

To the Editor:
Fragile X syndrome (FXS) is the most common inherited cause of intellectual disability. It is most often caused by an expansion of the CGG repeat in the 5’ untranslated region of the Fragile X Mental Retardation 1 (FMR1) gene. CGG repeat numbers ranging from 55 to 200 are defined as premutation alleles and repeat numbers greater than 200 lead to the clinical phenotype of FXS. In FXS, mRNA levels are severely decreased, as the gene is hypermethylated, therefore synthesis of the FMR1 protein (FMRP) is mostly absent. In the premutation range, FMR1 mRNA levels are higher than normal but mild to moderate deficits in FMRP may occur [Allen et al., 2004; Kenneson et al., 2001; Peprah et al., 2010; Tassone et al., 2000].
The fragile X genotype is varied, reflected by the wide range of clinical involvement observed in patients with FXS and premutation carriers. Differences in CGG repeat number, FMR1 mRNA levels, methylation status, and FMRP expression levels give rise to a broad spectrum of clinical involvement.
Here, we present a unique sibship, a brother and sister, in which FXS is present and the family history confirms consanguinity. The male sibling, identified as the proband, was nine-years, nine-months-old. DNA molecular testing, using PCR and Southern Blot analysis [Tassone et al., 2008], showed a partially methylated full mutation with methylated alleles of 273, 408, 810 CGG repeats and an unmethylated allele of 340 CGG repeats present in 15% of the cells. His FMR1 mRNA level was (0.9 ±0.04) and FMRP expression was barely detected by Western Blot analysis (<5%). His CYFIP1 mRNA levels were in the normal range. His mother had a normal pregnancy, his birth weight was nearly 4082g. The newborn period was unremarkable. His early developmental history is significant because he did not sit until 9 months, he never crawled, and he did not walk until 20 months. He used single words at two years and short phrases at six years. He had recurrent ear infections and two sets of pressure equalization (PE) tubes. His behaviors included excessive picking, hand flapping, poor eye contact, tactile defensiveness, perseveration, tantrums, shyness, self-talk, staring episodes, bed-wetting and encopresis. An EEG recording at five years of age was normal. He is overweight without a lack of satiation after meals. A physical evaluation at age nine-years, nine-months showed a height of 139.4 cm (61st centile), weight of 50.5 kg (98th centile), and head circumference of 54 cm (70th centile). His ears are mildly prominent and his palate is high arched. He has hyperextensible finger joints with MP extension to 90 degrees and double jointed thumbs, as well as flat feet with a mild degree of pronation. His testicular volume is 6ml bilaterally, within normal limits and his phallus is normal in size.
The subject also underwent neuropsychological and behavioral testing. The WASI [Wechsler 1999] indicated a verbal IQ (VIQ) of 55, performance IQ (PIQ) of 68, and a full-scale IQ (FSIQ) of 59. This FSIQ falls within the range of mild mental impairment, but is nearly two standard deviations higher than his sister’s FSIQ. On the ADOS, he met criteria for autism with his socialization score of 6 and repetitive behavior score of 3, with a total of 9, which is the autism cut-off on the revised algorithm [Gotham et al., 2007]. The SCQ revealed a score of 17, which is in the autism range and on DSM IV he also met criteria for autism. On the Vineland-II his communication score was 65, daily living skills was 74, socialization was 76, with an overall adaptive behavior composite of 71. The subject scored 50 on the PPVT, with an age equivalent of four-years, five-months. On the ADIS [Silverman and Albino 1996], he met criteria for social phobia.
The female was five-years, four-months-old. Molecular testing showed the presence of a premutation allele (73 CGG repeats) on one X chromosome and a full mutation on the other (CGG repeats = 670, 765, 855, 965). No normal allele was present. The premutation allele was active in 24 percent of cells. Her FMR1 mRNA levels were low (0.21 ±0.01) compared to normal values (1.42±0.25) [Tassone et al., 2000] and her FMR1 protein levels, measured by Western Blot analysis, were 15 percent of normal levels [Iwahashi et al., 2009]. CYFIP1 mRNA levels, which have been observed to be lower in subjects with FXS and the PWP, were within the normal range [Nowicki et al., 2007].
