

Abstract
Necrotizing enterocolitis (NEC) is the leading cause of gastrointestinal-related mortality in premature infants, and develops under conditions of exaggerated Toll-like receptor-4 (TLR4) signaling in the newborn intestinal epithelium. Since NEC does not develop spontaneously despite the presence of seemingly tonic stimulation of intestinal TLR4, we hypothesized that mechanisms must exist to constrain TLR4 signaling that become diminished during NEC pathogenesis, and focused on the intracellular stress response protein and chaperone Heat Shock Protein-70 (Hsp70). We now demonstrate that the induction of intracellular Hsp70 in enterocytes dramatically reduced TLR4 signaling as assessed by LPS-induced NFkB translocation, cytokine expression and apoptosis. These findings were confirmed in vivo, using mice that either globally lacked Hsp70 or which over-expressed Hsp70 within the intestinal epithelium. TLR4 activation itself significantly increased Hsp70 expression in enterocytes, which provided a mechanism of auto-inhibition of TLR4 signaling in enterocytes. In seeking to define the mechanisms involved, intracellular Hsp70-mediated inhibition of TLR4 signaling required both its substrate-binding EEVD-domain and association with the co-chaperone CHIP, resulting in ubiquitination and proteosomal degradation of TLR4. The expression of Hsp70 in the intestinal epithelium was significantly decreased in murine and human NEC compared to healthy controls, suggesting loss of Hsp70 protection from TLR4 could lead to NEC. In support of this, intestinal-Hsp70 overexpression in mice and pharmacologic upregulation of Hsp70 reversed TLR4-induced cytokines and enterocyte apoptosis, and prevented and treated experimental NEC. Thus, a novel TLR4 regulatory pathway exists within the newborn gut involving Hsp70 that may be pharmacologically activated to limit NEC severity.
INTRODUCTION
Necrotizing enterocolitis (NEC) is the leading cause of death from gastrointestinal disease in premature infants, and is characterized by the sudden onset of feeding intolerance that rapidly progresses to abdominal distention, systemic sepsis and death due to acute necrosis of the intestine (1). The intestinal epithelium in infants with NEC displays exaggerated enterocyte apoptosis and mucosal disruption, which is widely thought to lead to the trans-luminal passage of indigenous microbes and an unbridled activation of the host immune system(2). In seeking to determine the inciting molecular mechanisms leading to the development of this cascade, we (3, 4) and others (5, 6)have determined that activation of the innate immune receptor Toll like receptor 4 (TLR4) within the intestinal epithelium plays an important role in NEC pathogenesis. Specifically, TLR4 signaling in enterocytes leads to increased enterocyte apoptosis in vitro and in vivo, while inhibition of TLR4 signaling in the newborn intestinal epithelium prevents NEC development (3, 4, 7). While these studies have clearly placed the spotlight on the role of TLR4 in the pathogenesis of NEC, the observation that most premature infants do not develop NEC despite seemingly tonic activation of TLR4 within the gut raises the possibility that TLR4 signaling must somehow be curtailed within the newborn intestinal epithelium to limit disease development. Importantly however, the presence of negative regulatory strategies for TLR4 within the newborn intestinal epithelium and the degree to which such strategies may participate in the pathogenesis of NEC remains largely unexplored.
In the current studies, we test the hypothesis that the intracellular chaperone Heat shock protein 70 (Hsp70) could negatively regulate TLR4 signaling within enterocytes, and by extension that a loss of Hsp70 could lead to NEC development through unbridled TLR4 activation. The heat shock proteins – of which Hsp70 is a predominant member – represent a family of intracellular proteins that are activated by a variety of stressors, and that can assist in the delivery of target proteins to the ubiquitin-proteosome system for degradation through co-chaperone molecules such as CHIP, which stands for C-terminus of Hsp70 interacting Protein (8). An intracellular role for Hsp70 has not been linked to the pathogenesis of NEC nor to regulation of TLR4 within enterocytes, although Hsp70 has been shown to play an important role in the modulation of apoptosis after various forms of stress (9–12). Through its combined roles of both clearing proteins and modulating cell death, the net effect of Hsp70 induction within cells is to restore the host to a non-stressed environment (13–15). While cytoplasmic Hsp70 has not been linked to the regulation of TLR4 signaling inside the enterocyte, Hsp70 has been shown to serve a protective role in the intestine as demonstrated by Chang and colleagues (16) (17), although upstream regulatory pathways within the gut were not identified. Taken together, these findings in the literature now raise the exciting possibility that intracellular Hsp70 could represent a novel regulator of TLR4 signaling at baseline and in the development of NEC.
In support of this hypothesis, using enterocytes that either lack or are induced to express Hsp70 as well as by examining mice that either lack Hsp70 or that over-express Hsp70 within the intestinal epithelium, we now determine that intracellular Hsp70 limits TLR4 signaling in enterocytes, and moreover, that Hsp70 plays a central role in the pathogenesis of NEC. The mechanism by which Hsp70 limits TLR4 signaling in the gut involves an increase in CHIP-mediated ubiquitination and degradation of TLR4 via the ubiquitin-proteosomal pathway. Importantly, pharmacologic upregulation of Hsp70 within the intestinal mucosa led to a reduction in TLR4 signaling and a decrease in enterocyte apoptosis, leading to an attenuation in NEC severity. Taken together, these findings illustrate a novel pathway linking the regulation of Hsp70 with the negative control of TLR4 signaling within the gut, and provide evidence that the development of NEC results in part from exaggerated TLR4-induced enterocyte apoptosis due in part to reduced Hsp70 activity. Moreover, these results suggest that pharmacologic upregulation of Hsp70 could provide a novel approach to the prevention and/or treatment of NEC through the inhibition of TLR4 signaling in the newborn intestine.
MATERIALS AND METHODS
Cell culture and reagents
IEC-enterocytes were obtained from the American Type Culture Collection (ATCC, Manassas, VA). LPS (E coli 0111:B4 purified by gel-filtration chromatography, >99% pure) was from Sigma-Aldrich. The TLR2 ligand Pam3 and the tlr5 ligand flagellin were from Invivogen. Antibodies were as follows: p65 subunit of NF-κB -Santa Cruz Biotechnology; TLR4 – Imgenex, Santa Cruz (L14); cleaved-caspase 3 - Cell Signaling; the inducible isoform of Hsp70 - Stressgen SPA-810 and Santa Cruz (inducible, K20); the constitutive (control) isoform of Hsp70 i.e. Hsc70 - Stressgen SPA-815; Ubiquitin – Millipore; V5 – Genescript.
Where indicated, cells were pretreated with the proteosome inhibitor MG-132 (Calbiochem, 10uM) 2h prior to the indicated experimental condition. IEC-6 enterocytes were treated with LPS at concentrations that we have shown to be present in mice and humans with NEC i.e. 50μg/ml (3).
Preparation of lentiviruses and cell transfection
Lentiviruses expressing V5-tagged Hsp70 dominant negative C-terminal deletion mutant (ΔEEVD), wild-type Hsp70, siRNA to Hsp70, CHIP Hsp70 docking mutants (K30A), CHIP U-Box (H260Q) mutants, LacZ and were generated using a combination of ViraPower HiPerform Lentiviral and Lentiviral pLenti6.3/V5-DEST Gateway expression system (Invitrogen). In brief, the recombinant V5-tagged-pLenti6.3/V5-DEST expression plasmids expressing wild-type and dominant negative C-terminal deletion mutant (ΔEEVD) Hsp70, CHIP Hsp70 docking mutants (K30A), CHIP U-Box (H260Q) mutants, and LacZ were first generated using Gateway directional TOPO cloning systems (Invitrogen). V5-tagged pLenti6.3/V5-DEST expression plasmids are under the control of immediate early promoter human CMV (cytomegalovirus) and contains WPRE (Woodchunk Posttranscriptional Regulatory Element) and cPPT (polypurine tract) elements, which yields cell-specific, high performance expression of recombinant proteins. Recombinant V5-tagged pLenti6.3/V5-DEST expression plasmids were end-sequenced to verify the correct directional cloning. High expression lentiviral particles were next generated by co-transfection of recombinant V5-tagged pLenti6.3/V5-DEST plasmids and ViraPower packing mix in receptive 293FT cells, and used for transduction in destination IEC-6 cells for expression of recombinant proteins. mRNA and protein expression of recombinant proteins were verified by qRT-PCR, western blot and immunofluorescence staining. Cells were transduced with virus particles 48h prior to further treatment.
The authors are indebted to Dr. Richard Morimoto, for EEVD plasmids, Dr. Richard Mosser and Dr. Robin Anderson for Hsp70 plasmids, and Dr. Cam Patterson, for CHIP reagents, which were used to generate appropriate lentiviruses.
IEC-6 cells were pretreated with either Hsp70 siRNA (100nM, Dharmacon), via Lipofectamine LTX (Invitrogen) according to the manufacturer's directions, or a negative control siRNA that is directed at no known target for 48h in antibiotic free media at 37°C in a humidified chamber with 5% CO2. The media was changed and cells were treated with LPS, and assessed for apoptosis, NFkB translocation and/or cytokine expression. The NF-kB promoter GFP-reporter system was generated via the pPACKF1 Lentivector Packaging kit (System Biosciences) using the pSIF1-H1-siLuc-copGFP positive control expression plasmid and delivered via lentiviral transduction of IEC-6 cells 48h prior to stimulation with LPS and/or heat treatment as described above.
Hsp70 induction and knockdown
For the induction of Hsp70, IEC-6 cells were placed in an incubator at 42°C (5% CO2 and 23% O2) for 45min followed by either a 4h (LPS-mediated apoptosis analysis) or 8h (NF-kB translocation assay) recovery period at 37°C under otherwise the same ambient conditions, after which they were exposed to the indicated treatment. In parallel, IEC-6 cells were treated with Celastrol (3μM in DMSO) for 30min at 37°C and allowed an 8h recovery period at 37°C prior to treatment with LPS.
