

The advent of genome sequencing projects—culminating in the recent reports of the human sequence (1, 2)—has resulted in both the identification of novel genes and proteins as well as the proliferation of the “omes” that come from their analyses: the proteome (the complement of proteins), transcriptome (the complement of mRNA transcripts), metabolome (the complement of metabolites), and so on. These end products of global assays are needed to interpret the large fraction (typically close to half) of predicted proteins for which no proteins of similar structure exist or have been functionally characterized. The report by Ito et al. (3) is the largest contribution to date in the effort to generate the protein interactome, or map of protein–protein interactions, for the yeast Saccharomyces cerevisiae.
Yeast has been the major proving ground for functional genomics methods from the time its genome was sequenced in 1996 (4). Most such approaches use the underlying principle of “guilt by association” as the means of elucidating function. For example, genes that are coexpressed or proteins that are found in the same complex or in the same location are likely to be involved in the same or related cellular process. Theoretical methods to deduce function include bioinformatic analyses based on protein homology, phylogenetic relationships, and protein domain fusions (5). Empirical methods elucidate gene function by diverse approaches that include expression profiling, screens for biochemical activity, identification of proteins in macromolecular complexes by mass spectrometry, systematic gene disruptions, and determinations of protein interactions. The most popular means to carry out this last method on a genomewide basis is the yeast two-hybrid system (6), a genetic assay based on the properties of site-specific transcriptional activators. Hybrid proteins are generated in yeast composed of a DNA-binding domain fused with a protein X and a transcriptional activation domain fused with a protein Y; the interaction of X and Y leads to the expression of a reporter gene whose product is easily assayed, generally by growth of the yeast on a defined media.
This lack of overlap between the datasets indicates that the screens to date are far from saturating and that the yeast interactome may be larger than previously estimated.
Ito et al. (3) have followed up their earlier pilot study (7), by using a global two-hybrid approach in which they constructed a DNA-binding domain hybrid and an activation domain hybrid for each of the ≈6,000 predicted yeast proteins. They generated 62 pools of each type of yeast transformant, containing up to 96 independent hybrids each, followed by a systematic mating of the 62 × 62 pools to yield 3,844 sets of diploids. Subsequent recovery and sequencing of DNA from diploids positive for four different two-hybrid reporters identified the genes encoding the pairs of interacting proteins. This approach resulted in 4,549 two-hybrid positives among 3,278 proteins. An independent yeast two-hybrid project (8) used two other strategies: an individual DNA-binding domain hybrid tested against a library of all activation domain hybrids, and an individual DNA-binding domain hybrid tested against an array of ≈6,000 separate activation domain transformants. This independent study resulted in the identification of 957 putative interactions involving 1,004 proteins. Surprisingly, the overlap in the data among all three approaches is small, and neither of the two studies recapitulates more than ≈13% of the published interactions detected by the community of yeast biologists using conventional single protein analyses. The high fraction of false negatives may be explained by several factors such as the use of full-length proteins vs. the protein domains used in other studies, the differing levels of hybrid protein expression, the different reporter genes, and other variables in the two-hybrid assay. This lack of overlap between datasets indicates that the screens to date are far from saturating and suggests that the yeast interactome may be larger than estimates based on earlier studies.
These studies beg the question of what does it mean when a two-hybrid interaction has been detected in a genomewide approach. Ito et al. (3) focus on a core dataset of 806 interactions among 797 proteins instead of their complete dataset of 4,549 interactions among 3,278 proteins. The core dataset included cases in which the interactions were detected more than three times and excluded redundant interactions detected in both orientations of the two-hybrid assay. This focus is reasonable, given that roughly 3,000 of the interactions were identified only once or twice, and that a mere 15 proteins account for 1,504 (or ≈33%) of the interactions. Thus, this large-scale study likely contains a fraction of false positives, as is the case with the other recent two-hybrid efforts using proteins of S. cerevisiae (8), Helicobacter pylori (9), and Caenorhabditis elegans (10). Some of these are artifactual pairs in which a transcriptional signal occurs even though the two proteins do not interact with each other, and some are real two-hybrid interactions that do not correspond to interactions that occur in vivo. Such false positives also arise, of course, when individual researchers carry out two-hybrid searches with their favorite proteins, but they are far more likely to be discarded (or at least not reported) in the absence of any confirmatory data. Not only are these data downsizing a luxury that the genomic researcher cannot take advantage of, but individual researchers may upsize their data by additional experimentation. Thus, the hints to function in the datasets from these large-scale approaches may be best validated through conventional single protein analyses.
