

In the avalanche of recent information on the brain's control of energy homeostasis and the pathophysiology of obesity, there are two outstanding insights. One is enhanced knowledge of the neuroanatomy and neurochemistry of various components of the appetite regulation and energy expenditure networks in the hypothalamus, and the other is characterization of the neurobiology of leptin in regulating these two hypothalamic networks (1, 2). Leptin, an adipocyte-derived hormone normally transported across the blood brain barrier (BBB), was initially heralded as a major negative feedback signal relaying information of the body's energy stores to these hypothalamic networks on a moment to moment basis to maintain body weight around a set point. However, results of numerous investigations in rodents and humans have been disappointing because circulating leptin levels display marked variations among individuals and rise in direct proportion to the age-related increase in adipose tissue depots. Thus, leptin concentrations are greatly elevated in obese subjects (2). Consequently, despite the promising results of the leptin replacement therapy experiments in leptin-deficient mice and patients, endogenous hyperleptinemia could neither curb appetite nor augment energy expenditure in normal subjects experiencing an increase in the rate of weight gain. It soon became apparent that the ineffectiveness of endogenous leptin, appropriately termed leptin resistance, develops rapidly and that leptin therapy even at supraphysiological concentrations is largely ineffective in reducing the body weight of clinically obese patients (3). Although these revelations dampened the enthusiasm of clinicians and investigators in academia and industry alike, they presented a challenge to devise newer therapeutic strategies that would curtail the environmentally based increase in the rate of weight gain and the epidemic of obesity in most developed countries (4). The knowledge accrued over several years that cytokines readily cross the BBB and induce anorexia and weight loss by an action within the hypothalamus (5), and the serendipitous finding that ciliary neurotrophic factor (CNTF) treatment of amyotropic lateral sclerosis patients for neurotropic benefits produced severe anorexia and weight loss (6), an outcome later replicated in rodents (7, 8), presented a new avenue for therapeutic exploration. The paper by Lambert et al. (9) in this issue of PNAS has extended these findings by demonstrating the efficacy of a CNTF derivative, CNTFAx15, in correcting obesity and dependent metabolic disorders in mice. Results show that in leptin-deficient ob/ob mice and normal mice rendered obese and leptin-resistant by consumption of a high fat diet, CNTFAx15 treatment normalized the obese phenotypes. Furthermore, it is apparent that CNTFAx15 mobilizes intracellular signal transduction pathways in the hypothalamus that are similar to those activated by leptin, and not by interleukin-1, the prototype cytokine. This explains why the weight-reducing effects of CNTFAx15 are free of side effects, such as fever, taste aversion, and metabolic abnormalities that are generally evoked by cytokines.
How does CNTF/CNTFAx15 suppress appetite and reduce body weight? The urge to replenish the body's depleted energy stores is chemically coded in the appetite-regulating network resident in the arcuate and paraventricular nuclei (ARC-PVN) of the hypothalamus (ref. 1; Fig. 1). Basically, the drive to restore energy stores is elicited by the augmented release of appetite-stimulating signals, primarily the orexigenic signal, neuropeptide Y (NPY), and the restraint on appetite-inhibiting signals, such as the anorexigenic melanocortin, α-melanocyte stimulating hormone (α-MSH; refs. 1 and 10). Whereas the NPY- and α-MSH-producing neurons reside in the ARC, their receptors (Y1 and Y5 receptors for NPY and MC-4 receptors for α-MSH) are localized at their release sites in the magnocellular PVN (mPVN) and parvocellular PVN (pPVN), respectively (11–14). Interestingly, stimulation of feeding by NPY is supplemented by two additional signals that are coexpressed with NPY, agouti-related peptide (AgrP), which elicits feeding by competitive inhibition of α-MSH at MC-4 receptors, and γ-aminobutyric acid (GABA), which stimulates feeding by dampening the local restraint through GABAA receptors in the mPVN (14–16). Similarly, the peptide cocaine- and amphetamine-regulating transcript (CART), which is coexpressed with α-MSH in POMC producing neurons, supplements the α-MSH-induced restraint on appetite (1, 2). Additionally, morphological and experimental evidence suggests that NPY may directly modulate POMC neurons (1, 17, 18). These two connected, but functionally opposed neuronal systems, NPY and POMC, are leptin targets as evidenced by the presence of the functionally active, long isoform of the leptin receptor, leptin-Rb, on these neurons (1, 2).