Targeted knockdown of Hsp70 was accomplished via transfection with siRNA to Hsp70 or siRNA against no known target. Chemical inhibition of Hsp70 was performed via 2h pretreatment with Quercetin (20μM) (Sigma) in DMSO.
Induction of Hsp70 within cells was accomplished in vivo through the administration of Celastrol 1mg/kg as described below. To confirm that the effects of this compound on TLR4 signaling occurred through upregulation of Hsp70, all studies were performed in Hsp70 deficient mice after injection of Celastrol for comparison as described below.
Experimental endotoxemia and necrotizing enterocolitis
All experiments were approved by the Children's Hospital of Pittsburgh Animal Care Committee and the Institutional Review Board of the University of Pittsburgh. C57Bl-6, swiss-webster, Hsp70−/− and mice over-expressing Hsp70 on the villin promoter (Hsp70villin) and their appropriate wild-type controls were generated as described (16, 17) or obtained from Jackson Laboratories. All animals were age and sex-matched prior to use. Experimental NEC was induced in 10 day old mice as we have described (4, 7, 18) using formula gavage (Similac Advanced infant formula (Ross Pediatrics):Esbilac canine milk replacer 2:1) five times/day, and hypoxia (5%O2, 95%N2) for 10 minutes in a hypoxic chamber (Billups-Rothenberg) twice daily for 4 days. Where indicated, mice were administered Celastrol (1mg/kg) via intraperitoneal injections 1 day prior to the model and on day 3 of the model. Volume-matched DMSO was administered as a vehicle control in all Celastrol treatment models. The severity of disease was determined on histologic sections of the terminal ileum by a pediatric pathologist who was blinded to the study condition according to the work of of Radulescu et al (19) as follows: 0: normal intestine; 1: epithelial lifting or separation; 2: sloughing of epithelial cells to the mid villous level; 3: necrosis of the entire villous. This protocol results in the development of patchy necrosis involving the small intestine similar to human NEC, with an increase in circulating cytokines that mimics that seen in human NEC (18).
Preparation and processing of human intestinal tissue from infants with and without NEC
Intestinal samples were obtained from human neonates undergoing intestinal resection for NEC, for unrelated indications (control), or at time of stoma closure. All human tissue was obtained and processed as discarded tissue via waiver of consent with approval from the University of Pittsburgh Institutional Review Board and in accordance with the University of Pittsburgh anatomical tissue procurement guidelines.
Immunohistochemistry, immunoprecipitation and SDS-PAGE
The immuno-confocal microscopy of IEC-6 enterocytes, and mouse and human intestine was performed as previously described (20), and evaluated using an Olympus Fluoview 1000 confocal microscope under oil-immersion objectives. Images were assembled using Adobe Photoshop CS2 software (Adobe Systems Inc.). In parallel, Cryo-Gel (Cancer Diagnostics Inc.) frozen sections of terminal ileum were sectioned (6μm), rehydrated with PBS and fixed with 2% paraformaldehyde. Non-specific binding was blocked with 5% bovine serum albumin (BSA).
For immunoprecipitation, IEC-6 lysates were collected and 500ug of total protein was pre-cleared with 20ul/sample Agarose Protein A/G beads (Santa Cruz Biotechnology) for 30min at 4°C. Samples were centrifuged and supernatants were collected and treated with antibodies to Hsp70 (K20) or ubiquitin as indicated, or an isotype IgG control antibody, and then incubated with Agarose Protein A/G beads overnight at 4°C. Samples were then centrifuged, supernatants were discarded and beads were washed three times in PBS and resuspended in equal volumes of 2× gel loading buffer and boiled at 95°C for 3min. Lysates were then subjected to SDS-PAGE (21), and immunobloted with various antibodies (1:1000) as indicated. Blots were developed using the enhanced chemiluminescence reagent (ECL-Super Signal; Pierce), and developed on radiographic film.
Quantitative real-time polymerase chain reaction
Quantitative real-time PCR was performed as previously described using the Bio-Rad CFX96 Real-Time System (Biorad) (3) using the primers listed below. Where indicated, gene expression was assessed on 2.5% agarose gels using ethidium bromide staining. Images were obtained with a Kodak Gel Logic 100 Imaging System using Kodak Molecular Imaging software. The expression of the following genes by qRT-PCR was measured relative to the housekeeping genes β-actin and GAPDH.
| Gene | Species | Forward sequence | Reverse sequence | Amplicon Size (bP) |
| β-actin | Mouse/Rat | CCACAGCTGAGAGGGAAATC | TCTCCAGGGAGGAAGAGGAT | 108 |
| Human | TCCCTGGAGAAGAGCTACG | GTAGTTTCGTGGATGCCACA | 131 | |
| Hsp70 | Mouse/human | GCCAACAAGATCACCATCAC | TGTTGAAGGCATAGGATTCG | 154 |
| Rat | TTCAATATGAAGAGCGCCGTGGAG | TCCTCTTTCTCAGCCAGCCAGCGTGTTA | 134 | |
| GAPDH | Mouse | TGAAGCAGGCATCTGAGGG | CGAAGGTGGAAGAGTGGGAG | 102 |
| Human | TCTCCTCTGACTTCAACAGCGACA | CCCTGTTGCTGTAGCCAAATTCGT | 126 | |
| IL6 | Mouse/Rat | CCAATTTCCAATGCTCTCCT | ACCACAGTGAGGAATGTCCA | 182 |
| Human | TCTCCACAAGCGCCTTCG | CTCAGGGCTGAGATGCCG | 193 | |
| iNOS | Mouse/Rat | CTGCTGGTGGTGACAAGCACATTT | ATGTCATGAGCAAAGGCGCAGAAC | 167 |
| Human | AATGAGTCCCCGCAGCCCCT | AGTCATCCCGCTGCCCCAGT | 143 | |
| TLR4 | Mouse | TTTATTCAGAGCCGTTGGTG | CAGAGGATTGTCCTCCCATT | 186 |
| Human | AAGCCGAAAGGTGATTGTTG | CTGAGCAGGGTCTTCTCCAC | 153 | |
| Rat | TGCTCAGACATGGCAGTTTC | GCGATACAATTCGACCTGCT | 102 |
Determination of enterocyte signaling and mucosal injury in response to TLR4 activation
For the determination of NFkB translocation, IEC-6 cells were treated with LPS (50mg/ml, 45min). The extent of NFkB translocation was determined as in (18) in an adaptation of the methodology of Ding and Li.(22). In brief, a threshold limit was set based upon the emission signal for the nuclear stain DAPI in serial micrographs of cells that were co-stained for the p65 subunit of NFkB, while a corresponding cytoplasmic region of interest was defined by stenciling a circular region 12 pixels beyond the nucleus upon each cell. The average integrated pixel intensity pertaining to the corresponding NFkB emission within the cytoplasmic and nuclear regions was then determined for more than 200 cells per treatment group in at least four experiments per group, using MetaMorph software version 6.1 (Molecular Devices Corporation).
The release of IL-6 from IEC-6 cells was determined by enzyme-linked immunosorbent assay kit (R&D Systems) according to the manufacturer's instructions.
Enterocyte apoptosis was determined in IEC-6 after 16 hours of treatment with LPS under concentrations that we have measured in NEC (3) (50mg/ml) by immunostaining with antibodies to cleaved-caspase 3 and performing confocal immunofluorescent analysis. The number of cleaved-caspase 3 positive cells was identified in a blinded fashion using Metamorph software (Molecular Devices Corp) and expressed as a percentage of cleaved-caspase 3 positive cells per high-power field, with more than 100 fields/experiment studied, and more than 100 cells/field.
Enterocyte apoptosis in vivo was determined by measuring the percent of enterocytes that are positive for cleaved-caspase 3 by confocal microscopy per high-power field. Over 50 fields per sample are evaluated, as we have described (7).
Statistical Analysis
All experiments were repeated at least in triplicate, with more than 100 cells/high-power field. For mouse experiments of endotoxemia, at least 4 mice/group were assessed; for experiments of NEC, over 10 mouse pups per group were included and litter matched controls were included in all cases. Statistical analysis was performed using SPSS 13.0 software. ANOVA was used for comparisons for experiments involving more than two experimental groups. Two-tailed student's t-test was used for comparison for experiments consisting of two experimental groups. For analysis of the severity of NEC, chi-square analysis was performed. In all cases, statistical significance was accepted at p<0.05 between groups.
RESULTS
Hsp70 induction limits TLR4 signaling in enterocytes
To determine whether the induction of Hsp70 could limit TLR4 signaling, we first briefly exposed cultured enterocytes (IEC-6 cells) to conditions known to increase the expression of Hsp70 (42°C, 45 min Figure 1A), and then treated cells with the TLR4 agonist LPS at concentrations that we have previously measured from the stool of humans and mice with NEC (3). The extent of TLR4 signaling was determined by assessing (1) the extent of NFkB activation as reflected by the degree of translocation of the p65 subunit of NFkB from the cytoplasm to the nucleus as described in Methods, (2) the degree of induction of the pro-inflammatory cytokine IL-6, and (3) the degree of enterocyte apoptosis. As shown in Figure 1, LPS treatment caused a marked increase in NFkB activation (Figure 1 panel D i–ii) that was significantly reduced following heat treatment (Figure 1 panel C, Diii). Heat treatment also prevented the increase in IL-6 expression that occurred in LPS-treated IEC-6 cells (Figure 1 panel B), as well as a reduction in the extent of LPS-mediated apoptosis, both down to levels similar to untreated control cells (Figure 1 panel F i–iii). We have utilized the expression of cleaved caspases 3 as an estimate of apoptosis in these studies, as its increased expression has been shown to be a terminal event in the apoptosis cascade in enterocytes and provides a reliable and reproducible estimate of enterocyte apoptosis in both human and experimental NEC(7, 23–25). To determine whether a heat-induced increase in Hsp70 was required for the observed attenuation in TLR4 signaling, cells were exposed to heat after siRNA-mediated knockdown of Hsp70 in IEC-6 cells (Figure 1 panel E), which abrogated the protection in TLR4 signaling previously observed after heat treatment with regards to the induction in IL-6 expression (Figure 1 Panel B), NFkB translocation (Figure 1 panel C) and enterocyte apoptosis (Figure 1 panel F). Taken together, these findings indicate that Hsp70 can inhibit TLR4 signaling in enterocytes. We next sought to investigate the mechanisms mediating this effect, and focused initially on determining whether an association between Hsp70 and TLR4 was required for the negative effects of Hsp70 on TLR4 signaling.
