine production: MIP-2, TNF-α; Activation of p38, ERK, and NF-κβ | 100 |
| Hyaluronic Acid | TLR4 | DC | DC maturation | 101 |
| Heparan Sulfate | TLR4 | DC | DC maturation | 102 |
| S100A8/S100A9 | TLR4/MD2 | DC Macrophages | Chemokine/Cytokine production: IL-8, TNF-α; Upregulation of adhesion molecule ICAM-1 | 103, 104 |
| Fibronectin | TLR4 | Macrophages | Production of MMP-9; NF-κβ activation | 105 |
| Surfactant protein A | TLR4 | Macrophages | Cytokine production: TNF-α, IL- 10; NF-κβ activation | 106 |
| Versican | TLR2/TLR6/CD 14 | Macrophages | Chemokine/Cytokine production: TNF-α, IL-6, IL-1β, MIP-1α, MIP-1β, MIP-2 | 107, 108 |
| ATP | P2X7 | DC | Cytokine production: IL-1β; NLRP3 inflammasome activation | 109–112 |
| Mitochondria formyl peptides | FPR-1 | Neutrophils | MMP-8 and MMP-9 production; Activation of p44/42 MAPK; Ca+ flux | 114, 116 |
| Mitochondria DNA | TLR9 | Neutrophils | MMP-8 and MMP-9 production; Cytokine production: TNF-α, IL- 6; Activation of p38 MAPK | 115 |
| Uric acid | DC | Th17 Cytokine production: IL-1α/β, IL-6, and IL-23; Upregulation of co-stimulatory molecules | 117–121 | |
| NY-ESO-1 | CRT/TLR4 | DC | ND | 122, 125 |
Abbreviations used: FPR-1: formyl peptide receptor-1; MMP: matrix metalloproteinase; ND: not determined; TREM-1: triggering receptor expressed on myeloid cells-1; TRPM2: transient receptor potential protein M2.
The exact nature of these DAMP in the cancer microenvironment and their contributions to the cancer-associated inflammation and immunity are yet to be clearly understood, which are now an active area of investigation. Nevertheless, it is believed that cancer-derived DAMP and their partner receptors represent new molecular targets with potentially significant immunological outcomes upon intervention.
HSP
HSP are house-keeping proteins that are widely expressed in most cells, and are molecular chaperones under normal and stressed conditions. HSP from necrotic tumor cells display immunological properties characterized by induction of DC maturation, inflammatory cytokine production and stimulation of NK cell cytotoxicity.82 Some of these activities are related to promoting tumor growth;83 while others contribute to anti-tumor immunity. HSP90, Gp96, CRT, HSP70, HSP110, and Grp170 can function as chaperones of polypeptides in cancer. Tumor-derived HSP-peptide complexes can be taken up by antigen-presenting cells such as macrophages and DC and cross-presented by MHC class I molecules, which makes HSP excellent carriers for cancer vaccines. Scavenger receptors and CD91 are common recognition receptors for HSP on macrophage and DC surface.84 Among various HSP family members, HSP70, GRP78, and Gp96 have been found immunogenic in cancer patients, and also qualify as TAA.85–87 TLR2/TLR4 have been indicated as the major receptors involved in HSP70- and Gp96-mediated DC activation through the MyD88/NF-κB pathway,88 although conflicting data suggested that stimulation of TLR2 or TLR4 could be caused by microbial contaminants in the purified HSP preparations. Other cell-surface receptors are also indicated in HSP signaling, such as CD14 and CD40 in HSP70-mediated DC activation and scavenger receptor LOX-1 in HSP70-mediated antigen cross-presentation.89
HMGB1
HMGB1 is a widely-expressed protein normally located in the cell nucleus and functions as a DNA-binding transcriptional factor. However, it can be released as a secreted protein from necrotic and apoptotic cancer cells.90 In necrotic cell death, emitted HMGB1 contributes to inflammation in activating DC/macrophage to secret IFN-α, TNF-α, IL-12 and IFN-γ, up-regulate CD80 and CD86 co-stimulatory molecules, and induce adaptive CD8+ T cells.90, 91 In contrast, oxidized HMGB1 delivers tolerogenic signals during apoptosis. Extracellular HMGB1 usually associates with other molecules correlating with differential binding to DC/macrophage cell surface receptors. For example, HMGB1/DNA/RNA complex signals through RAGE (receptor for advanced glycation end products).92 HMGB1/IL-1β associates with the IL-1R/IL-1RAcP complex.93 HMGB1 and lipopolysaccharide (LPS) complex can activate TLR4;94, 95 while HMGB1/nucleosome preferentially engages TLR2.96 HMGB1/CXCL12 associates with receptors CXCR4, TLR4 and RAGE.97 HMGB1 has also been reported to directly bind to triggering receptor expressed on myeloid cells-1.98 Pro-inflammatory responses are usually caused by the above HMGB1 and associated partners; whereas several binding receptors of HMGB1 suppress its proinflammatory effects, such as CD24 and thrombospondin.99
ECM components
Multiple ECM components are upregulated or degraded in cancer, serving as pro-inflammatory mediators mostly through pattern recognition receptors TLR2 or TLR4 or both. Biglycan, an ECM proteoglycan liberated during inflammation, activates p38, ERK, and NF-κB signaling pathway through receptors TLR2 and TLR4 in macrophage and induces the production of inflammatory cytokines TNF-α and chemokine macrophage inflammatory protein-2 (MIP-2).100 ECM degradation product of polysaccharide fragments derived from hyaluronic acid101 and heparan sulfate102 have revealed new roles for immunomodulatory signals eliciting DC maturation via TLR4. S100A8/S100A9 proteins, another family of endogenous DAMP molecules, can specifically interact with the TLR4-MD2 complex on phagocytes, which results in elevated expression of TNF-α and stimulation of chemotactic response. This includes the secretion of pro-inflammatory chemokines IL-8, up-regulation of adhesion molecule ICAM-1 and adhesion receptor CD11b/CD18.103, 104 Fibronectin and surfactant protein A (SP-A) may also be recognized by TLR4 promoting expression of genes involved in the inflammatory response.105, 106 Recent studies suggest that versican, a large ECM proteoglycan that accumulates in the mouse Lewis lung carcinoma microenvironment, stimulates tumor infiltrating macrophages (via TLR2, and co-receptors TLR6 and CD14) to produce IL-6 and TNF-α, and accelerates LLC metastasis.107 Versican is also accumulated in stroma surrounding human skin tumors induced by UV, co-localizing with infiltrating neutrophils.108