The paper by Lambert et al. demonstrates the efficacy of CNTFAx15 in correcting obesity in leptin-resistant mice.
Figure 1.
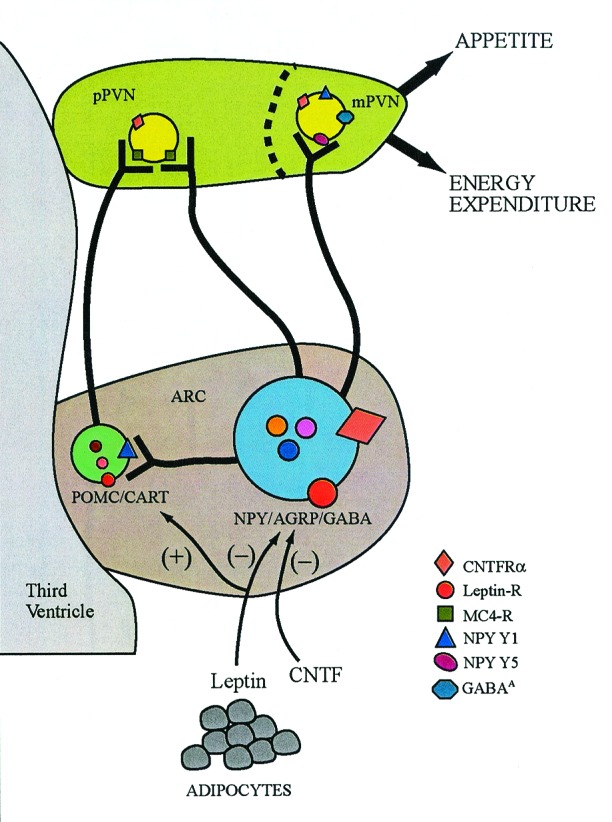
Schematic representation of hypothalamic signaling pathways in the regulation of appetite and energy expenditure. NPY, AgrP, and GABA, the appetite-stimulating signals, are coproduced in the perikarya located in the ARC. Likewise, the appetite-inhibiting peptides, α-MSH and CART, are coproduced in the POMC/CART coexpressing perikarya in the ARC. These distinct populations of neurons project into the two subdivisions of the PVN, the mPVN and pPVN, to activate their corresponding receptors for regulation of appetite and energy expenditure. NPY-producing neurons also contact the POMC/CART neurons locally in the ARC to curtail their tonic restraint on appetite. Leptin inhibits appetite through leptin-Rb located on the NPY/AgrP/GABA- and POMC/CART-expressing cell bodies in the ARC and at postsynaptic sites in the PVN where it regulates release of these signals and enhances energy expenditure through the sympathetic nervous system. The results show that CNTF/CNTFAx15, through activation of the CNTFRα located in the ARC and PVN, markedly diminish the availability of NPY for release in the PVN by suppressing its synthesis in the ARC and concurrently attenuating postsynaptic NPYergic signaling by decreasing NPY Y1 receptor and pCREB abundance.
Although CNTF is not a secretable protein it can readily penetrate the BBB in a manner similar to leptin (19). Whereas the CNTF receptor, CNTFRα, is abundant in feeding relevant sites, such as the ARC and PVN in the adult hypothalamus (20, Fig. 1), CNTF ligand expression is undetectable, leaving open the possibility of a CNTF-related ligand operating in these sites (21). Administration of CNTF to wild-type laboratory rats and mice rapidly induced anorexia and weight loss, primarily due to loss of body fat, that lasted for a few days even after cessation of treatment (20, 22). Importantly, CNTF/CNTFAx15 readily normalized the obese phenotype in leptin-deficient ob/ob mice, in db/db mice with impaired signaling due to mutated leptin receptor, in MC-4 receptor-deficient mice, and in normal wild-type mice rendered obese and leptin-resistant by a high-fat diet (9, 20, 22, 23). Thus, CNTF/CNTFAx15 appear to be more efficient than leptin because body weight was reduced in conditions where leptin was ineffective. That CNTF/CNTFAx15 are suitable substitutes for leptin is also implied by the findings that, like leptin, they diminish hypothalamic NPYergic signaling by suppressing NPY gene expression in the ARC (22) and NPY immunoreactivity in the PVN (ref. 9; Fig. 1). A second site of action is at the postsynaptic level in the PVN because CNTF/CNTFAx15 treatment suppressed NPY-induced feeding [likely a result of diminished NPY Y1 receptor abundance (22, 24)] and, as shown by Lambert et al. (9), diminished pCREB in the PVN.