Figure 1. Hsp70 induction limits TLR4 signaling in enterocytes.

A: Confocal micrographs showing the expression of Hsp70 (green), β-actin (red) and DAPI (blue) in IEC-6 enterocytes that were either untreated (i) or exposed to 42°C for 45 minutes (ii). B–C: quantitative RT-PCR showing the expression of IL-6 (B) or quantification of the extent of NFkB translocation (C) in IEC-6 cells that were either untreated (white bars) or exposed to heat (black bars), and were either untransfected or were transfected with Hsp70 siRNA. *p<0.05 vs untreated control; **p<0.05 vs heat-control, ***p<0.001 vs control cells transfected with Hsp70 siRNA. D: Confocal micrographs of IEC-6 enterocytes that were either untreated (i), treated with LPS (50μg/ml, 45 min, ii) or were treated with LPS after pre-treatment with heat (iii). Quantification in C is based upon over 50 cells per field and over 50 fields examined in 4 separate experiments. *p<0.05 vs untreated control; **p<0.05 vs heat-control. E: Representative SDS-PAGE showing lysates of IEC-6 that were untreated (control), incubated with PBS alone (vehicle), or were transfected with either control siRNA against no known substrate (non-targeted siRNA) or siRNA to Hsp70 (Hsp70 siRNA). Blot was stripped then re-probed with antibodies to β-actin. F: confocal micrographs (i–iv) and quantification (v) of IEC-6 enterocytes that were either untreated (i), treated with LPS in the absence (ii) or presence of pre-exposure to heat as above (iii), or pre-treated 48 hours prior with siRNA to Hsp70 as in (iv). *p<0.05 vs control; **p<0.005 vs LPS control; ***p<0.05 vs heat+hsp70sirna saline. Representative images are taken of over 50 fields examined with over 50 cells/field in 4 separate experiments. Size bar = 10μ. Representative apoptotic cells are indicated by arrows.
An EEVD-mediated association between TLR4 and Hsp70 is required for the inhibition of TLR4 signaling in enterocytes by Hsp70
The effects of Hsp70 on target proteins are largely influenced by interactions with accessory proteins called co-chaperones (26, 27). In a variety of cell types, Hsp70 regulates the intracellular function and fate of proteins through the formation of direct protein-protein interactions which occur largely through an EEVD binding domain in its C-terminus (28, 29) (30). To investigate whether an association between TLR4 and Hsp70 was required for the negative effects on TLR4 signaling, IEC-6 cells were stably transduced with lentivirus expressing either LacZ as a vector control or with Hsp70 bearing a truncation mutation in the EEVD domain while sparing the N-terminal ATPase domain as described (31) (Figure 2 panel Ai). After LPS treatment, cells were then subjected to immunoprecipitation using antibodies to Hsp70 in the absence or presence of heat exposure. As is shown in Figure 2 panel Ai, TLR4 was detected in immunoprecipitates obtained from IEC-6 lysates that had been transduced with control vector, and this association was increased after heat exposure. By contrast, the stable delivery via lentiviral transduction of a mutant Hsp70 lacking the C- terminus EEVD binding domain markedly reduced the ability to detect TLR4 in the immunoprecipitates after LPS treatment which did not vary after heat exposure, indicating that the EEVD mutation resulted in a reduction in the extent of association between Hsp70 and TLR4 (Figure 2 panel Ai). While heat treatment of IEC-6 cells that had been transduced with LacZ vector conferred significant protection from both LPS-induced NFkB translocation (Figure 2 panels Bi–iii) and enterocyte apoptosis (Figure 2 panel Ci–iii), this heat-mediated reduction in LPS signaling was lost in IEC-6 cells that had been transduced with the C-terminus EEVD binding mutant (Figure 2 panels B iv–vii and C iv–vii). As expected, LPS caused a significantly greater degree of IL-6 expression after transduction of delta EEVD Hsp70 as compared to non-transfected cells (Figure 2 B xii), consistent with the reduced effect of Hsp70 in inhibiting TLR4 signaling in these cells. To further investigate the link between Hsp70 expression and TLR4 signaling, IEC-6 cells were transduced with lentiviruses expressing wild-type Hsp70 which resulted in a significant increase in the degree of Hsp70 expression compared with non-transfected cells (Figure 2 Aii). Importantly, as shown in Figure 2 (panels viii–xi), as compared to lac-z transfected control cells, the addition of LPS led to a minimal degree of NFkB translocation in IEC-6 cells that over-express Hsp70, consistent with the notion that these cells are less responsive to TLR4 signaling due to the effects of Hsp70 on TLR4. Moreover, the degree of LPS-induced NFkB translocation in Hsp70-IEC-6 cells was minimally affected by heat exposure, consistent with the observation that these cells already over express Hsp70. Taken together, these findings demonstrate that the induction of Hsp70 leads to a reduction in TLR4 signaling, and that the effects of Hsp70 occur through an association between TLR4 and Hsp70 through its interaction the EEVD domain in its C-terminus. Given the role of Hsp70 in regulating the ubiquitin-mediated degradation of target proteins, we therefore next sought to investigate whether Hsp70 associations with TLR4 could subsequently alter the ubiquitination state of TLR4 in enterocytes.
Figure 2. An EEVD-mediated association between TLR4 and Hsp70 is required for the inhibition of TLR4 signaling in enterocytes by Hsp70.

Ai: Representative SDS-PAGE showing lysates of IEC-6 enterocytes that had been virally transduced with either LacZ or Hsp70 lacking the EEVD substrate binding domain (ΔEEVD), prior to treatment with LPS and maintained at 37°C or treated at 42°C for 45 min (heat) then immunoprecipitated with antibodies to Hsp70 and immunoblotted with anti-TLR4 antibodies; shown is IgG as a loading control. Aii. Quantitative RT-PCR showing the expression of Hsp70 in nontransfected control IEC-6 cells and IEC-6 cells that were transfected with full length Hsp70. B–C: representative confocal micrographs of IEC-6 enterocytes that were either transduced with LacZ (B: i–iii, C: i–iii) or ΔEEVD-Hsp70 (B: iv-vi, C: iv-vi) then untreated (B i and iv, C i and iv), or LPS treated (B: ii and v, C: ii and v) or treated with LPS plus pre-treatment with heat (B: iii and vi, C: iii and vi). Cells were then stained with for NFkB (green in B), or cleaved-caspase 3 (green), β-actin (red) and DAPI (blue) in C. NFkB translocation (B xi) and % apoptosis (C vii) based upon at least 50 fields with over 50 cells/field; *p<0.05 LPS open and solid bars vs control open and solid bar; **p<0.05 LPS+HS vs LPS open bar, ***p<0.05 control black bar vs LPS+HS black bar. Diamond: p<0.05 LPS black bar vs LPS open bar; summary of 4 separate experiments. Shown in panels B viii–x are confocal micrographs of IEC-6 cells that were transfected with Hsp70 and treated as indicated; in xi Δ=no significant difference between untreated, LPS-treated or heat exposed LPS-treated Hsp70-IEC-6 cells. Shown in panel B xii: Quantitative RT-PCR showing IL-6 expression in IEC-6 cells that were transfected with Lac-Z or delta EEVD then treated with LPS as in Methods (6h, 50μg/ml). *p<0.05 vs ctrl, **p<0.05 vs LPS in Lac-Z transfected cells. Representative of 3 separate experiments. Size bar=10μ. Representative apoptotic cells are indicated by arrows.
The induction of Hsp70 leads to the ubiquitination and degradation of TLR4 via the co-chaperone CHIP
In various cell types, Hsp70 serves to regulate the fate of target proteins in part by critically influencing their ubiquitination state and subsequent degradation through the proteosomal system (32). The degree of ubiquitination effected by Hsp70 is directly influenced by the activity of a co-chaperone molecule named C-terminus of Hsp70 Interacting Protein (CHIP), an E3-ligase bearing a ubiquitin ligase U-box domain and a Hsp70 docking domain (33, 34). We now postulate that Hsp70 induction limits TLR4 signaling by promoting its ubiquitination state via CHIP. To determine whether Hsp70 induction affected TLR4 expression via effects on the extent of ubiquitination of TLR4, we first immunoprecipitated lysates of IEC-6 cells using anti-ubiquitin antibodies in the absence or presence of heat, and then performed SDS-PAGE using antibodies to TLR4. As shown in Figure 3 panel Ai, heat exposure increased the degree of TLR4 ubiquitination, which was associated with a reduction in TLR4 expression (Figure 3 panel A ii). Further evidence of the importance of Hsp70 in regulating TLR4 expression after heat treatment is shown in Figure 3 panel Aiii. After the administration of siRNA for Hsp70, the expression of TLR4 was significantly greater as detected by SDS-PAGE than in wild-type cells that express Hsp70 (please see Figure 1E for expression of Hsp70 in the siRNA treated cells, confirming knockdown of Hsp70 by the siRNA approach). Importantly, heat treatment did not cause a reduction in TLR4 in IEC-6 cells after knockdown of Hsp70, consistent with the important role of heat-induced Hsp70 in mediating the regulation of TLR4 (Figure 3 Aiii), which is lost in these cells after Hsp70 knockdown. Furthermore, the importance of ubiquitination in mediating the effects of Hsp70 on TLR4 expression and signaling is demonstrated as the treatment of IEC-6 cells with the proteosomal inhibitor MG-132 prevented the loss of TLR4 expression in response to heat (not shown) while also reversing the protective effects of heat shock on TLR4-induced enterocyte apoptosis (Figure 3 B and C). To explore whether CHIP/Hsp70-mediated interactions were required for the protective effects of Hsp70 on TLR4 signaling and function via CHIP-mediated docking and ubiquitination of TLR4, we next introduced, via lentiviral-mediated transduction, two dominant negative forms of CHIP into IEC-6 cells: H260Q - the E3-ligase U-box mutant - that impairs the ability of Hsp70/CHIP to ubiquitinate target proteins, and K30A - a docking domain mutant - that impairs the ability of CHIP to interact with Hsp70 and target proteins (33, 35). We then assessed effects on protein stability, ubiquitination state and TLR4 signaling. Introduction of the H260Q ubiquitin ligase mutant prevented the increase in the ubiquitination of TLR4 in response to heat exposure as compared to IEC-6 cells that were infected with empty vector (“LacZ” Figure 3 panel Ai), and significantly reduced the protective effects of heat exposure on LPS-mediated enterocyte apoptosis (Figure 3 panels B, D–E). Furthermore, transduction of IEC-6 cells with the K30A docking mutant also reversed the protection in LPS-induced enterocyte apoptosis that was previously provided from heat exposure (Figure 3 panels B, F). Taken together, these findings demonstrate that Hsp70 serves to limit TLR4 activation through ubiquitin-mediated protein degradation via CHIP. We next sought to determine whether TLR4 could limit its own signaling in part through upregulation of Hsp70.