ATP
Recent evidence show that high levels of extracellular ATP can function as an endogenous danger signal and pro-inflammatory factor.109 High concentrations of extracellular ATP are quickly detected after tumor death induced by stress and chemotherapeutic agents.110 ATP is believed to play an important role in rendering the “immunogenic” death of tumor (late stage apoptosis and necrosis) and induction of anti-cancer immune response accompanied with chemotherapy.111 Following chemotherapy, ATP emitted from dying cancer cells engages the purinergic receptor P2X7 on immature DC, activating the NOD-like receptor family, pyrin domain containing-3 protein (NLRP3) inflammasome and driving the secretion of IL-1β. IL-1β then contributes to adaptive immunity against cancers, including priming IFNγ-producing CD8+ T cells.112
Mitochondrial DAMP
Mitochondrial DAMP are newly identified intracellular DAMP that can be released into the circulation from shock-injured tissues, which can elicit significant immune consequences.113 Among them, mitochondrial formyl peptides activate human PMN through formyl peptide receptor-1;114 mitochondrial DNA, which are evolutionarily derived from bacteria, is recognized by innate immune system via TLR9, that similarly binds bacteria DNA. Mitochondrial DAMP promote PMN Ca2+ flux, activate p38 MAPK115 and p44/42 MAPK,116 and induce PMN to secrete IL-8 and matrix metalloproteinase-9. This has lead to PMN migration, degranulation and contribute to systemic inflammatory responses in vivo. Dying tumor cells may also release mitochondria debris containing formyl peptides and DNA, producing similar immune outcomes.
Uric Acid
Uric acid is a by-product of nucleic acid metabolism, which can be released from dying tumor cells and serve as a DAMP alert, shaping both the innate and adaptive immune responses.117 First, uric acid crystals may form in tumor cells with high contents of nucleic acids, which are able to up-regulate co-stimulatory molecules on immature DC and subsequently prime CD8+ T cells.118 Second, in cooperation with NF-kB activation (such as that caused by LPS), uric acid crystals have recently been shown to induce DC to secrete IL-1α/β, IL-6, and IL-23, which subsequently drive pro-inflammatory Th17 differentiation of naive CD4+ T cells.119 IL-1 then binds to the IL-1R and signals through MyD88 to amplify pro-inflammatory responses, including neutrophil recruitment.120 The effect of Th17 differentiation is dependent on the NLRP3 inflammasome, and cytokines IL-1α/β and IL-18. The receptor that identifies uric acid crystals is not clear. The binding of uric acid crystals with immature DC seems not to be mediated by a specific receptor on the cell surface, but instead depends on directly engaging the cholesterol-rich membrane lipid rafts and Syk kinase activation.121, 122
TAA and DAMP
TAA are usually defined based on their recognition by spontaneous T cell and antibody responses in cancer patients. When encountering antigen presenting cells, TAA themselves are generally perceived as by-standers that rely on the above-referenced “danger signals” to initiate adaptive immune responses. According to this paradigm, TAA will be mostly resulted from the neo-peptides of genetic mutations in cancer cells. However, human TAA identified to date are commonly seen as non-mutated self-proteins.3 It is speculated that direct interactions may exist between some TAA and the innate immune cells, which may play a role in the initiation of adaptive anti-tumor immunity in vivo. In search of intrinsic factors derived from TAA that contribute to anti-tumor immune responses, our laboratory has been focused on NY-ESO-1, a non-mutated cancer/testis antigen with distinctively strong immunogenicity.123 Spontaneous antibody and T cell immune responses against NY-ESO-1 are readily detectable in a wide spectrum of cancer patients with NY-ESO-1-expressing tumors, including older patients with late stage cancers, whose immune systems are known to be less responsive. The immunogenicity of NY-ESO-1 is not due to its higher level of expression compared to other TAA. Indeed, at least in melanoma, the expression of NY-ESO-1 is much lower than that of melanocyte differentiation antigens such as gp100, MART-1, TRP-1, and TRP-2, as well as other cancer/testis antigens, such as MAGE-1 and MAGE-3.124 Our recent investigation of the specific interaction between polymeric NY-ESO-1 and TLR4/CRT on the surface of immature DC, macrophages, and monocytes, indicates a unique interaction between NY-ESO-1 and the innate immune system.122, 125 Although the exact signaling events of NY-ESO-1/DC interactions still need to be elucidated, NY-ESO-1 is shown to serve as an endogenous molecular adjuvant in anti-tumor immune responses. Expression plasmids encoding NY-ESO-1 fused with TAA carbonic anhydrase 9 generated robust antibody responses against the otherwise non-immunogenic protein in mice.125
NY-ESO-1 thus represents the first example of a cancer/testis antigen that is also a DAMP. On the other hand, antibody (and maybe T cell) responses against well-known protein DAMP, such as HSP70, GRP78, and HMGB1 are present in various cancer patients.85–87 These DAMP are thus also TAA, supporting the cross-over roles between TAA and DAMP, i.e. certain TAA may serve as DAMP and certain protein DAMP may serve as TAA.