Despite complications from redundancy and false positives, the useful information from these protein interaction projects falls into at least four categories. First, interactions of an uncharacterized protein with proteins of defined function can lead to a tentative assignment of function for the novel protein. For example, Ito et al. (3) suggest from the interaction data that the protein Ydr016c is involved in the yeast spindle pole body, a prediction supported by localization and genetic data. Indeed, the greater the connectivity among a group of proteins, the greater the likelihood of a common function. Second, multiple interactions among a set of proteins can support molecular mechanisms. A set of linked proteins involved in autophagy is consistent with two of the proteins catalyzing the conjugation of a third protein to a fourth one (3). Third, interactions between yeast proteins may occur between the orthologous proteins of another organism, a situation in which the protein pairs have been termed interologs (10). The yeast Ufd1-Npl4-cdc48 interactions also occur among the orthologous mammalian proteins (11). Fourth, the datasets allow the generation of interaction maps (a small segment of which is shown in Fig. 1) that connect huge numbers of proteins. Global network maps reveal nexus points representing central players in a pathway and crosstalk between pathways. Intriguingly, the analyses of both the core data of 806 interactions and the 2,209 interactions from the yeast community's two-hybrid screens (3), as well as those from other large two-hybrid datasets (12, 13), reveal a single network comprised of more than half the proteins and interactions.
Figure 1.
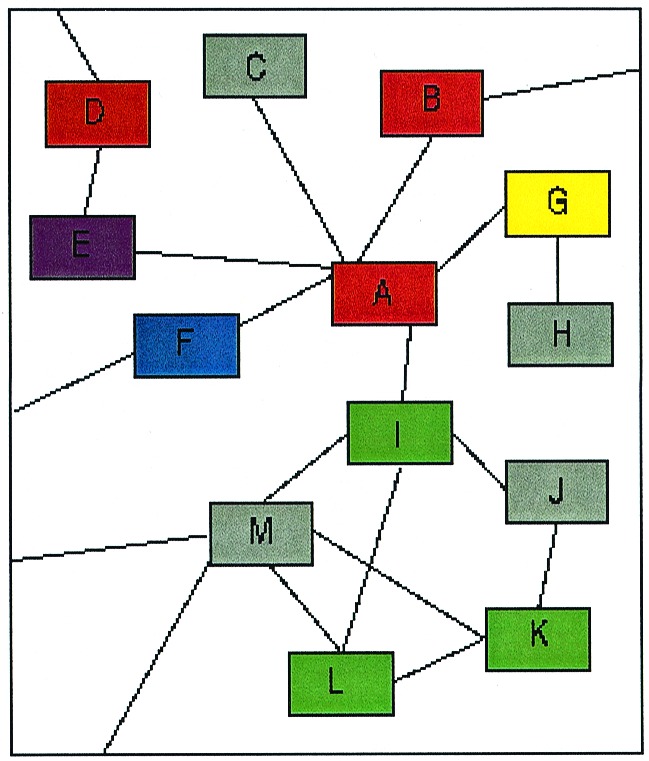
A small region of an idealized protein interaction network. Each letter represents a yeast protein, and each color a different functional category, with gray being uncharacterized proteins. M and J are likely to be involved in the same function as I, L, and K because all are multiply connected in the network. Predicting the functions of C and H is less clear cut.
This then leads to the question of what is the size of the yeast interactome. The answer is difficult to determine, with the mean number of interactions/protein identified in different studies varying from 0.1 to 24 (14). This is a wide variance derived from larger-scale studies that tend to give lower numbers of interactions per protein and studies on specific complexes that tend to give higher numbers. The core dataset from Ito et al. (3) of 806 interactions adds another measurement to these estimates, but their number of 1.0 interactions/protein, given the lack of overlap between their results and conventional single protein analyses, is probably an underestimate. The total number of protein interactions in S. cerevisiae has been independently extrapolated to be between 10,000 and 40,000 (14, 15).