CNTF belongs to the class 1 superfamily of cytokines well known for their pleiotropic actions (25, 26). Cytokines are causal factors for anorexia, weight loss, and metabolic breakdown leading to cachexia, the typical symptoms associated with cancer, chronic infection, and prolonged immune reaction (27). This intrinsic pleiotropic nature of cytokines is due to the common signal transduction sequalae in target cells. However, it is now obvious that these severe symptoms are rarely attributed to one single cytokine, but rather to the concerted action of several cytokines. Nevertheless, it is reasonable to suspect that administration of CNTF or CNTFAx15 may elicit some, if not all, cytokine-like symptoms. Untoward side effects, such as fever, taste aversion, and metabolic effects, were not observed when CNTF or CNTFAx15 were administered in doses effective in reducing weight (9, 20). Concomitant central infusion of NPY with CNTF completely reversed the anorexic and weight-reducing effects of CNTF (28), reiterating the specificity and safety of CNTF. Despite these assurances, careful evaluation of the proinflammatory and cytotoxic effects of chronic systemic CNTF/CNTFAx15 treatment is clearly warranted.
Experimental evidence indicates that leptin activates cytokine-like signal transduction pathways by stimulating JAK-STAT via the leptin-Rb in hypothalamic sites implicated in energy homeostasis (9, 20, 29). Additionally, leptin activates Suppressor of Cytokine Signaling-3 (SOCS-3), which reduces intracellular signaling by inhibiting JAK activity. Excessive expression of SOCS-3 has been proposed as a potential mechanism underlying leptin resistance by switching off or diminishing leptin signaling, thereby permitting overeating and reduction in energy expenditure. However, CNTF also markedly increased SOCS-3 along with the continued severe reduction in appetite and weight (29). Consequently, SOCS-3 activation is unlikely to be the major signal transduction event underlying leptin resistance. Although a few pathways activated by CNTF in the hypothalamus may parallel the pathways activated by leptin, it is highly likely that independent, and yet to be defined, CNTFRα-induced cellular and intracellular signal transduction events may underlie appetite and weight reduction. That disparate hypothalamic mechanisms operate after CNTF and leptin is also evident by the fact that, whereas leptin reduces weight by decreasing appetite through modulation of the appetite-regulating network and by augmenting energy loss through modulation of the energy expenditure network linked with sympathetic nervous system (refs. 1 and 2; Fig. 1), so far only selective diminution of appetite via NPYergic signaling has been identified as the mechanism responsible for the profound weight-reducing effects of CNTF/CNTFAx15.
With the increase in the incidence of obesity and abnormal weight gain and associated metabolic disorders such as type II diabetes and cardiovascular diseases, the need for newer therapeutic strategies to stem this epidemic continues to grow. A rational approach may be to exploit the anorectic and weight-reducing properties of the naturally occurring cytokines that readily gain access to hypothalamic sites by saturable transport across the BBB. Because of the pleiotropic nature of cytokines and leptin, overlapping signal transduction pathways, and the cross talk among cytokines, redesigning the chemical structure of the cytokines to reduce or eliminate side effects is highly desirable. Application of CNTF and its derivative, CNTFAx15, as weight-reducing therapy is a step in that direction. An alternative approach that deserves serious consideration and has the potential for sustained outcome is gene therapy to deliver the transgenes that encode desirable cytokines. Indeed, one-time delivery of viral vectors encoding transgenes for leptin, CNTF, and leukemia inhibitory factor, either systemically or directly into the hypothalamus in a site-specific manner to minimize the undesirable side effects of cytokines at peripheral targets, has proven to be effective in reducing body weight for extended periods (30, 31).†,‡ Admittedly, the interventional strategies that have begun to exploit the diversity in cytokine action are in their infancy, and many hurdles remain to be overcome before successful application to control human obesity is contemplated.
