

Abstract
Chronic obstructive pulmonary disease (COPD) is characterized by peribronchial and perivascular inflammation and largely irreversible airflow obstruction. Acute disease exacerbations, due frequently to viral infections, lead to enhanced disease symptoms and contribute to long-term progression of COPD pathology. Previously, we demonstrated that NK cells from cigarette smoke (CS)-exposed mice exhibit enhanced effector functions in response to stimulating cytokines or toll-like receptor ligands. Here, we show that the activating receptor NKG2D is a key mediator for CS stimulated NK cell hyperresponsiveness as CS-exposed NKG2D-deficient mice (Klrk1-/-) did not exhibit enhanced effector functions as assessed by cytokine responsiveness. NK cell cytotoxicity against MHC class I-deficient targets was not affected in a COPD model. However, NK cells from CS-exposed mice exhibit greater cytotoxic activity towards cells that express the NKG2D ligand RAET1ε. We also demonstrate that NKG2D-deficent mice exhibit diminished airway damage and reduced inflammation in a model of viral COPD exacerbation without affecting viral clearance. Furthermore, adoptive transfer of NKG2D+ NK cells into CS-exposed, influenza-infected NKG2D-deficient mice recapitulated the phenotypes observed in CS-exposed, influenza-infected WT mice. Our findings indicate that NKG2D stimulation during long term CS-exposure is a central pathway in the development of NK cell hyperresponsiveness and influenza-mediated exacerbations of COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by peribronchial and perivascular inflammation and largely irreversible airflow obstruction (1). Long term cigarette smoking (CS) is the major cause of COPD and while the severity of the disease may vary, all long term smokers will develop some degree of lung function impairment (2). In addition to CS exposure, acute disease exacerbations due to infection contribute significantly to COPD progression. COPD exacerbations are characterized by a worsening of the patient's condition including changes in symptoms such as dyspnea, cough, and sputum production (3). These exacerbations are typically marked by a visit to healthcare providers often resulting in long hospital stays at the cost of billions of dollars a year in direct costs (4). The majority of exacerbations are believed to be caused by viral infections which result in more severe symptoms and longer recovery times (5, 6).
The potential role of NK cells in the development and progression of COPD pathologies have received little attention although NK cells are known to contribute to tissue remodeling through their potent cytolytic potential and elaboration of high levels of pro-inflammatory cytokines (7). NK cell functions are tightly controlled by a balance of activating and inhibitory receptors. Of the activating receptors, NKG2D (Klrk1) is distinguished as it is present on virtually all NK cells, recognizes stress- and infection-induced ligands, and can overcome inhibitory signals to evoke cytotoxicity and cytokine release (8). NKG2D is expressed almost exclusively on cytotoxic lymphocytes (i.e., NK cells, NK T cells, CD8+ T cells, γδ+ T cells) and recognizes a diverse array of ligands on the cell surface. NKG2D ligands include MICA, MICB, and the UL-16 binding proteins in humans and the retinoic acid–inducible early genes (Raet1α–Raet1ε), H60, and Mult1 in mice (8). In the case of NK cells, engagement of NKG2D causes lysis of ligand expressing cells and cytokine production, most notably IFN-γ (8). NKG2D ligand expression is absent on cell surfaces under normal conditions. However, ligands are induced under conditions of stress, DNA damage, infection and transformation (9, 10). In the context of CS exposure, we showed that NKG2D ligands are induced on stressed human airway epithelial cells (11), and persistently expressed on pulmonary epithelial cells of smokers with and without COPD (12). RAET1 is also expressed in the alveolar and airway epithelium in a mouse model of COPD (12). Utilizing data obtained from COPD patients and a mouse model of lung specific NKG2D ligand expression, we also confirmed that the activation of the NKG2D receptor by CS-induced ligands is a potentially important contributor to lung inflammation and tissue destruction in COPD (12).
Recently, we demonstrated that chronic cigarette smoke exposure “primes” NK cells to produce more IFN-γ after stimulation by ligands for Toll-like receptors (TLR) 3,7,9 as well as with IL-12, IL-18, or both (13). Excessive NK cell activation after infection could worsen the outcomes associated with CS exposure. In the context of COPD, the presence of NK cells hyperresponsive to viral provocation could increase inflammation and contribute to symptomatic exacerbations and disease progression. In this study, we examined the roles of NK cells and the NKG2D activating receptor in viral exacerbations of COPD. We show that purified NK cells from CS-exposed mice are more cytotoxic towards NKG2D ligand-expressing target cells. Furthermore, we show that NKG2D is necessary for the development of enhanced NK cell hyperresponsiveness to activating cytokines. Moreover, we demonstrate that NKG2D is required for exaggerated pathologies resulting from influenza-infection in a mouse model of COPD. These studies reveal potentially important roles for NK cells in development and progression of COPD and indicate that these mechanisms are controlled by the presence and function of NKG2D.