Clinical development based on interactions of cancer-derived DAMP and their receptors
Targeting TLR
Current strategies in clinical development include (1) TLR functional blockade using neutralizing antibodies and antagonists, (2) TLR signaling pathway inhibitors, and (3) the use of TLR agonists alone or as vaccine adjuvants.126–129 We emphasize on TLR agonists in immunotherapy of solid cancers in the following paragraph.
Due to complicated and sometimes adverse immune effects of TLR agonists, their overall use as cancer monotherapies is limited locally but not systematically. So far, TLR agonists approved by the FDA for clinical use in cancer treatment consist of the classic Bacillus Calmette-Guein (mycobacterium mixture) targeting TLR2, TLR4, and TLR9 for bladder cancer,130 Imiquimod (small-molecule single-stranded RNA) targeting TLR7 for superficial basal cell carcinoma,131, 132 and the AS04 adjuvant system (detoxified lipid A on aluminum hydroxide) targeting TLR4 for human papillomavirus as a prophylactic cervical cancer vaccine.127 Several other TLR agonists, such as CpG oligo-deoxynucleotides targeting TLR7, polyriboinsinic-polyribocytidylic acid targeting TLR9, and flagellin-protein fusions targeting TLR5 are being actively evaluated as adjuvants in multiple cancer indications.133 For example, a small single stranded RNA molecule based TLR7 agonist, 852A, stimulates immature DC to produce multiple cytokines including IFNα in vitro and in vivo. It is now being evaluated in a Phase II clinical trial for treatment of inoperable melanoma.134 There are also numerous efforts to discover new TLR agonists with low toxicities and improved systemic anti-tumor effects from natural product extracts analysis and structural modifications. TLR agonists are being exploited as adjuvants in cancer vaccines based on their ability to induce maturation of antigen presenting cells.133 They can also combine with chemotherapy, radiotherapy or monoclonal antibodies to improve efficacy.
Molecular adjuvant effect of HSP and other DAMP
HSP have been applied as carriers/adjuvants for cancer vaccines in clinical trials. The most commonly used approaches include autologous tumor-derived HSP-polypeptide complexes and chimeric HSP-TAA fusion proteins. Promising effects are being obtained in clinical trials using Gp96 complex purified from patients’ own cancers including glioma, renal cell carcinoma, melanoma and pediatric neurological cancer patients. For example, in a phase II trial carried out in stage IV melanoma patients treated with autologous tumor-derived Gp96, twenty eight among 39 patients had residual measurable disease; whereas 11 were disease free after surgery.135 In another Phase II study of HSP polypeptide complex for patients with metastatic renal cell carcinoma, two patients had a partial remission, one had a complete remission and 18 had stable disease, among 61 patients treated. These HSP-based vaccines exhibit minimal toxicity and promising antitumor activity.89 Phase III clinical trials have been initiated in advanced melanoma and kidney cancer with earlier stage disease.136
Pre-clinical studies have indicated potential advantages in cancer vaccine-induced helper T cells and cytotoxic T cells generated through activating immature DC directly with DAMP rather than indirectly via pro-inflammatory or activating cytokines provided by neighboring cells.137, 138 In particular, following the recognition of the mechanism of immunogenicity, HMGB1 and NY-ESO-1 are being studied in preclinical investigations as immune adjuvants with perspectives as potential vaccine adjuvants in human trials in the future.125, 139 DAMP, due to its limited toxicity comparing with bacterial and viral products, are attractive candidates of molecular adjuvant development.
Other areas of clinical development exploiting cancer/innate immune cell interactions, such as blocking DAMP that are associated with chronic inflammation for the prevention and treatment of cancer, blocking or enhancing cytokines/chemokines in cancer biotherapy, utilization of growth factors to increase the number of DC and other antigen presenting cells, have been the subject of other review articles 1, 2, 140 and not explicitly discussed here.