Figure 3. The induction of Hsp70 leads to the ubiquitination and degradation of TLR4 via the co-chaperone CHIP.

A i: Representative immunoblots in which LacZ and H260Q-transfected IEC-6 enterocytes were exposed to heat or were maintained at 37°C, then immunoprecipitated with anti-ubiquitin antibodies and immunoblotted with anti-TLR4 antibodies, displaying poly-ubiquitinated species (“pUB-TLR4”). The location of TLR4 on the gel is shown. ii: Representative SDS-PAGE of IEC-6 cells probed with anti-TLR4 antibodies that were either non-transfected or transfected with H260Q then maintained at 37°C or exposed to heat, in which heat exposure leads to a reduction in TLR4 expression that is not seen in H260Q-transfected cells. iii: SDS-PAGE showing expression of TLR4 and loading protein control in either wild-type (“WT”) IEC-6 cells or IEC-6 cells treated with siRNA to Hsp70 (“siHSP70”) that were either untreated (“Ctrl”) or treated with heat as in Methods (“Heat”). B–F: Representative confocal micrographs of IEC-6 enterocytes treated with MG-132 (C), or transduced with LacZ (D) or H260Q-CHIP (E) or K30A-CHIP (F) and treated as indicated and immuno-stained with cleaved-caspase 3 (green), β-actin (red) and DAPI (blue). Size bar = 10μ. Shown in (B) % apoptosis per HPF > 50 fields with over 50 cells/field.; *p<0.05 control (all bars) versus LPS (all bars); **p<0.05 LPS (open bar) versus LPS+Heat (open bar); vs LPS open bar, ***p<0.05 Control versus LPS + Heat (black, red and blue bars) in 3 separate experiments. Size bar=10μ. Arrows point to apoptotic cells in each group under the indicated treatment.
TLR4 induces Hsp70 expression which then negatively effects TLR4 signaling
During the inflammatory response, the activation of TLR4 in response to LPS must be carefully controlled; the failure to negatively regulate a TLR4 response would necessarily result in a pro-inflammatory cytokine storm each time a TLR4 signal was initiated. Having shown that Hsp70 induction can inhibit TLR4 signaling in enterocytes, we next considered the possibility that TLR4 activation itself could also lead to an induction of Hsp70, which could then serve to negatively regulate (and therefore to self limit) TLR4 responsiveness. To do so, we first sought to evaluate the time dependency of the signaling response of TLR4 in IEC-6 cells using two separate techniques. IEC-6 cells were transiently transduced with a NFkB-promotor driven GFP-reporter construct as a readout of TLR4 activation, then treated with LPS and assessed for the expression of GFP by confocal microscopy over time. Cells were also stained for Hsp70 to assess its induction in response to LPS. Second, the induction of the pro-inflammatory molecule iNOS was assessed by RT-PCR over time after treatment with LPS. As shown in Figure 4 panels A (red staining), B (solid bars), LPS caused a time-dependent signaling response in IEC-6 cells that was maximum at 4–6 hours, then decreased by 16h. LPS also caused a time-dependent increase in the expression of Hsp70 in IEC-6 cells (Figure 4 panels A, B, C) that peaked at the time at which TLR4 signaling decreased (Figure 4 panels B, C). As a positive control for the effects of Hsp70 on the confocal based assay, heat exposure resulted in a marked inhibition of TLR4 signaling, as revealed by reduced GFP expression and an increase in Hsp70 expression (Figure 4 panel A iv–vi). Of note, the effects of LPS and heat on the intracellular induction of Hsp70 in IEC-6 cells is shown further in Figure 4 panel D.
Figure 4. TLR4 induces Hsp70 expression which then negatively effects TLR4 signaling.

A: Representative confocal micrographs of IEC-6 enterocytes transduced with GFPNFkB then treated with LPS (t=0) in the absence of heat (i–iii) or LPS after pre-treatment with heat (iv-vi); cells were stained for Hsp70(red) and assessed for GFP (green) at the indicated time point. B: Quantification of iNOS mRNA by RT-PCR (checkered bars) and Hsp70 protein (solid bars) relative to β-actin. *p<0.05 vs t=0 checkered bars; ** p<0.05 16h vs 8h, 6h and 4h, checkered bars. ***p<0.05 t=16h vs other time points, solid bars. In 4 separate experiments. C: Representative SDS-PAGE showing Hsp70 in IEC-6 cells treated with LPS. D: Representative confocal micrographs of IEC-6 enterocytes under the indicated conditions and stained for Hsp70 (green), β-actin (red) and DAPI (blue). E: Representative SDS-PAGE of IEC-6 lysates treated with LPS, immunoprecipitated with anti-Hsp70 antibodies and immunoblotted with TLR4 (upper bands) and Hsp70. Fi: Fold increase of IL-6 release by ELISA over media alone in IEC-6 treated as indicated. *p<0.005 solid bars vs open bars indicated point. Diamond: p<0.05 open bars vs 4h time point. Representative of 3 separate experiments ii: iNOS RT-PCR in IEC-6 cells treated with LPS as indicated. Representative of 4 separate experiments iii: Apoptosis in IEC-6 cells treated as indicated. *p<0.05 no siRNA solid vs open bars; **p<0.05 Hsp70 siRNA vs no siRNA solid bars; ***p<0.05 vs Hsp70 siRNA vs control siRNA, solid bars; based upon 4 separate experiments >50 fields/experiment and over 50 cells/field.
Several lines of evidence next indicate that the intracellular increase in Hsp70 expression that was observed to occur in response to LPS could serve to limit TLR4 signaling. First, treatment of IEC-6 with the Hsp70 inhibitor Quercetin resulted in an increase in the extent of TLR4-induced IL-6 expression (Figure 4, panel Fi). Second, the increased expression of Hsp70 that was noted in response to LPS resulted in an increase in the degree to which TLR4 could be detected in lysates of IEC-6 cells that had been immunoprecipitated with antibodies to Hsp70 (Figure 4 panel E), indicating that the increased Hsp70 expression in response to TLR4 activation also resulted in increased association of Hsp70 with TLR4 consistent with the mechanism of action of Hsp70 in reducing TLR4 signaling shown in Figures 2 and 3. Third, inhibition of Hsp70 using siRNA resulted in a marked exaggeration in the extent of TLR4 signaling, as measured by an increase in both LPS-induced iNOS expression (Figure 4 panel Fii) and LPS-induced enterocyte apoptosis (Figure 4 panel Fiii) compared with control cells that had been transfected with control siRNA or no siRNA. To further define a link between TLR4 and Hsp70 signaling, we performed flow cytometry on either wild-type IEC-6 cells or Hsp70-deficient IEC-6 cells that we had generated through lentiviral transduction of Hsp70 siRNA, in the presence of heat and LPS stimulation. As shown in Figure 4 panel G, LPS caused an increase in TLR4 expression that was reduced upon heat exposure in wild-type cells. By contrast, TLR4 expression was increased in Hsp70-deficient IEC-6 cells, that was not reduced further upon heat exposure. Importantly, heat exposure did not prevent NFkB translocation in IEC-6 cells that were treated with the TLR2 ligand Pam(3)CSK(4) and the TLR5 ligand flagellin, and these findings were not affected by the presence of Quercetin, suggesting the effect of heat induced TLR4 suppression is specific for TLR4 signaling (not shown). Taken together, these findings reveal that TLR4 can induce the expression of Hsp70, which serves to inhibit LPS signaling in enterocytes, and by extension, that Hsp70 can exert a physiological role in constraining the effect of TLR4 signaling that occurs. We therefore next sought to investigate the role of Hsp70 on LPS-induced TLR4 signaling in vivo.
Hsp70 negatively regulates TLR4 signaling in the intestinal epithelium
The systemic administration of LPS to newborn mice is known to cause a significant inflammatory response in the small intestine that includes an increase in the expression of iNOS and an induction in enterocyte apoptosis (36–38). Based upon the above findings in which Hsp70 was found to inhibit enterocyte TLR4 in vitro, we next sought to evaluate the effects of Hsp70 on enterocyte TLR4 signaling in vivo. To do so, we injected saline or LPS into wild-type mice, or into two additional mouse strains: mice that were globally deficient in Hsp70 (Hsp70−/−), and mice that selectively over-express Hsp70 within the intestinal epithelium (Hsp70villin). As shown in Figure 5 panel A, the injection of LPS into wild-type mice caused a time-dependent increase in the mucosal expression of iNOS, as well as a significant induction in enterocyte apoptosis (Figure 5 panels Bi–ii, C). By contrast, injection of LPS into mice that selectively over-express Hsp70 within the intestinal epithelium resulted in a marked reduction in LPS-mediated iNOS expression within the intestinal epithelium compared to wild-type mice (Figure 5 panel A) and a reduction in enterocyte apoptosis (Figure 5 panels Biii–iv, C), demonstrating that Hsp70 could negatively regulate TLR4 signaling in vivo, and supporting the in vitro data shown in Figure 4. Moreover, the injection of LPS into Hsp70−/− mice resulted in a significantly increased degree of apoptosis compared to LPS-injected wild-type mice (Figure 5 panels Bv–vi, H), providing further evidence that Hsp70 negatively regulates TLR4 signaling in vivo. We therefore next sought to determine the physiological relevance of these findings in the pathogenesis of NEC.