Footnotes
See companion article on page 4652.
Dhillon, H., Minter, R., Topping, D., Prima, V., Moldawer, L., Muzyczka, N., Kalra, P., Kalra, S. & Zolotnkhin, S., 2nd Annual Meeting of the American Society for Gene Therapy, Washington, DC, June 9–13, 1999, abstr. 178.
Beretta, E., Prima, V., Dhillon, H., Moldawer, L.L., Muzyczka, N., Zolotnkhin, S., Kalra, P. S. & Kalra, S. P., 3rd Annual Meeting of the American Society of Gene Therapy, Denver, CO, May 31–June 4, 2000, abstr. 186.
References
- 1.Kalra S P, Dube M G, Pu S, Xu B, Horvath T L, Kalra P S. Endocr Rev. 1999;20:68–100. doi: 10.1210/edrv.20.1.0357. [DOI] [PubMed] [Google Scholar]
- 2.Friedman J M, Halaas J L. Nature (London) 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 3.Heymsfield S B, Greenberg A S, Fujioka K, Dixon R M, Kushner R, Hunt T, Lubina J A, Patane J, Self B, Hunt P, et al. J Am Med Assoc. 1999;282:1568–1575. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 4.Mokdad A H, Serdula M K, Dietz W H, Bowman B A, Marks J S, Koplan J P. J Am Med Assoc. 1999;282:1519–1522. doi: 10.1001/jama.282.16.1519. [DOI] [PubMed] [Google Scholar]
- 5.Plata-Salaman C R. Semin Oncol. 1998;25:64–72. [PubMed] [Google Scholar]
- 6.Miller R G, Petajan J H, Bryan W W, Armon C, Barohn R J, Goodpasture J C, Hoagland R J, Parry G J, Ross M A, Stromatt S C. Ann Neurol. 1996;39:256–260. doi: 10.1002/ana.410390215. [DOI] [PubMed] [Google Scholar]
- 7.Espat N J, Auffenberg T, Rosenberg J J, Rogy M, Martin D, Fang C H, Hasselgren P O, Copeland E M, Moldawer L L. Am J Physiol. 1996;271:R185–R190. doi: 10.1152/ajpregu.1996.271.1.R185. [DOI] [PubMed] [Google Scholar]
- 8.Martin D, Merkel E, Tucker K K, McManaman J L, Albert D, Relton J, Russell D A. Am J Physiol. 1996;271:R1422–R1428. doi: 10.1152/ajpregu.1996.271.5.R1422. [DOI] [PubMed] [Google Scholar]
- 9.Lambert P D, Anderson K D, Sleeman M W, Tan J, Hijarunguru A, Corcoran T L, Murray J D, Thabet K E, Yancopoulous G D, Wiegan S J. Proc Natl Acad Sci USA. 2001;98:4652–4657. doi: 10.1073/pnas.061034298. . (First Published March 20, 2001; 10.1073/pnas.061034298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huszar D, Lynch C A, Fairchild-Huntress V, Dunmore J H, Fang Q, Berkemeier L R, Gu W, Kesterson R A, Boston B A, Cone R D, et al. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 11.Lu D, Willard D, Patel I R, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik R P, Wilkison W O, et al. Nature (London) 1994;371:799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- 12.Yokosuka M, Kalra P S, Kalra S P. Endocrinology. 1999;140:4494–4500. doi: 10.1210/endo.140.10.7058. [DOI] [PubMed] [Google Scholar]
- 13.Yokosuka M, Dube M, Kalra P S, Kalra S P. Peptides. 2001;22:507–514. doi: 10.1016/s0196-9781(01)00360-6. [DOI] [PubMed] [Google Scholar]