Conclusions
Spontaneous immune responses against cancer are complex and can be well summarized in the immune editing model.5 In most patients present at the clinic, chronic inflammation and immune suppression are the dominant effects in the tumor microenvironment. However, this does not exclude the existence of cancer-derived intrinsic factors that may have a powerful activation effect to the immune system. By dissecting the molecular details of cancer and innate immune system interactions as summarized in Fig. 1 and Table 1, we hope to individually identify cancer-derived intrinsic factors involved in this complex network and point to areas with the potential of tipping the balance through immunological interventions. These factors are composed of certain cancer-derived DAMP as well as their partner receptors on the immature DC, which represent new molecular targets for immunotherapy of cancer in the future.
Acknowledgments
The authors are thankful to the support of the NIHR21CA137651 grant under the American Recovery and Reinvestment Act and the Research Scholar Award (#RSG-08-070-01-LIB) from the American Cancer Society. Robert M. Prins, Ph.D. and David H. Nguyen, Ph.D. of UCLA provided helpful discussions for the draft of this manuscript.
Footnotes
Conflicts of Interest and Source of Funding: The authors declare no conflict of interests to this work. Source of funding are from the National Institutes of Health (NIHR21CA137651) and the American Cancer Society (#RSG-08-070-01-LIB).
References
- 1.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 2.Ostrand-Rosenberg S. Immune surveillance: a balance between protumor and antitumor immunity. Curr Opin Genet Dev. 2008;18:11–18. doi: 10.1016/j.gde.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 4.Scanlan MJ, Gure AO, Jungbluth AA, et al. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32. doi: 10.1034/j.1600-065x.2002.18803.x. [DOI] [PubMed] [Google Scholar]
- 5.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 6.Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349:1543–1554. doi: 10.1056/NEJMra023009. [DOI] [PubMed] [Google Scholar]
- 7.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang JC, Haworth L, Sherry RM, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349:427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baselga J, Cortes J, Kim SB, et al. Pertuzumab plus Trastuzumab plus Docetaxel for Metastatic Breast Cancer. N Engl J Med. 2012;366:109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 12.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11–22. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 14.Yokoyama WM, Plougastel BF. Immune functions encoded by the natural killer gene complex. Nat Rev Immunol. 2003;3:304–316. doi: 10.1038/nri1055. [DOI] [PubMed] [Google Scholar]
- 15.Bauer S, Groh V, Wu J, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. [PubMed] [Google Scholar]
- 16.Salih HR, Antropius H, Gieseke F, et al. Functional expression and release of ligands for the activating immunoreceptor NKG2D in leukemia. Blood. 2003;102:1389–1396. doi: 10.1182/blood-2003-01-0019. [DOI] [PubMed] [Google Scholar]
- 17.Vetter CS, Groh V, thor Straten P, et al. Expression of stress-induced MHC class I related chain molecules on human melanoma. J Invest Dermatol. 2002;118:600–605. doi: 10.1046/j.1523-1747.2002.01700.x. [DOI] [PubMed] [Google Scholar]
- 18.Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev. 2010;235:267–285. doi: 10.1111/j.0105-2896.2010.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diefenbach A, Jamieson AM, Liu SD, et al. Ligands for the murine NKG2D receptor: expression by tumor cells and activation of NK cells and macrophages. Nat Immunol. 2000;1:119–126. doi: 10.1038/77793. [DOI] [PubMed] [Google Scholar]
- 20.Carayannopoulos LN, Naidenko OV, Kinder J, et al. Ligands for murine NKG2D display heterogeneous binding behavior. Eur J Immunol. 2002;32:597–605. doi: 10.1002/1521-4141(200203)32:3<597::aid-immu597>3.3.co;2-5. [DOI] [PubMed] [Google Scholar]
- 21.Cerwenka A, Bakker AB, McClanahan T, et al. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12:721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 22.Diefenbach A, Hsia JK, Hsiung MY, et al. A novel ligand for the NKG2D receptor activates NK cells and macrophages and induces tumor immunity. Eur J Immunol. 2003;33:381–391. doi: 10.1002/immu.200310012. [DOI] [PubMed] [Google Scholar]
- 23.Pende D, Parolini S, Pessino A, et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J Exp Med. 1999;190:1505–1516. doi: 10.1084/jem.190.10.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vitale M, Bottino C, Sivori S, et al. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J Exp Med. 1998;187:2065–2072. doi: 10.1084/jem.187.12.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sivori S, Pende D, Bottino C, et al. NKp46 is the major triggering receptor involved in the natural cytotoxicity of fresh or cultured human NK cells. Correlation between surface density of NKp46 and natural cytotoxicity against autologous, allogeneic or xenogeneic target cells. Eur J Immunol. 1999;29:1656–1666. doi: 10.1002/(SICI)1521-4141(199905)29:05<1656::AID-IMMU1656>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 26.Ryan JC, Turck J, Niemi EC, et al. Molecular cloning of the NK1.1 antigen, a member of the NKR-P1 family of natural killer cell activation molecules. J Immunol. 1992;149:1631–1635. [PubMed] [Google Scholar]