Figure 5. Hsp70 negatively regulates TLR4 signaling in the intestinal epithelium.
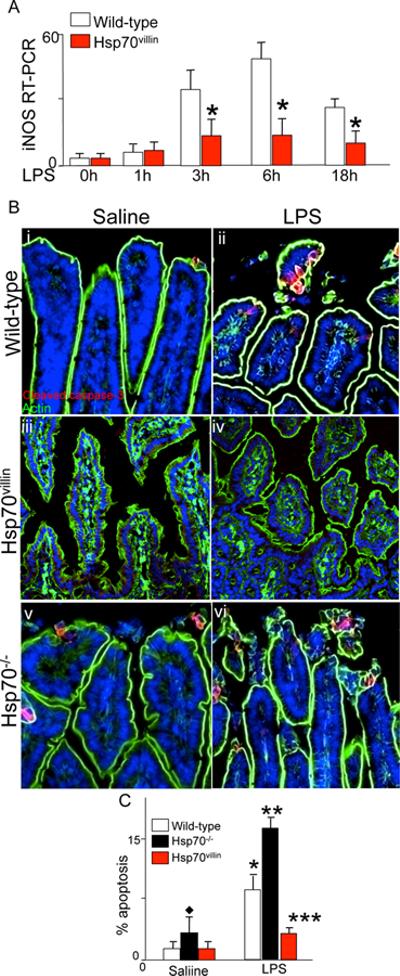
A: RT-PCR for iNOS in the intestinal epithelium in wild-type (open bars) or Hsp70villin mice treated (red bar) with LPS for the time points indicated. *p<0.05 red bar vs open bar for each point indicated. B: confocal micrographs of newborn intestine obtained from the terminal ileum after injection with saline (B i, iii, iv) or LPS (5mg/kg, 16h B ii, iv, vi) in mice that were either wild-type (B i–ii), Hsp70villin (B iii–iv), or Hsp70−/− (B v–vi). C:
Quantification of apoptosis in the small intestine of newborn mice as in panels B after injection with saline or LPS as indicated. Based upon 4 separate experiments with over 4 mice per group and over 50 fields examined per group. *p<0.05 saline vs LPS for open and black bars; ** p<0.005 LPS white bar vs LPS solid bar; ***p<0.005 LPS red bar vs LPS solid bar; diamond: saline treated black bar versus open bar or red bar.
Hsp70 signaling negatively regulates the development of NEC
We and others have demonstrated that NEC is a disease characterized by TLR4-mediated apoptosis within the newborn small intestine (3) (5, 24). Having now shown that Hsp70 can limit TLR4 signaling in enterocytes both in vivo and in vitro, we next sought to evaluate whether a lack of Hsp70 may lead to an increase in the severity of NEC. As shown in Figure 6 panel A, the expression of Hsp70 in both mice (Figure 6 panels Ai–ii) and humans (Figure 6 panels Aiii–iv) with NEC was significantly decreased compared to control bowel, indicating the possibility that a lack of Hsp70 may play a role in NEC development. To investigate directly whether Hsp70 could regulate the development of this disease, NEC was induced in wild-type, Hsp70−/− and Hsp70villin mice using a combination of formula gavage and intermittent hypoxia. As shown in Figure 6B, the induction of NEC in wild-type mice resulted in enterocyte apoptosis (Panels B i–ii and E i), mucosal disruption (Panel B iii–iv), and an increase in the expression of iNOS in the intestinal mucosa (Panel E ii). Importantly, the induction of NEC in Hsp70−/− mice showed a significant increase in the extent of enterocyte apoptosis (Panels C i–ii and E i), mucosal disruption (Panels iii–iv), iNOS expression (Panel E ii) and increased disease severity (Panel E iii), while the induction of NEC in the Hsp70villin mouse that over-expresses Hsp70 in the intestinal epithelium resulted in a marked reduction in each of these measures (Panels D–E). Taken together, these findings illustrate that Hsp70 plays a key role in the regulation of NEC. We next sought to evaluate whether pharmacologic induction of Hsp70 could inhibit TLR4 signaling and affect NEC severity.
Figure 6. Hsp70 signaling negatively regulates the development of NEC.

A: i: RT-PCR showing Hsp70 on each day of the 4-day NEC model; *p<0.05 vs day 0; *p<0.05 vs day 1. Representative of over 4 separate experiments, 10 mice/group; ii: Representative confocal micrographs showing the expression of Hsp70 (red) and DAPI (blue) in the terminal ileum of mice without (“control”) and with NEC (“NEC”). iii: PCR showing the expression of Hsp70 in intestine from infants without (open bar) and with NEC (solid bar); *p<0.05 solid vs open, based upon 9 separate samples per group; iv: representative SDS-PAGE from infant without (ctrl) and with NEC blotted with antibodies to Hsp70 then probed for Hsc70 and β-actin; B–D: Representative confocal and H&E micrographs of sections of the terminal ileum from newborn mice with and without NEC that were either wild-type (B), Hsp70−/− (C) and Hsp70villin (D). In panels B i–ii, C i–ii, D i–ii, slides were stained for cleaved-caspase 3 (red) and DAPI (blue). E: i apoptosis, ii: iNOS RT-PCR in the terminal ileum; iii: NEC severity. *p<0.05 NEC in wild-type vs control; **p<0.005 NEC in Hsp70−/− vs wild type; *** NEC in Hsp70villin vs wild-type and Hsp70−/−. Based upon at least 4 experiments with over 15 mice per strain per group. Size bar=250μm.
Pharmacologic induction of Hsp70 limits TLR4 signaling in enterocytes in vitro and in vivo, and attenuates the severity of experimental necrotizing enterocolitis
In the final series of studies, we sought to evaluate whether the pharmacologic induction of Hsp70 could inhibit TLR4 signaling in vitro and in vivo, and thus attenuate the severity of NEC. To do so, we utilized the small molecule Celastrol (39, 40), a novel cell permeable triterpenoid antioxidant that has been shown to induce Hsp70 expression and activity in a variety of cells (41, 42). As shown in Figure 7 panel Ai, treatment of IEC-6 cells with Celastrol led to a rapid induction of Hsp70 as determined by SDS-PAGE. In parallel, the injection of Celastrol into mice on each of three consecutive days resulted in an increase in the expression of Hsp70 within the intestinal mucosa on each day (Figure 7 panel Aii). Importantly, the exposure of IEC-6 cells to Celastrol resulted not only in a marked increase in cytoplasmic Hsp70 expression (red staining in Figure 7 panels Bi–iv) but also to a significant reduction in the extent of TLR4 signaling, as measured by a reduction in the extent of LPS-induced apoptosis (Figure 7 panel Biv versus Bii, Ei), a reduction in the extent of LPS-induced NFkB translocation (Figure 7 panels C, Eii), and a significant reduction in the extent of LPS-induced IL-6 expression (Figure 7 panel Eiii). There were no effects of Celastrol treatment alone on enterocyte apoptosis, NFkB translocation or IL-6 expression (Figure 7 panels Biii, Ciii, Ei). Having shown that the injection of Celastrol into mice can induce Hsp70 expression within the intestinal mucosa (Figure 7Aii), we next sought to determine whether Celastrol could inhibit TLR4 signaling in the intestinal epithelium via effects on Hsp70 induction. To do so, wild-type and Hsp70−/− mice were injected with Celastrol 24h prior to LPS, then assessed for the extent of enterocyte apoptosis and iNOS expression in the intestinal mucosa. As shown in Figure 7 panels Di–iv and Fi, the administration of Celastrol to wild-type mice led to a significant reduction in the extent of LPS-induced enterocyte apoptosis compared to the effects of LPS in wild-type mice that did not receive Celastrol, as well as to a significant reduction in the extent of LPS-induced expression of iNOS in the intestinal mucosa compared to wild-type mice (Figure 7 panel W). Importantly, there was no protective benefit of Celastrol when it was administered to Hsp70−/− mice, confirming that its protective effects required Hsp70 induction (Figure 7 panels Dv–viii, F). The specificity of Celastrol for TLR4-mediated enterocyte apoptosis was confirmed in vitro. While LPS caused a significant increase in enterocyte apoptosis in Hsp70-deficient IEC-6 cells, the addition of Celastrol did not confer protection, and in fact, these Hsp70-deficient enterocytes were significantly more susceptible to apoptosis than their non-transfected counterparts (Supplementary Figure 1).
Figure 7. Pharmacologic induction of Hsp70 limits TLR4 signaling in enterocytes in vitro and in vivo.

A: i: Representative SDS-PAGE of IEC-6 enterocytes treated with Celastrol or DMSO and blotted with Hsp70 then re-probed for β-actin; ii: Representative SDS-PAGE of mucosal scrapings from terminal ileum of newborn mice administered Celastrol daily for 3 days. B–C: Representative confocal micrographs of IEC-6 enterocytes treated as indicated and immunostained for Hsp70 (red in B) and DAPI (blue in B) or NFkB (green in C) and β-actin (red in C). Size bar=10 mm. D: Representative confocal micrographs of terminal ileum in newborn wild-type (i–iv) or Hsp70−/− mice (v–viii) that were treated as indicated as indicated and stained for cleaved-caspase 3 (green), DAPI (blue) and β-actin (red). Size bar=250 mm. E: quantification in IEC-6 as indicated. *p<0.05 control vs LPS open bars; ** LPS open bars vs LPS+Celastrol closed bars in 4 separate experiments. F: Quantification of apoptosis (i) and iNOS expression (ii) by RTPCR in the terminal ileum of the newborn wild-type (open bars) or Hsp70−/− mice mouse as indicated. *p<0.05 open bars LPS vs control, **p<0.05 LPS + Celastrol vs LPS open bars; representative of 4 separate experiments, over 10 mice/group. Representative apoptotic cells are indicated by arrows.