Methods
Mice
C57BL/6J and C57BL/6J B2m-/- mice (female, 8 to 10 wk old) were purchased from The Jackson Laboratory (Bar Harbor, ME). NKG2D-deficient (Klrk1−/−) mice were generated as described (14). All of the experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Cincinnati Medical Center.
Reagents
The following antibodies and immunological reagents were used: NKp46 (29A1.4), NK1.1 (PK136), CD49b (DX5), CD94 (18d3), Streptavidin-APC, NKG2D (CX5), 2B4 (eBio244F4), 7-AAD, CFSE, and IFNγ ELISA (eBioscience); IL-2 and IL-12 (Peprotech); IL-18 (R&D systems); Dynabead CD49b+ NK cell isolation kit (Invitrogen); poly(I:C) (HMW) (Invivogen); anti-asialo GM1 (Wako); α-CCSP polyclonal antibody (Seven Hills Bioreagents).
Mouse Model of COPD
Mice were exposed to either filtered, room air (FA) or cigarette smoke (CS) generated from 3R4F Kentucky Reference Cigarettes (University of Kentucky, Lexington, KY) as previously described (15). CS exposures were carried out with a TE-10z smoking machine attached to an exposure chamber (Teague Enterprises, Woodland, CA). The concentration of the smoke/air mixture was maintained at 150 ± 15 mg/m3 total suspended particulates. Particulate concentrations were determined by weighing vacuum-drawn total particulate deposition onto filters connected to the chambers. Mice were exposed whole body in the exposure chamber for 4 h/d, 5 d/wk for 6 months.
Leukocyte isolation
Mice were euthanized with an i.p. injection of sodium pentobarbital followed by exsanguination. Lungs were perfused with 6 ml 1× PBS containing 0.6 mM EDTA. Lungs and spleens were withdrawn aseptically, and leukocytes were isolated as previously described (15).
NK purification and cell stimulation
Spleens from five mice were pooled and splenocytes isolated as described above and resuspended in cRPMI (RPMI 1640 with 2.05 mM l-glutamine [HyClone, Waltham, MA] containing 10% FBS, 1% sodium pyruvate, 100 U/ml penicillin, 100 μg/ml Streptomycin, and 1× nonessential amino acids [MP Biomedicals, Solon, OH]). Cell resuspension was followed by a 20 min plastic adherence plating step at 37°C and 5% CO2 which greatly reduces the presence of contaminating adherent cells. After plating, remaining leukocytes were enriched for NK cells by positive selection following the manufacturers' protocol for the Dynabead FlowComp Mouse CD49b NK isolation kit (Invitrogen). After enrichment NK cells were >60% pure. The remaining cells were stained with NKp46 and sorted by flow cytometry for NK cells resulting in a purity >99%. A total of 2.0 × 105 cells in 100 μl of cRPMI containing 20 U/ml mouse rIL-2 (PeproTech, Rocky Hill, NJ) were aliquoted per well into a 96-well round-bottom culture plate (Costar, Cambridge, MA) and cultured at 37°C and 5% CO2. Cells were rested 2 h then stimulated for 16 h with IL-18 (10 μg/ml), or IL-12 + IL-18 (10 μg/ml). Supernatants were harvested and assayed for IFN-γ production by ELISA (ebioscience).
In Vivo Cytotoxicity
In vivo cytotoxicity was assed according to a modified procedure previously described (16). Splenocytes were isolated from wild-type C57BL/6J mice or C57BL/6J B2m-/- mice deficient in MHC Class I molecule expression. Cells were washed twice with PBS + 0.2% horse serum (HS) and re-suspended at 10 × 106 cells/ml. The C57BL/6 B2m-/- cells were stained with 5 μM CFSE (ebioscience) and the wild-type C57BL/6 cells were stained with 0.5 μM CFSE. Cells were incubated for 8 min at 37°C and the reaction was stopped by adding ice-cold PBS + 20% HS. Cells were washed 2× with PBS + 0.2% HS and re-suspended at 3×107 cells/ml in PBS. Equal numbers of cells (3.0 × 106 in 100 μl each) were combined and administered by tail vein injection into FA- or CS-exposed mice. After 16 hrs, ∼200 μl of blood was collected via mandibular bleed using a 4 mm animal lancet (Goldenrod). RBCs were lysed using 1× RBC Lysis solution (Qiagen) then fixed in 1% paraformaldehyde for 20 min at 4°C. Cells were spun at 300 × g and the supernatant discarded. Pellets were resuspended in 100 μl of a 2.5% saponin solution. The cells were analyzed by flow cytometry. Cytotoxicity was calculated as follows: (number of CFSE high cells in sample/ number of CFSE low cells in sample)/(number of CFSE high cells injected/number of CFSE low cells injected). Flow cytometry was performed using a FACSCalibur (BD Biosciences, San Jose, CA). The data were analyzed using the FlowJo software (Tree Star, Ashland, OR).