- 27.Plougastel B, Matsumoto K, Dubbelde C, et al. Analysis of a 1-Mb BAC contig overlapping the mouse Nkrp1 cluster of genes: cloning of three new Nkrp1 members, Nkrp1d, Nkrp1e, and Nkrp1f. Immunogenetics. 2001;53:592–598. doi: 10.1007/s002510100367. [DOI] [PubMed] [Google Scholar]
- 28.Mason LH, Anderson SK, Yokoyama WM, et al. The Ly-49D receptor activates murine natural killer cells. J Exp Med. 1996;184:2119–2128. doi: 10.1084/jem.184.6.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith HR, Chuang HH, Wang LL, et al. Nonstochastic coexpression of activation receptors on murine natural killer cells. J Exp Med. 2000;191:1341–1354. doi: 10.1084/jem.191.8.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bellon T, Heredia AB, Llano M, et al. Triggering of effector functions on a CD8+ T cell clone upon the aggregation of an activatory CD94/kp39 heterodimer. J Immunol. 1999;162:3996–4002. [PubMed] [Google Scholar]
- 31.Chan CJ, Andrews DM, McLaughlin NM, et al. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J Immunol. 2010;184:902–911. doi: 10.4049/jimmunol.0903225. [DOI] [PubMed] [Google Scholar]
- 32.Long EO. Regulation of immune responses through inhibitory receptors. Annu Rev Immunol. 1999;17:875–904. doi: 10.1146/annurev.immunol.17.1.875. [DOI] [PubMed] [Google Scholar]
- 33.Moretta L, Biassoni R, Bottino C, et al. Human NK-cell receptors. Immunol Today. 2000;21:420–422. doi: 10.1016/s0167-5699(00)01673-x. [DOI] [PubMed] [Google Scholar]
- 34.Karlhofer FM, Ribaudo RK, Yokoyama WM. MHC class I alloantigen specificity of Ly-49+ IL-2-activated natural killer cells. Nature. 1992;358:66–70. doi: 10.1038/358066a0. [DOI] [PubMed] [Google Scholar]
- 35.Stoneman ER, Bennett M, An J, et al. Cloning and characterization of 5E6(Ly-49C), a receptor molecule expressed on a subset of murine natural killer cells. J Exp Med. 1995;182:305–313. doi: 10.1084/jem.182.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mason LH, Ortaldo JR, Young HA, et al. Cloning and functional characteristics of murine large granular lymphocyte-1: a member of the Ly-49 gene family (Ly-49G2) J Exp Med. 1995;182:293–303. doi: 10.1084/jem.182.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plougastel B, Jones T, Trowsdale J. Genomic structure, chromosome location, and alternative splicing of the human NKG2A gene. Immunogenetics. 1996;44:286–291. doi: 10.1007/BF02602558. [DOI] [PubMed] [Google Scholar]
- 38.Braud VM, Allan DS, O’Callaghan CA, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391:795–799. doi: 10.1038/35869. [DOI] [PubMed] [Google Scholar]
- 39.Beutler B, van Huffel C. Unraveling function in the TNF ligand and receptor families. Science. 1994;264:667–668. doi: 10.1126/science.8171316. [DOI] [PubMed] [Google Scholar]
- 40.Gruss HJ. Molecular, structural, and biological characteristics of the tumor necrosis factor ligand superfamily. Int J Clin Lab Res. 1996;26:143–159. doi: 10.1007/BF02592977. [DOI] [PubMed] [Google Scholar]
- 41.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 42.Anderson DM, Maraskovsky E, Billingsley WL, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- 43.Smith CA, Farrah T, Goodwin RG. The TNF receptor superfamily of cellular and viral proteins: activation, costimulation, and death. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 44.Wang YG, Kim KD, Wang J, et al. Stimulating lymphotoxin beta receptor on the dendritic cells is critical for their homeostasis and expansion. J Immunol. 2005;175:6997–7002. doi: 10.4049/jimmunol.175.10.6997. [DOI] [PubMed] [Google Scholar]
- 45.Yu P, Fu YX. Targeting tumors with LIGHT to generate metastasis-clearing immunity. Cytokine Growth Factor Rev. 2008;19:285–294. doi: 10.1016/j.cytogfr.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan Z, Yu P, Wang Y, et al. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood. 2006;107:1342–1351. doi: 10.1182/blood-2005-08-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakamura A, Kubo T, Takai T. Fc receptor targeting in the treatment of allergy, autoimmune diseases and cancer. Adv Exp Med Biol. 2008;640:220–233. doi: 10.1007/978-0-387-09789-3_17. [DOI] [PubMed] [Google Scholar]
- 48.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 49.Anderson P, Caligiuri M, O’Brien C, et al. Fc gamma receptor type III (CD16) is included in the zeta NK receptor complex expressed by human natural killer cells. Proc Natl Acad Sci U S A. 1990;87:2274–2278. doi: 10.1073/pnas.87.6.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jeannin P, Jaillon S, Delneste Y. Pattern recognition receptors in the immune response against dying cells. Curr Opin Immunol. 2008;20:530–537. doi: 10.1016/j.coi.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 51.Gardai SJ, Bratton DL, Ogden CA, et al. Recognition ligands on apoptotic cells: a perspective. J Leukoc Biol. 2006;79:896–903. doi: 10.1189/jlb.1005550. [DOI] [PubMed] [Google Scholar]