- 14.Cowley M A, Pronchuk N, Fan W, Dinulescu D M, Colmers W F, Cone R D. Neuron. 1999;24:155–163. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- 15.Horvath T L, Bechmann I, Naftolin F, Kalra S P, Leranth C. Brain Res. 1997;756:283–286. doi: 10.1016/s0006-8993(97)00184-4. [DOI] [PubMed] [Google Scholar]
- 16.Pu S, Jain M R, Horvath T L, Diano S, Kalra P S, Kalra S P. Endocrinology. 1999;140:933–940. doi: 10.1210/endo.140.2.6495. [DOI] [PubMed] [Google Scholar]
- 17.Horvath T L, Naftolin F, Kalra S P, Leranth C. Endocrinology. 1992;131:2461–2467. doi: 10.1210/endo.131.5.1425443. [DOI] [PubMed] [Google Scholar]
- 18.Jegou S, Blasquez C, Delbende C, Bunel D T, Bunel H. Ann NY Acad Sci. 1993;680:260–278. doi: 10.1111/j.1749-6632.1993.tb19689.x. [DOI] [PubMed] [Google Scholar]
- 19.Pan W, Kastin A J, Maness L M, Maness J M. Neurosci Lett. 1999;263:69–71. doi: 10.1016/s0304-3940(99)00083-x. [DOI] [PubMed] [Google Scholar]
- 20.Gloaguen I, Costa P, Demartis A, Lazzaro D, Di Marco A, Graziani R, Paonessa G, Chen F, Rosenblum C I, Van der Ploeg L H, et al. Proc Natl Acad Sci USA. 1997;94:6456–6461. doi: 10.1073/pnas.94.12.6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elson G C, Lelievre E, Guillet C, Chevalier S, Plun-Favreau H, Froger J, Suard I, de Coignac A B, Delneste Y, Bonnefoy J Y, et al. Nat Neurosci. 2000;3:867–872. doi: 10.1038/78765. [DOI] [PubMed] [Google Scholar]
- 22.Xu B, Dube M G, Kalra P S, Farmerie W G, Kaibara A, Moldawer L L, Martin D, Kalra S P. Endocrinology. 1998;139:466–473. doi: 10.1210/endo.139.2.5723. [DOI] [PubMed] [Google Scholar]
- 23.Marsh D J, Hollopeter G, Huszar D, Laufer R, Yagaloff K A, Fisher S L, Burn P, Palmiter R D. Nat Genet. 1999;21:119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 24.Xu B, Kalra P S, Moldawer L L, Kalra S P. Regul Pept. 1998;75–76:391–395. doi: 10.1016/s0167-0115(98)00093-7. [DOI] [PubMed] [Google Scholar]
- 25.Stahl N, Yancopoulos G D. J Neurobiol. 1994;25:1454–1466. doi: 10.1002/neu.480251111. [DOI] [PubMed] [Google Scholar]
- 26.Tartaglia L A. J Biol Chem. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- 27.Matthys P, Billiau A. Nutrition. 1997;13:763–770. doi: 10.1016/s0899-9007(97)00185-8. [DOI] [PubMed] [Google Scholar]
- 28.Pu S, Dhillon H, Moldawer L L, Kalra P S, Kalra S P. J Neuroendocrinol. 2000;12:827–832. doi: 10.1046/j.1365-2826.2000.00526.x. [DOI] [PubMed] [Google Scholar]
- 29.Bjorbaek C, Elmquist J K, El-Haschimi K, Kelly J, Ahima R S, Hileman S, Flier J S. Endocrinology. 1999;140:2035–2043. doi: 10.1210/endo.140.5.6736. [DOI] [PubMed] [Google Scholar]
- 30.Dhillon H, Ge Y, Minter R M, Prima V, Moldawer L L, Muzyczka N, Zolotukhin S, Kalra P S, Kalra S P. Regul Pept. 2000;92:97–105. doi: 10.1016/s0167-0115(00)00155-5. [DOI] [PubMed] [Google Scholar]
- 31.Dhillon H, Kalra S, Prima V, Zolotukhin S, Scarpace P, Moldawer L, Muzyczka N, Kalra P. Regul Pept. 2001;99:69–77. doi: 10.1016/s0167-0115(01)00237-3. [DOI] [PubMed] [Google Scholar]