Based upon the above findings, we next sought to evaluate whether the induction of Hsp70 that occurs with the administration of Celastrol could attenuate the severity of experimental NEC. To test this directly, we first administered either DMSO or Celastrol by intraperitoneal injection to newborn pups on day 0 and day 1 of the experimental model, and assessed the effects on extent of mucosal disruption, enterocyte apoptosis, induction of iNOS and disease severity. As shown in Figure 8, the administration of Celastrol markedly reduced the degree of mucosal disruption (Figure 8, Panels A i–iii), enterocyte apoptosis (Figure 8, Panels A v–vii and C i), mucosal iNOS expression (Figure 8, Panel C ii) and disease severity (Figure 8, Panel C iii) compared with mice that had been administered DMSO. It is noteworthy that the injection of Celastrol resulted in a marked increase in the expression of Hsp70 and a reduction in TLR4 compared to mice with NEC that received DMSO alone, consistent with the mechanism of action for Celastrol shown in Figure 7.
Figure 8. Pharmacologic induction of Hsp70 prevents and also treats experimental necrotizing enterocolitis in mice.

A: Representative H&E (i–iv) or confocal images (v–viii, cleaved-caspase 3 (green), β-actin (red) DAPI (blue)) of sections from terminal ileum of newborn mice that were either breast fed (“control”, Ai and Av), or induced to develop NEC and administered either DMSO (Aii, Avi) or Celastrol (1mg/kg on day 0 and 1 of the 4 day model, Aiii and Avii). In parallel, mice that had NEC for 2 days were administered Celastrol (1mg/kg for 2 days, Aiv and Aviii). Size bar=250μm. B: SDS-PAGE of mucosal scrapings from mice subjected to experimental NEC and injected with either DMSO or Celastrol on the first two days of the model; blots were probed for Hsp70 then stripped and re-probed for TLR4 and β-actin. C: Quantification of enterocyte apoptosis (Ci), iNOS by RT-PCR in the terminal ileum (Cii), and NEC severity (Ciii) under the indicated condition. *p<0.05 NEC Ctrl (solid bar) vs control (open bar), **p<0.05 NEC Celastrol – prevention (solid bar) vs NEC control (solid bar); ***p<0.005 NEC Celastrol-treatment (checkered bar) vs NEC control (solid bar); representative of 4 separate experiments> 10 mice/group.
Having shown that the induction of Hsp70 through the administration of Celastrol could prevent the development of NEC when administered prior to the start of the experimental model, we next sought to determine whether the administration of Celastrol could reduce the severity of NEC once the disease had been established in mice. To do so, we injected mice with Celastrol on the last two days of the four-day model, at a time in which significant inflammation is already established. Strikingly, as shown in Figure 8, mice with NEC that received Celastrol after disease induction showed restoration of mucosal architecture (Figure 8, Panel A iv), a significant reduction in enterocyte apoptosis (Figure 8, Panel A viii and C i) and a reduction in NEC severity (Figure 8, Figure C iii), all to levels that were similar to that of mice without NEC, and which were comparable to levels observed in mice receiving Celastrol as a prevention strategy (compare checkered to solid bars in Figure 8 Panel C). Taken together, these findings suggest that the pharmaceutical induction of Hsp70 with compounds such as Celastrol may be used as a novel approach to the prevention or treatment of NEC through effects on the inhibition of TLR4 signaling in the newborn small intestine.
DISCUSSION
We now define a novel mechanism by which TLR4 is regulated in the newborn intestinal epithelium that has important implications in the pathogenesis of necrotizing enterocolitis, a disease that is characterized by exaggerated TLR4 signaling within the intestinal mucosa (3–5). Specifically, we identify that the induction of Hsp70 leads to a reduction in TLR4-induced signaling in enterocytes as measured by a reduction in NFkB activation, cytokine induction and apoptosis, and that induction of Hsp70 either pharmacologically or genetically leads to a reduction in TLR4 signaling and a marked inhibition in the severity of NEC. The current results identify a novel pathway that links cytoplasmic Hsp70 induction with TLR4 regulation, and demonstrate that impaired Hsp70 expression or function may in part underlie the causes of this devastating disease. These findings represent a novel departure from current thinking in the field, by revealing that future treatments for NEC may involve not only non-specific immunological approaches such as the elimination of microbial pathogens or the administration of particular feeding regimens (43), but rather, may involve the pharmacologic induction of an intracellular chaperone such as Hsp70 to limit disease progression through inhibitory effects on the innate immune receptor TLR4.
One of the most important findings of the current study involves the proposed mechanism of action of Hsp70 in limiting TLR4 signaling within enterocytes. As a molecular chaperone, Hsp70 can associate with co-chaperone proteins through an EEVD motif in its C-terminus (28, 29). As shown in Figure 2, we now demonstrate that the inhibition of TLR4 signaling in response to Hsp70 induction required this EEVD-binding motif (30), as the introduction of a mutant lacking this domain prevented the association between TLR4 and Hsp70, and also reversed the protection of Hsp70 induction on TLR4 signaling (Figure 2). This mechanism of action for Hsp70 is in agreement with recent work of Chow et al, who showed that Hsp70 mutants with a functional EEVD motif but lacking N-terminus ATPase activity were still capable of protecting L929 fibroblasts from apoptosis induced by pro-inflammatory cytokines(44). We also determined that the association between Hsp70 and TLR4 results in enhanced ubiquitination and degradation of TLR4, a process that we have now determined to require the co-chaperone CHIP (Figure 3). CHIP has not previously been linked to TLR4 signaling, and we further reveal that mutations in both its docking (K30A) and ubiquitination domains (H260Q) prevented the protective effects of Hsp70 induction on TLR4. These findings clarify the mechanism by which CHIP acts to mediate the inhibitory effects of Hsp70 on TLR4 signaling and are in agreement with the known function of CHIP in regulating the activity of other Hsp70 targets through ubiquitination (45, 46), yet represent the first direct link of CHIP to an intestinal inflammatory disease. It is noteworthy that in the original description of the CHIP-deficient mouse, attention was drawn to the intestinal phenotype that was observed when mice were subjected to a brief hyperthermic stress, characterized by friability of the small intestine with marked apoptosis of the intestinal epithelium (47), although potential CHIP targets that could mediate this effect on the small intestine during stress were not identified. It is tempting to now speculate that CHIP may play a central role in the maintenance of intestinal homeostasis in part by preventing the unbridled activation of immune targets of CHIP such as TLR4.
It should be noted that the current findings in which cytoplasmic Hsp70 serves to curtail the signaling of TLR4 within the intestinal epithelium lie in distinction to a growing and somewhat controversial body of work concerning the extracellular role of Hsp70 and other heat shock proteins in activating the innate immune system via TLR4 (48–53). In this regard, Retzlaff et al showed that the exogenous administration of Hsp70 could increase IL-1, IL-6 and TNF in cultured macrophages (54), while Wheeler et al have shown that the extracellular exposure of Hsp70 to neutrophils from wild-type mice leads to the release of IL-8, yet this effect is not observed in neutrophils from C3H/HeJ mice that have inhibitory mutations in TLR4 (55). While very exciting, such studies have recently been called into question by concerns that the observed effects might actually result not from the heat shock proteins themselves, but rather from contaminants such as LPS which could inadvertently be present within the protein preparations, or which could be bound specifically to the heat shock proteins (56). For example, Wallin et al (57), Bausinger et al (58) and Gao and Tsan (59) have shown that the activation of immune cells previously attributed to Hsp70 were lost when highly purified (i.e. contaminant free) recombinant proteins were used, although these results have been recently and convincingly rebutted in a review article on this topic (60, 61). In contrast to studies in the field of extracellular Hsp70 biology, the novelty and importance of the current findings lie in the newly discovered link between TLR4 and Hsp70 within the enterocyte both in vitro and in vivo, and the potential etiological relevance to the development of NEC. And while they represent an extension of the classic role of Hsp70 in modulating the fate of cytoplasmic proteins, the relevance – if any – to the body of literature surrounding the fate of Hsp70 outside the cells is unknown.
The current findings in support of a role for Hsp70 in the protection from the development of NEC through the inhibition of TLR4 signaling in the small intestine may or may not apply to other diseases of intestinal inflammation including ulcerative colitis and Crohn's disease, in which TLR4 signaling may play a lesser or perhaps even opposite role. Although we (3, 4) and others (5, 6) have shown that the development of NEC requires TLR4 activation, it has been shown that TLR4 plays a protective role in experimental colitis (62, 63). Several reasons may account for this apparent discrepancy that have relevance to the current study. TLR4 activation leads to intestinal injury in a well-defined and physiologically relevant context, namely the newborn small intestine. In support of this concept, we have recently demonstrated that TLR4 activation with LPS leads to increased enterocyte apoptosis in the terminal ileum of newborn mice but not adult mice, and in the small intestine but not the newborn colon (7). Further, reports that demonstrate a protective role for TLR4 in models of colitis have typically been based upon the use of global TLR4 knockout mice, in which TLR4 signaling is disrupted in enterocytes as well as T-cells and myeloid cells. We have recently shown that TLR4 signaling within the enterocyte itself is important for the induction of intestinal injury leading to NEC, using enterally administered adenoviral constructs that bear inhibitory mutations in TLR4 whose expression is largely favored within the small bowel mucosa (4, 18). It is therefore reasonable to conclude that the protective effects attributed to TLR4 signaling in the gut by previous authors may reflect in part the mitigating effects of TLR4 signaling on other cells. In support of this possibility, we note that Fukata and colleagues have recently shown in an elegant study using chimeric mice that TLR4 signaling in colonic epithelial cells worsened intestinal inflammation (64). These findings argue that the effects of TLR4 in the development of intestinal inflammation are strongly influenced by a variety of factors, including the effector cells involved, developmental factors and involved region of the intestine. The precise effects of Hsp70 at these varying stages of development and within these different cell types remains to be explored in further detail, but are likely to provide important clues to the underlying causes of these diseases.