- 52.Gardai SJ, McPhillips KA, Frasch SC, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 53.Kobayashi N, Karisola P, Pena-Cruz V, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miyanishi M, Tada K, Koike M, et al. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 55.Nakayama M, Akiba H, Takeda K, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009;113:3821–3830. doi: 10.1182/blood-2008-10-185884. [DOI] [PubMed] [Google Scholar]
- 56.Moffatt OD, Devitt A, Bell ED, et al. Macrophage recognition of ICAM-3 on apoptotic leukocytes. J Immunol. 1999;162:6800–6810. [PubMed] [Google Scholar]
- 57.Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10:328–343. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klechevsky E, Morita R, Liu M, et al. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity. 2008;29:497–510. doi: 10.1016/j.immuni.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palucka K, Ueno H, Zurawski G, et al. Building on dendritic cell subsets to improve cancer vaccines. Curr Opin Immunol. 2010;22:258–263. doi: 10.1016/j.coi.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Albert ML, Pearce SF, Francisco LM, et al. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med. 1998;188:1359–1368. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lai C, Lemke G. An extended family of protein-tyrosine kinase genes differentially expressed in the vertebrate nervous system. Neuron. 1991;6:691–704. doi: 10.1016/0896-6273(91)90167-x. [DOI] [PubMed] [Google Scholar]
- 62.Stitt TN, Conn G, Gore M, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80:661–670. doi: 10.1016/0092-8674(95)90520-0. [DOI] [PubMed] [Google Scholar]
- 63.Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8:327–336. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanayama R, Tanaka M, Miwa K, et al. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 65.Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol. 2006;16:189–197. doi: 10.1016/j.tcb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 66.Regnault A, Lankar D, Lacabanne V, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. 1999;189:371–380. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 2011;71:2411–2416. doi: 10.1158/0008-5472.CAN-10-2583. [DOI] [PubMed] [Google Scholar]
- 68.Amulic B, Cazalet C, Hayes GL, et al. Neutrophil Function: From Mechanisms to Disease. Annu Rev Immunol. 2012 doi: 10.1146/annurev-immunol-020711-074942. [DOI] [PubMed] [Google Scholar]
- 69.Scapini P, Laudanna C, Pinardi C, et al. Neutrophils produce biologically active macrophage inflammatory protein-3alpha (MIP-3alpha)/CCL20 and MIP-3beta/CCL19. Eur J Immunol. 2001;31:1981–1988. doi: 10.1002/1521-4141(200107)31:7<1981::aid-immu1981>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 70.Hicks AM, Riedlinger G, Willingham MC, et al. Transferable anticancer innate immunity in spontaneous regression/complete resistance mice. Proc Natl Acad Sci U S A. 2006;103:7753–7758. doi: 10.1073/pnas.0602382103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Riedlinger G, Adams J, Stehle JR, Jr, et al. The spectrum of resistance in SR/CR mice: the critical role of chemoattraction in the cancer/leukocyte interaction. BMC Cancer. 2010;10:179. doi: 10.1186/1471-2407-10-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gasser S, Orsulic S, Brown EJ, et al. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Armeanu S, Bitzer M, Lauer UM, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005;65:6321–6329. doi: 10.1158/0008-5472.CAN-04-4252. [DOI] [PubMed] [Google Scholar]
- 74.Hallett WH, Ames E, Motarjemi M, et al. Sensitization of tumor cells to NK cell-mediated killing by proteasome inhibition. J Immunol. 2008;180:163–170. doi: 10.4049/jimmunol.180.1.163. [DOI] [PubMed] [Google Scholar]
- 75.Armeanu S, Krusch M, Baltz KM, et al. Direct and natural killer cell-mediated antitumor effects of low-dose bortezomib in hepatocellular carcinoma. Clin Cancer Res. 2008;14:3520–3528. doi: 10.1158/1078-0432.CCR-07-4744. [DOI] [PubMed] [Google Scholar]
- 76.Romagne F, Vivier E. Natural killer cell-based therapies. F1000 Med Rep. 2011;3:9. doi: 10.3410/M3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Terme M, Ullrich E, Delahaye NF, et al. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat Immunol. 2008;9:486–494. doi: 10.1038/ni1580. [DOI] [PubMed] [Google Scholar]
- 78.Sheridan C. First-in-class cancer therapeutic to stimulate natural killer cells. Nat Biotechnol. 2006;24:597. doi: 10.1038/nbt0606-597. [DOI] [PubMed] [Google Scholar]
- 79.Robak T, Robak E. New anti-CD20 monoclonal antibodies for the treatment of B- cell lymphoid malignancies. BioDrugs. 2011;25:13–25. doi: 10.2165/11539590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 80.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 82.Suto R, Srivastava PK. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science. 1995;269:1585–1588. doi: 10.1126/science.7545313. [DOI] [PubMed] [Google Scholar]
- 83.Lee CH, Wu CL, Shiau AL. Toll-like receptor 4 signaling promotes tumor growth. J Immunother. 2010;33:73–82. doi: 10.1097/CJI.0b013e3181b7a0a4. [DOI] [PubMed] [Google Scholar]