What is the future in the construction of protein networks? It is clear that Ito et al. (3) have added many pieces to the vast puzzle that is the yeast protein interaction map. Now there are two studies in which effectively all yeast proteins have been searched by the two-hybrid assay to generate a total of more than ≈1,800 interactions, as well as the ≈2,200 deposited by individual researchers into the Yeast Proteome Database (16). Further contributions to the yeast interactome need to come from diverse sources, with a major role played by the single protein analyses that will be essential to fill in the gaps in the map. Genomewide techniques such as mass spectrometry-based methods (17, 18) and newly developed protein chips (19, 20) will likely play a role. Integration of data from different functional genomics analyses examining diverse parameters—transcription; protein localization, concentration, and modification; phenotypes of deletion strains; and other gene and protein properties—will allow the validity of the two-hybrid interactions to be assessed and enable assembly of more accurate protein networks. The interaction maps from work like that of Ito et al. (3) provide a basic framework on which the difficult task of adding more dynamic protein properties remains. Understanding how these protein networks function will continue to be an interesting challenge.
Acknowledgments
We thank Joseph Gera, Chandra Tucker, and Peter Uetz for comments. S.F. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
See companion article on page 4569.
References
- 1.International Human Genome Sequencing Consortium. Nature (London) 2001;409:860–921. [Google Scholar]
- 2.Venter J C, Adams M D, Myers E W, Li P W, Mural R J, Sutton G G, Smith H O, Yandell M, Evans C A, Holt R A, et al. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. Proc Natl Acad Sci USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. . (First Published March 13, 2001; 10.1073/pnas.061034498) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goffeau A, Barrell B G, Bussey H, Davis R W, Dujon B, Feldmann H, Galibert F, Hoheisel J D, Jacq C, Johnston M, et al. Science. 1996;274:563–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 5.Eisenberg D, Marcotte E M, Xenarios I, Yeates T O. Nature (London) 2000;405:823–826. doi: 10.1038/35015694. [DOI] [PubMed] [Google Scholar]
- 6.Fields S, Song O. Nature (London) 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 7.Ito T, Tashiro K, Muta S, Ozawa R, Chiba T, Nishizawa M, Yamamoto K, Kuhara S, Sakaki Y. Proc Natl Acad Sci USA. 2000;97:1143–1147. doi: 10.1073/pnas.97.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uetz P, Giot L, Cagney G, Mansfield T A, Judson R S, Knight J R, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. Nature (London) 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 9.Rain J-C, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, Schachter V, et al. Nature (London) 2001;409:211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- 10.Walhout A J, Sordella R, Lu X, Hartley J L, Temple G F, Brasch M A, Thierry-Mieg N, Vidal M. Science. 2000;287:116–122. doi: 10.1126/science.287.5450.116. [DOI] [PubMed] [Google Scholar]
- 11.Meyer H H, Shorter J G, Seemann J, Pappin D, Warren G. EMBO J. 2000;19:2181–2192. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwikowski B, Uetz P, Fields S. Nat Biotechnol. 2000;18:1257–1261. doi: 10.1038/82360. [DOI] [PubMed] [Google Scholar]
- 13.Fellenberg M, Albermann K, Zollner A, Mewes H M, Hani J. Proc Int Conf Intell Syst Mol Biol. 2000;8:152–161. [PubMed] [Google Scholar]
- 14.Walhout A J, Boulton S J, Vidal M. Yeast. 2000;17:88–94. doi: 10.1002/1097-0061(20000630)17:2<88::AID-YEA20>3.0.CO;2-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tucker C L, Gera J F, Uetz P. Trends Cell Biol. 2001;11:102–106. doi: 10.1016/s0962-8924(00)01902-4. [DOI] [PubMed] [Google Scholar]
- 16.Costanzo M C, Hogan J D, Cusick M E, Davis B P, Fancher A M, Hodges P E, Kondu P, Lengieza C, Lew-Smith J E, Lingner C, et al. Nucleic Acids Res. 2000;28:73–76. doi: 10.1093/nar/28.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pandey A, Mann M. Nature (London) 2000;405:837–846. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- 18.Yates J R. Trends Genet. 2000;16:5–8. doi: 10.1016/s0168-9525(99)01879-x. [DOI] [PubMed] [Google Scholar]
- 19.MacBeath G, Schreiber S L. Science. 2000;289:1760–1763. doi: 10.1126/science.289.5485.1760. [DOI] [PubMed] [Google Scholar]
- 20.Zhu H, Klemic J F, Chang S, Bertone P, Casamayor A, Klemic K G, Smith D, Gerstein M, Reed M A, Snyder M. Nat Genet. 2000;26:283–289. doi: 10.1038/81576. [DOI] [PubMed] [Google Scholar]