Her mother had a normal pregnancy except for a medication in the first trimester that was taken to eliminate a possible miscarriage. Her developmental milestones were delayed; she never crawled, walked at 18 months, talked at 28 months, and used phrases at 4 years. Her parents noticed breast development at age 2 years. At age 3.5 years she was treated with monthly injections of Lupron for premature thelarche, which decreased her breast development. She has never had vaginal bleeding or significant pubic hair indicating that precocious puberty was not present. There is no history of cardiac, immunological, or ophthalmological problems. The subject’s parents reported that she picks and scratches excessively and hand flaps. She is shy, socially anxious with poor eye contact and poor relatedness, tactilly defensive, hyperactive, aggressive at times, and has outbursts and tantrums daily. She began eating excessively around age 4 years, developing significant obesity, which with hyperphagia and lack of satiation fit the Prader Willi-like phenotype (PWP) of FXS [de Vries et al., 1996].
A physical evaluation at age 5-years, 4-months showed a height of 114 cm (80th centile), weight of 30 kg (98th centile), and head circumference of 53 cm (97th centile). Her ears are mildly prominent, palate is high and narrow, and she has hyperextensible finger joints with metacarpophalangeal (MP) extension to 90 degrees and double-jointed thumbs. Her feet are flat with a mild degree of pronation and her fourth toe is short bilaterally with a hint of a partial two-three toe syndactyly similar to her grandmother.
The subject underwent neuropsychological and behavioral testing. The WPPSI-III [Wechsler 2002] indicated a verbal IQ (VIQ) of 48, performance IQ (PIQ) of 45, and a full-scale IQ (FSIQ) of 40. The FSIQ falls within the moderate range of intellectual impairment. However, during the testing, she was easily distracted and did not cooperate well. Cognitive testing was repeated one year later, using the abbreviated Stanford Binet-V [Roid 2003]. This measure indicated an FSIQ of 61. On the ADOS [Lord et al., 1999], she received a socialization score of 5 and a restrictive and repetitive behavior score of 2, with a total of 7 which is just below the ASD range. The SCQ [Rutter et al., 2003] revealed a score of 13, which is just below the autism spectrum cutoff. On the Vineland-II [Sparrow et al., 2005] her communication score was 74, daily living skills was 66, socialization was 68, motor was 84. Overall adaptive behavior composite was 69. This score is in the mildly intellectually impaired range and significantly higher than her WPPSI-III scores, although it is in line with the abbreviated IQ obtained from the Stanford Binet. This indicates that her overall cognitive ability is in the mildly intellectually impaired range.
The mother and father of the sibling pair are first cousins and confirmed carriers of the premutation. The mother (40 and 99 CGG repeats) reports fibromyalgia, swelling and numbness in her legs, mood swings, depression, shyness, and social anxiety. The father (64 CGG repeats) is a graduate student with a history of social anxiety. The proband’s older brother has a normal allele, with 43 CGG repeats. The paternal and maternal grandmothers are sisters and are both obligate premutation carriers. The family history is significant because of consanguinity, as illustrated in Figure 1.
Figure 1.