- 84.Basu S, Binder RJ, Ramalingam T, et al. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- 85.Mintz PJ, Kim J, Do KA, et al. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat Biotechnol. 2003;21:57–63. doi: 10.1038/nbt774. [DOI] [PubMed] [Google Scholar]
- 86.Kijanka G, Hector S, Kay EW, et al. Human IgG antibody profiles differentiate between symptomatic patients with and without colorectal cancer. Gut. 2010;59:69–78. doi: 10.1136/gut.2009.178574. [DOI] [PubMed] [Google Scholar]
- 87.Zhang J, Wang K, Liu SS, et al. Using proteomic approach to identify tumor-associated proteins as biomarkers in human esophageal squamous cell carcinoma. J Proteome Res. 2011;10:2863–2872. doi: 10.1021/pr200141c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Castelli C, Rivoltini L, Rini F, et al. Heat shock proteins: biological functions and clinical application as personalized vaccines for human cancer. Cancer Immunol Immunother. 2004;53:227–233. doi: 10.1007/s00262-003-0481-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 91.Sims GP, Rowe DC, Rietdijk ST, et al. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 92.Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 93.Sha Y, Zmijewski J, Xu Z, et al. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J Immunol. 2008;180:2531–2537. doi: 10.4049/jimmunol.180.4.2531. [DOI] [PubMed] [Google Scholar]
- 94.Youn JH, Oh YJ, Kim ES, et al. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-alpha production in human monocytes. J Immunol. 2008;180:5067–5074. doi: 10.4049/jimmunol.180.7.5067. [DOI] [PubMed] [Google Scholar]
- 95.Hreggvidsdottir HS, Ostberg T, Wahamaa H, et al. The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol. 2009;86:655–662. doi: 10.1189/jlb.0908548. [DOI] [PubMed] [Google Scholar]
- 96.Urbonaviciute V, Furnrohr BG, Meister S, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–3018. doi: 10.1084/jem.20081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Campana L, Bosurgi L, Bianchi ME, et al. Requirement of HMGB1 for stromal cell-derived factor-1/CXCL12-dependent migration of macrophages and dendritic cells. J Leukoc Biol. 2009;86:609–615. doi: 10.1189/jlb.0908576. [DOI] [PubMed] [Google Scholar]
- 98.El Mezayen R, El Gazzar M, Seeds MC, et al. Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol Lett. 2007;111:36–44. doi: 10.1016/j.imlet.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen GY, Tang J, Zheng P, et al. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science. 2009;323:1722–1725. doi: 10.1126/science.1168988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schaefer L, Babelova A, Kiss E, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115:2223–2233. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Termeer C, Benedix F, Sleeman J, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Johnson GB, Brunn GJ, Kodaira Y, et al. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- 103.Vogl T, Tenbrock K, Ludwig S, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 104.Foell D, Wittkowski H, Vogl T, et al. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81:28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- 105.Okamura Y, Watari M, Jerud ES, et al. The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 106.Guillot L, Balloy V, McCormack FX, et al. Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J Immunol. 2002;168:5989–5992. doi: 10.4049/jimmunol.168.12.5989. [DOI] [PubMed] [Google Scholar]
- 107.Kim S, Takahashi H, Lin WW, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kunisada M, Yogianti F, Sakumi K, et al. Increased Expression of Versican in the Inflammatory Response to UVB- and Reactive Oxygen Species-Induced Skin Tumorigenesis. Am J Pathol. 2011;179:3056–3065. doi: 10.1016/j.ajpath.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aymeric L, Apetoh L, Ghiringhelli F, et al. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res. 2010;70:855–858. doi: 10.1158/0008-5472.CAN-09-3566. [DOI] [PubMed] [Google Scholar]
- 110.Di Virgilio F. Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol Sci. 2007;28:465–472. doi: 10.1016/j.tips.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 111.Martins I, Tesniere A, Kepp O, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle. 2009;8:3723–3728. doi: 10.4161/cc.8.22.10026. [DOI] [PubMed] [Google Scholar]
- 112.Ghiringhelli F, Apetoh L, Tesniere A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 113.Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Raoof M, Zhang Q, Itagaki K, et al. Mitochondrial peptides are potent immune activators that activate human neutrophils via FPR-1. J Trauma. 2010;68:1328–1332. doi: 10.1097/TA.0b013e3181dcd28d. [DOI] [PubMed] [Google Scholar]
- 115.Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock. 2010;34:55–59. doi: 10.1097/SHK.0b013e3181cd8c08. [DOI] [PubMed] [Google Scholar]