Based upon the current findings, we now propose a model by which Hsp70 limits TLR4 signaling and plays a key role in influencing the development of enterocyte apoptosis and the development of NEC (Figure 9). Under healthy conditions, the relationship between the indigenous flora of the host and the baseline activation of TLR4 exists in homeostatic balance, which we now attribute in part to a constitutive role of Hsp70 in limiting the extent of TLR4 signaling by controlling its degradation through proteosomal pathways. The interaction between TLR4 and Hsp70 may occur within intracellular compartments such as the Golgi apparatus, where TLR4 signaling has been shown to occur within the enterocyte(65). By contrast, under the conditions of stress that favor the development of NEC (increased LPS, hypoxia and prematurity), the exhaustion of Hsp70 signaling accompanied by the relative increase in TLR4 expression leads to exaggerated TLR4 activation and the development of the increased enterocyte apoptosis and pro-inflammatory cytokine expression in the newborn intestine that leads to NEC. It is also notable that the pharmacologic induction of Hsp70 can curtail TLR4 signaling and both prevent and treat experimental NEC.
Figure 9. Proposed model: Hsp70 regulates TLR4 signaling in enterocytes in the pathogenesis of NEC.

As described in the text, under healthy conditions (left cell), TLR4 is activated by host microbes. The degree of activation is limited by Hsp70 through effects on TLR4 degradation through proteosomal pathways via CHIP. By contrast, under the conditions of stress that favor the development of NEC (right cell), the reduction in Hsp70 expression accompanied by the increase in TLR4 expression leads to exaggerated TLR4 activation and the development of the increased enterocyte apoptosis and pro-inflammatory cytokine expression in the newborn intestine. This leads to the development of NEC. Moreover, pharmacologic induction of Hsp70 can curtail TLR4 signaling and both prevent and treat experimental NEC.
In summary, we have now identified a novel pathway by which through Hsp70 that serves to limit TLR4 signaling in the intestinal epithelium, and moreover, have shown that factors that increase Hsp70 signaling can attenuate NEC severity through inhibition of TLR4. Such findings, we believe, offer new insights not only into the molecular requirements that lead to NEC development, but also to offer novel therapeutic approaches for this devastating disease.
Supplementary Material
ACKNOWLEDGMENTS
The authors are indebted to Dr. Richard Morimoto, for EEVD plasmids, Dr. Richard Mosser and Dr. Robin Anderson for Hsp70 plasmids, and Dr. Cam Patterson, for CHIP reagents, which were used to generate appropriate lentiviruses. We also thank Dr. Jeffrey Brodsky and Dr. Timothy Billiar (University of Pittsburgh) for insightful comments in the planning of the experiments described in this work.
Footnotes
Grant support: DJH is supported by R01GM078238 and RO1DK08752 from the National Institutes of Health. AA is supported by F30DK085930 from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) of the National Institutes of Health
REFERENCES
- 1.Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med. 2011;363:255–264. doi: 10.1056/NEJMra1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afrazi A, Sodhi CP, Richardson W, Neal M, Good M, Siggers R, Hackam DJ. New insights into the pathogenesis and treatment of necrotizing enterocolitis: toll-like receptors and beyond. Pediatr Res. 2011;69:183–188. doi: 10.1203/PDR.0b013e3182093280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leaphart CL, Cavallo JC, Gribar SC, Cetin S, Li J, Branca MF, Dubowski TD, Sodhi CP, Hackam DJ. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J. Immunol. 2007;179:4808–4820. doi: 10.4049/jimmunol.179.7.4808. [DOI] [PubMed] [Google Scholar]
- 4.Sodhi CP, Shi XH, Richardson WM, Grant ZS, Shapiro RA, Prindle TJ, Branca M, Russo A, Gribar SC, Ma C, Hackam DJ. Toll-like Receptor-4 Inhibits Enterocyte Proliferation via Impaired beta-Catenin Signaling in Necrotizing Enterocolitis. Gastroenterology. 2010;138:185–196. doi: 10.1053/j.gastro.2009.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jilling T, Simon D, Lu J, Meng FJ, Li D, Schy R, Thomson RB, Soliman A, Arditi M, Caplan MS. The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J Immunol. 2006;177:3273–3282. doi: 10.4049/jimmunol.177.5.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolfs TG, Derikx JP, C.M. H, Vanderlocht J, Driessen A, de Bruïne AP, Bevins CL, Lasitschka F, Gassler N, van Gemert WG, Buurman WA. Localization of the lipopolysaccharide recognition complex in the human healthy and inflamed premature and adult gut. Inflamm Bowel Dis. 2010;16:68–75. doi: 10.1002/ibd.20995. [DOI] [PubMed] [Google Scholar]
- 7.Richardson WM, Sodhi CP, Russo A, Siggers RH, Afrazi A, Gribar SC, Neal MD, Dai S, Prindle TJ, Branca M, Ma C, Ozolek J, Hackam DJ. Nucleotide-binding Oligomerization Domain-2 Inhibits Toll Like Receptor-4 Signaling in the Intestinal Epithelium. Gastroenterology. 2010;139:904–917. doi: 10.1053/j.gastro.2010.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabai VL, Meriin AB, Mosser DD, Caron AW, Rits S, Shifrin VI, Sherman MY. Hsp70 prevents activation of stress kinases. A novel pathway of cellular thermotolerance. J Biol Chem. 1997;272:18033–18037. doi: 10.1074/jbc.272.29.18033. [DOI] [PubMed] [Google Scholar]
- 9.Jiang B, Liang P, Deng G, Tu Z, Liu M, Xiao X. Increased stability of Bcl-2 in HSP70-mediated protection against apoptosis induced by oxidative stress. Cell Stress Chaperones. 2011;16:143–152. doi: 10.1007/s12192-010-0226-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka K, Mizushima T. Protective role of HSF1 and HSP70 against gastrointestinal diseases. Int J Hyperthermia. 2009;25:668–676. doi: 10.3109/02656730903213366. [DOI] [PubMed] [Google Scholar]
- 11.Jiang B, Wang K, Liang P, Xiao W, Wang H, Xiao X. ATP-binding domain of heat shock protein 70 is essential for its effects on the inhibition of the release of the second mitochondria-derived activator of caspase and apoptosis in C2C12 cells. FEBS J. 2009;276:2615–2624. doi: 10.1111/j.1742-4658.2009.06989.x. [DOI] [PubMed] [Google Scholar]
- 12.Carrizo LC, Ruete CM, Manucha WA, Ciocca DR, Valles PG. Heat shock protein 70 expression is associated with inhibition of renal tubule epithelial cell apoptosis during recovery from low-protein feeding. Cell Stress Chaperones. 2006;11:309–324. doi: 10.1379/CSC-199.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Didelot C, Schmitt E, Brunet M, Maingret L, Parcellier A, Garrido C. Heat shock proteins: endogenous modulators of apoptotic cell death. Handb Exp Pharmacol. 2006:171–198. doi: 10.1007/3-540-29717-0_8. [DOI] [PubMed] [Google Scholar]
- 14.Beere HM, Green DR. Stress management - heat shock protein-70 and the regulation of apoptosis. Trends Cell Biol. 2001;11:6–10. doi: 10.1016/s0962-8924(00)01874-2. [DOI] [PubMed] [Google Scholar]
- 15.Joly AL, Wettstein G, Mignot G, Ghiringhelli F, Garrido C. Dual role of heat shock proteins as regulators of apoptosis and innate immunity. J Innate Immun. 2010;2:238–247. doi: 10.1159/000296508. [DOI] [PubMed] [Google Scholar]
- 16.Tao Y, Hart J, Lichtenstein L, Joseph LJ, Ciancio MJ, Hu S, Chang EB, Bissonnette M. Inducible heat shock protein 70 prevents multifocal flat dysplastic lesions and invasive tumors in an inflammatory model of colon cancer. Carcinogenesis. 2009;30:175–182. doi: 10.1093/carcin/bgn256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu S, Zhu X, Triggs JR, Tao Y, Wang Y, Lichtenstein L, Bissonnette M, Musch MW, Chang EB. Inflammation-induced, 3'UTR-dependent translational inhibition of Hsp70 mRNA impairs intestinal homeostasis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1003–1011. doi: 10.1152/ajpgi.00027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gribar SC, Sodhi CP, Richardson WM, Anand RJ, Gittes GK, Branca MF, Jakub A, Shi XH, Shah S, Ozolek JA, Hackam DJ. Reciprocal expression and signaling of TLR4 and TLR9 in the pathogenesis and treatment of necrotizing enterocolitis. J Immunol. 2009;182:636–646. doi: 10.4049/jimmunol.182.1.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radulescu A, Zorko NA, Yu X, Besner GE. Preclinical neonatal rat studies of heparin-binding EGF-like growth factor in protection of the intestines from necrotizing enterocolitis. Pediatr Res. 2009;65:437–442. doi: 10.1203/PDR.0b013e3181994fa0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leaphart CL, Qureshi F, Cetin S, Li J, Dubowski T, Batey C, Beer-Stolz D, Guo F, Murray SA, Hackam DJ. Interferon-[gamma] inhibits intestinal restitution by preventing gap junction communication between enterocytes. Gastroenterology. 2007;132:2395–2411. doi: 10.1053/j.gastro.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 21.Cetin S, Ford HR, Sysko LR, Agarwal C, Wang J, Neal MD, Baty C, Apodaca G, Hackam DJ. Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J Biol Chem. 2004;279:24592–24600. doi: 10.1074/jbc.M313620200. [DOI] [PubMed] [Google Scholar]