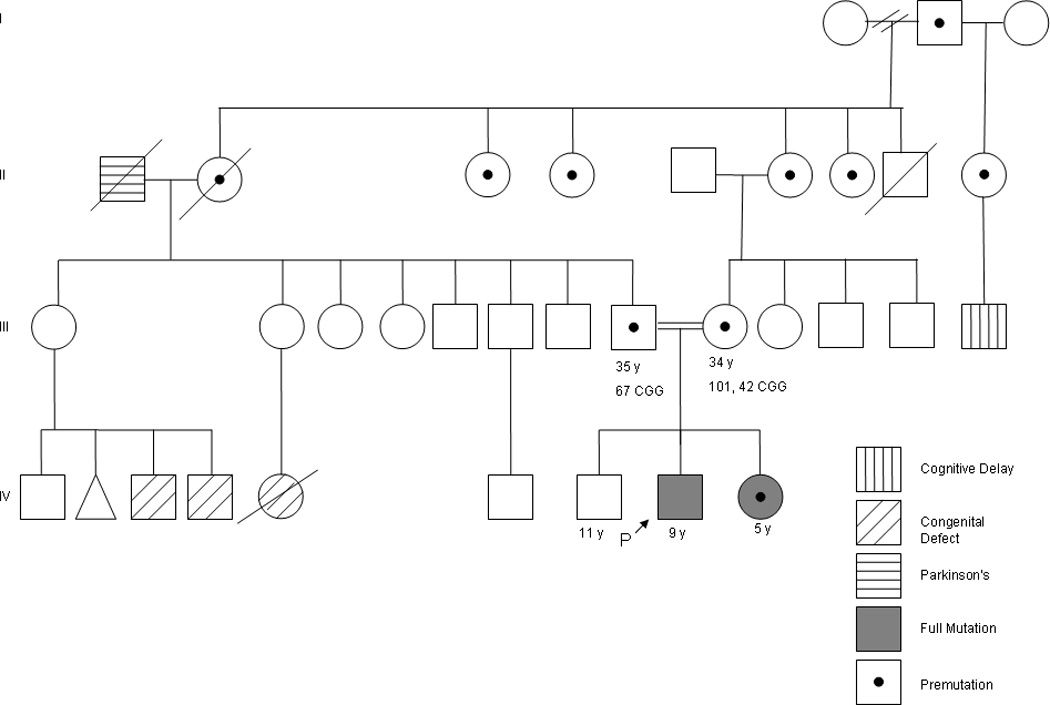
Family Pedigree
This is a study of a unique sibling pair consisting of a nine-year, nine-month-old male with a partially methylated full mutation and a five-year, four-month old female with a full mutation and a premutation (73) allele, a compound heterozygote with features of the PWP of FXS. There are several reports of females with compound heterozygous mutations [Hegde et al., 2001; Heine-Suner et al., 2003; Linden et al., 1999; Martorell et al., 2011; Mila et al., 1996; Russo et al., 1998]. The female sibling reported here differs from the previously reported females because she is more severely affected, having a lower FSIQ than the reported females carrying premutation and full mutation alleles [Linden et al., 1999; Martorell et al., 2011; Russo et al., 1998]. However, one study reported on four sisters, all compound heterozygous, one of whom showed a complete inactivation of the chromosome carrying the premutation allele with consequent absence of FMRP and severe mental retardation [Martorell et al., 2011]. The lower IQ observed in the female sibling is likely due to the presence of a full mutation allele on the active X chromosome in 76% of her cells, the pronounced FMRP deficit resulting from the lack of a normal allele as well as possible background gene effects. However, FMRP levels and the X inactivation ratio were measured in peripheral blood leukocytes and may not reflect the levels in brain. The FSIQs of the previously reported females fell within the borderline range, and approximately 70% of females with a full mutation have cognitive deficits in the borderline to mentally retarded range [de Vries et al., 1996]. The female reported here also exhibits more severe behavioral characteristics than those seen in previously reported compound heterozygous and other females with FXS. For example, she displays some characteristics of the PWP of FXS including hyperphagia, obesity, and some autistic-like behaviors, such as spinning, hand-flapping, and severe language delay. The features of the PWP are thought to relate to hypothalamic dysfunction that is more severe than what is typically seen in individuals with FXS and her precocious thelarche is consistent with hypothalamic dysfunction [Nowicki et al., 2007], however our patient does not have lowered CYFIP levels as reported by Nowicki et al in males with FXS and PWS. Prader Willi-like phenotype has been observed in both genders in patients with different chromosomal abnormalities including a 6q16.2 deletion [Varela et al., 2006], maternal uniparental disomy 14 syndrome [Hosoki et al., 2009], in 1p36 deletion syndrome [D'Angelo et al., 2006; Tsuyusaki et al., 2010]. To our knowledge this is the first pediatric female case of PWP in FXS, as a previous case of an adult female with XXX and FXS recently was reported to present with complex neuro-behavioral involvement, severe hyperphagia, and affective disorder consistent with the Prader Willi-like Phenotype in FXS [Vandersteen et al., 2009].