- 116.Hauser CJ, Sursal T, Rodriguez EK, et al. Mitochondrial damage associated molecular patterns from femoral reamings activate neutrophils through formyl peptide receptors and P44/42 MAP kinase. J Orthop Trauma. 2010;24:534–538. doi: 10.1097/BOT.0b013e3181ec4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kono H, Chen CJ, Ontiveros F, et al. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J Clin Invest. 2010;120:1939–1949. doi: 10.1172/JCI40124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 119.Conforti-Andreoni C, Spreafico R, Qian HL, et al. Uric Acid-Driven Th17 Differentiation Requires Inflammasome-Derived IL-1 and IL-18. J Immunol. 2011;187:5842–5850. doi: 10.4049/jimmunol.1101408. [DOI] [PubMed] [Google Scholar]
- 120.Chen CJ, Shi Y, Hearn A, et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262–2271. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ng G, Sharma K, Ward SM, et al. Receptor-independent, direct membrane binding leads to cell-surface lipid sorting and Syk kinase activation in dendritic cells. Immunity. 2008;29:807–818. doi: 10.1016/j.immuni.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zeng G, Aldridge ME, Tian X, et al. Dendritic cell surface calreticulin is a receptor for NY-ESO-1: direct interactions between tumor-associated antigen and the innate immune system. J Immunol. 2006;177:3582–3589. doi: 10.4049/jimmunol.177.6.3582. [DOI] [PubMed] [Google Scholar]
- 123.Gnjatic S, Nishikawa H, Jungbluth AA, et al. NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res. 2006;95:1–30. doi: 10.1016/S0065-230X(06)95001-5. [DOI] [PubMed] [Google Scholar]
- 124.Riker AI, Kammula US, Panelli MC, et al. Threshold levels of gene expression of the melanoma antigen gp100 correlate with tumor cell recognition by cytotoxic T lymphocytes. Int J Cancer. 2000;86:818–826. doi: 10.1002/(sici)1097-0215(20000615)86:6<818::aid-ijc10>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 125.Liu Y, Tian X, Leitner WW, et al. Polymeric Structure and Host Toll-like Receptor 4 Dictate Immunogenicity of NY-ESO-1 Antigen in Vivo. J Biol Chem. 2010;286:37077–37084. doi: 10.1074/jbc.M111.280123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Duthie MS, Windish HP, Fox CB, et al. Use of defined TLR ligands as adjuvants within human vaccines. Immunol Rev. 2011;239:178–196. doi: 10.1111/j.1600-065X.2010.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Steinhagen F, Kinjo T, Bode C, et al. TLR-based immune adjuvants. Vaccine. 2011;29:3341–3355. doi: 10.1016/j.vaccine.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Adams S. Toll-like receptor agonists in cancer therapy. Immunotherapy. 2009;1:949–964. doi: 10.2217/imt.09.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Maruyama K, Selmani Z, Ishii H, et al. Innate immunity and cancer therapy. Int Immunopharmacol. 2011;11:350–357. doi: 10.1016/j.intimp.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 130.Sylvester RJ, Oosterlinck W, van der Meijden AP. A single immediate postoperative instillation of chemotherapy decreases the risk of recurrence in patients with stage Ta T1 bladder cancer: a meta-analysis of published results of randomized clinical trials. J Urol. 2004;171:2186–2190. doi: 10.1097/01.ju.0000125486.92260.b2. [DOI] [PubMed] [Google Scholar]
- 131.Geisse J, Caro I, Lindholm J, et al. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: results from two phase III, randomized, vehicle-controlled studies. J Am Acad Dermatol. 2004;50:722–733. doi: 10.1016/j.jaad.2003.11.066. [DOI] [PubMed] [Google Scholar]
- 132.Bong AB, Bonnekoh B, Franke I, et al. Imiquimod, a topical immune response modifier, in the treatment of cutaneous metastases of malignant melanoma. Dermatology. 2002;205:135–138. doi: 10.1159/000063904. [DOI] [PubMed] [Google Scholar]
- 133.Dunne A, Marshall NA, Mills KH. TLR based therapeutics. Curr Opin Pharmacol. 2011;11:404–411. doi: 10.1016/j.coph.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 134.Dudek AZ, Yunis C, Harrison LI, et al. First in human phase I trial of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced cancer. Clin Cancer Res. 2007;13:7119–7125. doi: 10.1158/1078-0432.CCR-07-1443. [DOI] [PubMed] [Google Scholar]
- 135.Eton O, Ross MI, East MJ, et al. Autologous tumor-derived heat-shock protein peptide complex-96 (HSPPC-96) in patients with metastatic melanoma. J Transl Med. 2010;8:9. doi: 10.1186/1479-5876-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Parmiani G, Testori A, Maio M, et al. Heat shock proteins and their use as anticancer vaccines. Clin Cancer Res. 2004;10:8142–8146. doi: 10.1158/1078-0432.CCR-04-1194. [DOI] [PubMed] [Google Scholar]
- 137.Sporri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nat Immunol. 2005;6:163–170. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- 138.Kratky W, Reis e Sousa C, Oxenius A, et al. Direct activation of antigen-presenting cells is required for CD8+ T-cell priming and tumor vaccination. Proc Natl Acad Sci U S A. 2011;108:17414–17419. doi: 10.1073/pnas.1108945108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Fagone P, Shedlock DJ, Bao H, et al. Molecular adjuvant HMGB1 enhances anti-influenza immunity during DNA vaccination. Gene Ther. 2011;18:1070–1077. doi: 10.1038/gt.2011.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Demaria S, Pikarsky E, Karin M, et al. Cancer and inflammation: promise for biologic therapy. J Immunother. 2010;33:335–351. doi: 10.1097/CJI.0b013e3181d32e74. [DOI] [PMC free article] [PubMed] [Google Scholar]