- 22.Ding LA, Li JS. Effects of glutamine on intestinal permeability and bacterial translocation in TPN-rats with endotoxemia. World J Gastroenterol. 2003;9:1327–1332. doi: 10.3748/wjg.v9.i6.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siggers RH, Hackam DJ. The role of innate immune-stimulated epithelial apoptosis during gastrointestinal inflammatory diseases. Cell Mol Life Sci. 2011;68:3623–3634. doi: 10.1007/s00018-011-0821-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khailova L, Mount Patrick SK, Arganbright KM, Halpern MD, Kinouchi T, Dvorak B. Bifidobacterium bifidum reduces apoptosis in the intestinal epithelium in necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1118–1127. doi: 10.1152/ajpgi.00131.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jilling T, Lu J, Jackson M, Caplan MS. Intestinal epithelial apoptosis initiates gross bowel necrosis in an experimental rat model of neonatal necrotizing enterocolitis. Pediatr Res. 2004;55:622–629. doi: 10.1203/01.PDR.0000113463.70435.74. [DOI] [PubMed] [Google Scholar]
- 26.Houry WA. Chaperone-assisted protein folding in the cell cytoplasm. Curr Protein Pept Sci. 2001;2:227–244. doi: 10.2174/1389203013381134. [DOI] [PubMed] [Google Scholar]
- 27.Naylor DJ, Hartl FU. Contribution of molecular chaperones to protein folding in the cytoplasm of prokaryotic and eukaryotic cells. Biochem Soc Symp. 2001:45–68. doi: 10.1042/bss0680045. [DOI] [PubMed] [Google Scholar]
- 28.Brinker A, Scheufler C, Von Der Mulbe F, Fleckenstein B, Herrmann C, Jung G, Moarefi I, Hartl FU. Ligand discrimination by TPR domains. Relevance and selectivity of EEVD-recognition in Hsp70 x Hop x Hsp90 complexes. J Biol Chem. 2002;277:19265–19275. doi: 10.1074/jbc.M109002200. [DOI] [PubMed] [Google Scholar]
- 29.Liu FH, Wu SJ, Hu SM, Hsiao CD, Wang C. Specific interaction of the 70-kDa heat shock cognate protein with the tetratricopeptide repeats. J Biol Chem. 1999;274:34425–34432. doi: 10.1074/jbc.274.48.34425. [DOI] [PubMed] [Google Scholar]
- 30.Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 31.Freeman BC, Myers MP, Schumacher R, Morimoto RI. Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. EMBO J. 1995;14:2281–2292. doi: 10.1002/j.1460-2075.1995.tb07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535–4545. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001;2:1133–1138. doi: 10.1093/embo-reports/kve246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature. 2006;440:551–555. doi: 10.1038/nature04600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickinson E, Tuncer R, Nadler E, Boyle P, Alber S, Watkins S, Ford H. NOX, a novel nitric oxide scavenger, reduces bacterial translocation in rats after endotoxin challenge. Am J Physiol Gastrointest Liver Physiol. 1999;277:G1281–1287. doi: 10.1152/ajpgi.1999.277.6.G1281. [DOI] [PubMed] [Google Scholar]
- 37.Unno N, Wang H, Menconi MJ, Tytgat S, Larkin V, Smith M, Morin MJ, Chavez A, Hodin R, Fink MP. Inhibition of inducible nitric oxide synthase ameliorates endotoxin-induced gut mucosal barrier dysfunction in rats. Gastroenterology. 1997;113:1246–11257. doi: 10.1053/gast.1997.v113.pm9322519. [DOI] [PubMed] [Google Scholar]
- 38.Cetin S, Leaphart CL, Li J, Ischenko I, Hayman M, Upperman J, Zamora R, Watkins S, Ford HR, Wang J, Hackam DJ. Nitric oxide inhibits enterocyte migration through activation of RhoA-GTPase in a SHP-2-dependent manner. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1347–1358. doi: 10.1152/ajpgi.00375.2006. [DOI] [PubMed] [Google Scholar]
- 39.Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI. Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem. 2004;279:56053–56060. doi: 10.1074/jbc.M409267200. [DOI] [PubMed] [Google Scholar]
- 40.Trott A, West JD, Klaic L, Westerheide SD, Silverman RB, Morimoto RI, Morano KA. Activation of heat shock and antioxidant responses by the natural product celastrol: transcriptional signatures of a thiol-targeted molecule. Mol Biol Cell. 2008;19:1104–1112. doi: 10.1091/mbc.E07-10-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paimela T, Hyttinen JM, Viiri J, Ryhanen T, Karjalainen RO, Salminen A, Kaarniranta K. Celastrol regulates innate immunity response via NF-kappaB and Hsp70 in human retinal pigment epithelial cells. Pharmacol Res. 2011 doi: 10.1016/j.phrs.2011.05.027. [DOI] [PubMed] [Google Scholar]
- 42.Kalmar B, Greensmith L. Activation of the heat shock response in a primary cellular model of motoneuron neurodegeneration-evidence for neuroprotective and neurotoxic effects. Cell Mol Biol Lett. 2009;14:319–335. doi: 10.2478/s11658-009-0002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan JA, Young L, McGuire W. Pathogenesis and prevention of necrotizing enterocolitis. Curr Opin Infect Dis. 2011;24:183–189. doi: 10.1097/QCO.0b013e328345d5b5. [DOI] [PubMed] [Google Scholar]
- 44.Chow AM, Steel R, Anderson RL. Hsp72 chaperone function is dispensable for protection against stress-induced apoptosis. Cell Stress Chaperones. 2009;14:253–263. doi: 10.1007/s12192-008-0079-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Hohfeld J, Patterson C. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem. 2001;276:42938–42944. doi: 10.1074/jbc.M101968200. [DOI] [PubMed] [Google Scholar]
- 46.Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol. 2001;3:100–105. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- 47.Dai Q, Zhang C, Wu Y, McDonough H, Whaley RA, Godfrey V, Li HH, Madamanchi N, Xu W, Neckers L, Cyr D, Patterson C. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003;22:5446–5458. doi: 10.1093/emboj/cdg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 49.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 50.Pockley AG, Muthana M, Calderwood SK. The dual immunoregulatory roles of stress proteins. Trends Biochem Sci. 2008;33:71–79. doi: 10.1016/j.tibs.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 51.Chase MA, Wheeler DS, Lierl KM, Hughes VS, Wong HR, Page K. Hsp72 induces inflammation and regulates cytokine production in airway epithelium through a TLR4- and NF-kappaB-dependent mechanism. J Immunol. 2007;179:6318–6324. doi: 10.4049/jimmunol.179.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen T, Guo J, Han C, Yang M, Cao X. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J Immunol. 2009;182:1449–1459. doi: 10.4049/jimmunol.182.3.1449. [DOI] [PubMed] [Google Scholar]
- 53.Gong J, Zhu B, Murshid A, Adachi H, Song B, Lee A, Liu C, Calderwood SK. T cell activation by heat shock protein 70 vaccine requires TLR signaling and scavenger receptor expressed by endothelial cells-1. J Immunol. 2009;183:3092–3098. doi: 10.4049/jimmunol.0901235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Retzlaff C, Yamamoto Y, Hoffman PS, Friedman H, Klein TW. Bacterial heat shock proteins directly induce cytokine mRNA and interleukin-1 secretion in macrophage cultures. Infect Immun. 1994;62:5689–5693. doi: 10.1128/iai.62.12.5689-5693.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wheeler DS, Chase MA, Senft AP, Poynter SE, Wong HR, Page K. Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4. Respir Res. 2009;10:31. doi: 10.1186/1465-9921-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsan MF, Gao B. Heat shock proteins and immune system. J Leukoc Biol. 2009;85:905–910. doi: 10.1189/jlb.0109005. [DOI] [PubMed] [Google Scholar]
- 57.Wallin RP, Lundqvist A, More SH, von Bonin A, Kiessling R, Ljunggren HG. Heat-shock proteins as activators of the innate immune system. Trends Immunol. 2002;23:130–135. doi: 10.1016/s1471-4906(01)02168-8. [DOI] [PubMed] [Google Scholar]
- 58.Bausinger H, Lipsker D, Ziylan U, Manie S, Briand JP, Cazenave JP, Muller S, Haeuw JF, Ravanat C, de la Salle H, Hanau D. Endotoxin-free heat-shock protein 70 fails to induce APC activation. Eur J Immunol. 2002;32:3708–3713. doi: 10.1002/1521-4141(200212)32:12<3708::AID-IMMU3708>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 59.Gao B, Tsan MF. Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J Biol Chem. 2003;278:174–179. doi: 10.1074/jbc.M208742200. [DOI] [PubMed] [Google Scholar]
- 60.Henderson B, Calderwood SK, Coates AR, Cohen I, van Eden W, Lehner T, Pockley AG. Caught with their PAMPs down? The extracellular signalling actions of molecular chaperones are not due to microbial contaminants. Cell Stress Chaperones. 2010;15:123–141. doi: 10.1007/s12192-009-0137-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Wijk F, Prakken B. Heat shock proteins: Darwinistic immune modulation on dangerous grounds. J Leukoc Biol. 2010;88:431–434. doi: 10.1189/jlb.0410236. [DOI] [PubMed] [Google Scholar]
- 62.Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, Nast CC, Lechago J, Xu R, Naiki Y, Soliman A, Arditi M, Abreu T. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1055–1065. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- 63.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of Commensal Microflora by Toll-Like Receptors Is Required for Intestinal Homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 64.Fukata M, Hernandez Y, Conduah D, Cohen J, Chen A, Breglio K, Goo T, Hsu D, Xu R, Abreu MT. Innate immune signaling by Toll-like receptor-4 (TLR4) shapes the inflammatory microenvironment in colitis-associated tumors. Inflamm Bowel Dis. 2009 doi: 10.1002/ibd.20880. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hornef MW, Normark BH, Vandewalle A, Normark S. Intracellular Recognition of Lipopolysaccharide by Toll-like Receptor 4 in Intestinal Epithelial Cells. J. Exp. Med. 2003;198:1225–1235. doi: 10.1084/jem.20022194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.