ACKNOWLEDGMENTS
Grant sponsor: This work was supported by the National Institute of Health, Grant numbers: HD036071, HD02274.
This work is dedicated to the memory of Matteo.
REFERENCES
- Allen EG, He W, Yadav-Shah M, Sherman SL. A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Hum Genet. 2004;114:439–447. doi: 10.1007/s00439-004-1086-x. [DOI] [PubMed] [Google Scholar]
- D'Angelo CS, Da Paz JA, Kim CA, Bertola DR, Castro CI, Varela MC, Koiffmann CP. Prader-Willi-like phenotype: investigation of 1p36 deletion in 41 patients with delayed psychomotor development, hypotonia, obesity and/or hyperphagia, learning disabilities and behavioral problems. Eur J Med Genet. 2006;49:451–460. doi: 10.1016/j.ejmg.2006.02.001. [DOI] [PubMed] [Google Scholar]
- de Vries BB, Wiegers AM, Smits AP, Mohkamsing S, Duivenvoorden HJ, Fryns JP, Curfs LM, Halley DJ, Oostra BA, van den Ouweland AM, Niermeijer MF. Mental status of females with an FMR1 gene full mutation. Am J Hum Genet. 1996;58(5):1025–1032. [PMC free article] [PubMed] [Google Scholar]
- Gotham K, Risi S, Pickles A, Lord C. The Autism Diagnostic Observation Schedule: revised algorithms for improved diagnostic validity. J Autism Dev Disord. 2007;37:613–627. doi: 10.1007/s10803-006-0280-1. [DOI] [PubMed] [Google Scholar]
- Hegde MR, Fawkner M, Chong B, McGaughran J, Gilbert D, Love DR. Compound heterozygosity at the FMR1 gene. Genet Test. 2001;5:135–138. doi: 10.1089/109065701753145600. [DOI] [PubMed] [Google Scholar]
- Heine-Suner D, Torres-Juan L, Morla M, Busquets X, Barcelo F, Pico G, Bonilla L, Govea N, Bernues M, Rosell J. Fragile-X syndrome and skewed X-chromosome inactivation within a family: a female member with complete inactivation of the functional X chromosome. Am J Med Genet A. 2003;122:108–114. doi: 10.1002/ajmg.a.20160. [DOI] [PubMed] [Google Scholar]
- Hosoki K, Kagami M, Tanaka T, Kubota M, Kurosawa K, Kato M, Uetake K, Tohyama J, Ogata T, Saitoh S. Maternal uniparental disomy 14 syndrome demonstrates prader-willi syndrome-like phenotype. J Pediatr. 2009;155:900–903. e1. doi: 10.1016/j.jpeds.2009.06.045. [DOI] [PubMed] [Google Scholar]
- Iwahashi C, Tassone F, Hagerman RJ, Yasui D, Parrott G, Nguyen D, Mayeur G, Hagerman PJ. A quantitative ELISA assay for the fragile x mental retardation 1 protein. J Mol Diagn. 2009;11:281–289. doi: 10.2353/jmoldx.2009.080118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenneson A, Zhang F, Hagedorn CH, Warren ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001;10:1449–1454. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- Linden MG, Tassone F, Gane LW, Hills JL, Hagerman RJ, Taylor AK. Compound heterozygous female with fragile X syndrome. Am J Med Genet A. 1999;83(4):318–321. [PubMed] [Google Scholar]
- Lord C, Rutter M, DiLavore PC, Risi S. Autism Diagnostic Observation Schedule. Los Angeles, CA: Western Psychological Services; 1999. [Google Scholar]
- Martorell L, Nascimento MT, Colome R, Genoves J, Naudo M, Nascimento A. Four sisters compound heterozygotes for the pre- and full mutation in fragile X syndrome and a complete inactivation of X-functional chromosome: implications for genetic counseling. J Hum Genet. 2011;56:87–90. doi: 10.1038/jhg.2010.140. [DOI] [PubMed] [Google Scholar]
- Mila M, Castellvi Bel S, Gine R, Vazquez C, Badenas C, Sanchez A, Estivill X. A female compound heterozygote (pre- and full mutation) for the CGG FMR1 expansion. Hum Genet. 1996;98:419–421. doi: 10.1007/s004390050232. [DOI] [PubMed] [Google Scholar]
- Nowicki ST, Tassone F, Ono MY, Ferranti J, Croquette MF, Goodlin-Jones B, Hagerman RJ. The Prader-Willi phenotype of fragile X syndrome. J Dev Behav Pediatr. 2007;28:133–138. doi: 10.1097/01.DBP.0000267563.18952.c9. [DOI] [PubMed] [Google Scholar]
- Peprah E, He W, Allen E, Oliver T, Boyne A, Sherman SL. Examination of FMR1 transcript and protein levels among 74 premutation carriers. J Hum Genet. 2010;55:66–68. doi: 10.1038/jhg.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roid GH. Stanford Binet intelligence scales. Itasca, IL: Riverside Publishing; 2003. [Google Scholar]
- Russo S, Briscioli V, Cogliati F, Macchi M, Lalatta F, Larizza L. An unusual fragile X sibship: female compound heterozygote and male with a partially methylated full mutation. Clin Genet. 1998;54:309–314. doi: 10.1034/j.1399-0004.1998.5440408.x. [DOI] [PubMed] [Google Scholar]
- Rutter M, Bailey A, Berument SK, Lord C, Pickles A. Social Communication Questionnaire (SCQ) Los Angeles: Western Psychological Services; 2003. [Google Scholar]
- Silverman WK, Albino AM. The Anxiety Disorders Interview Schedule for Children for DSM-IV: Child and Parent Versions. San Antonio, TX: Psychological Corporation; 1996. [Google Scholar]
- Sparrow SS, Cicchetti DV, Balla DA. Vineland Adaptive Behavior Scales. Second edition. Circle Pines: AGS Publishing; 2005. [Google Scholar]
- Tassone F, Hagerman RJ, Taylor AK, Gane LW, Godfrey TE, Hagerman PJ. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am J Hum Genet. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10:43–49. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuyusaki Y, Yoshihashi H, Furuya N, Adachi M, Osaka H, Yamamoto K, Kurosawa K. 1p36 deletion syndrome associated with Prader-Willi-like phenotype. Pediatr Int. 2010;52:547–550. doi: 10.1111/j.1442-200X.2010.03090.x. [DOI] [PubMed] [Google Scholar]
- Vandersteen AM, Moore D, MacFarlane N, Josifova D. Genetic diagnosis in clinical psychiatry: A case report of a woman with a 47,XXX karyotype and Fragile X syndrome. Eur J Psychiat. 2009;23:31–36. [Google Scholar]
- Varela MC, Simoes-Sato AY, Kim CA, Bertola DR, De Castro CI, Koiffmann CP. A new case of interstitial 6q16.2 deletion in a patient with Prader-Willi-like phenotype and investigation of SIM1 gene deletion in 87 patients with syndromic obesity. Eur J Med Genet. 2006;49:298–305. doi: 10.1016/j.ejmg.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Abbreviated Scale of Intelligence (WASI) San Antonio: Harcourt Assessment, Inc.; 1999. [Google Scholar]
- Wechsler D. Wechsler Preschool and Primary Scale of Intelligence. 3rd Edition. San Antonio: The Psychological Corporation; 2002. [Google Scholar]