Ex Vivo Cytotoxicity
For NK cell enrichment, spleen leukocytes from two mice were isolated, pooled and enriched using Dynabead FlowComp Mouse CD49b NK isolation kit described above (Invitrogen). Cells were resuspended in cRPMI containing 20 U/ml mouse rIL-2 at effector numbers described in the text and added to 5 ml polystyrene round-bottom tubes. Cells were allowed to rest for 2 h at 37°C and 5% CO2. RMA-Mock and RMA-Raet1ε cells (kindly provided by Dr. Lewis Lanier, UCSF) were stained with 0.5 μM CFSE (eBioscience) as described above. RMA cells were resuspended in cRPMI and ∼20,000 stained cells were added to tubes containing purified NK cells for a total volume of 400 μl. Combined cells were spun at 300 × g and allowed to incubate at 37°C and 5% CO2 for 4 h. After incubation, cells were washed in FACS buffer and resuspended in 300 μl buffer. Prior to flow cytometry analysis, 2.5 μl of 7-AAD (eBioscience) was added to each tube. Cytotoxicity was calculated with the following equation using % 7-AAD(-) cells for each group: ((Background-Test)/Background) × 100= % Cytotoxicity.
RT-PCR
Frozen tissue was homogenized using a Tissumizer (Tekmar Co.), and total RNA was isolated with Trizol reagent (Invitrogen). DNase treatment to remove residual DNA was performed using the Turbo DNA-free kit (Ambion). Reverse transcription of total RNA was performed using the high-capacity cDNA Archive kit (Applied Biosystems). FAM labeled probes used for RT-PCR were Ulbp1 (Mult1) (Mm01180648_m1), Raet1 (Mm04206137_gh), and Rpl32 (Mm02528467_g1) (Applied Biosystems). Quantitative reverse transcription-PCR (RT-PCR) was performed using TaqMan universal PCR master mix (Applied Biosystems) on an Applied Biosystems 7300 real-time PCR system. Expression of mRNA was quantified by the ΔΔCT method using Rpl32 as the endogenous control.
Virus infection
The influenza A virus HK×31 (H3N2) was used in this study (17). The virus was passaged in embryonated chicken eggs by standard procedures and titrated on Madin-Darby canine kidney (MDCK) cells. Mice were infected with 2 × 103 pfu of virus through noninvasive oral aspiration as described previously (18). Infection was allowed to proceed for 4 days at which time the mice were euthanized. Lungs were clamped at the left bronchus and the left lung was frozen for viral titer while the right lung was inflation formalin-fixed and paraffin-embedded as previously described (11).
Pathology Assessment and Immunohistochemistry
To assess pulmonary inflammation, paraffin sections of lungs were stained with hematoxylin and eosin. Histological scores for alveolar, peribronchial, and perivascular inflammation was semi-quantitatively scored as the sum of the severity [absent (0), minimal (1), slight (2), moderate (3), strong (4), or severe (5)] and distribution [no inflammation (0) to diffuse (5)] by two analysts blinded to the treatment groups. At least four non-sequential sections were used for each mouse. Airways obstruction was quantitated as the percentage of intrapulmonary airways exhibiting any degree of coalesced luminal debris (i.e., epithelial cells, leukocytes, mucus). Clara cell secretory protein (CCSP) immunohistochemistry was performed on slides to assess airway damage after influenza infection using a polyclonal antibody to mouse CCSP (Seven Hills Bioreagents). The primary antibody was used at 1:20,000 dilution with a goat anti-rabbit biotinylated secondary antibody at 1:200 dilution (Vector Laboratories) after citrate antigen retrieval.
NK Cell Transfer
For NK cell enrichment, spleen and lung leukocytes from six FA or CS exposed mice were isolated, pooled and enriched using Dynabead FlowComp Mouse CD49b NK isolation kit described above (Invitrogen). Cells were then stained and sorted for CD3- CD49b+ populations by flow cytometry. Cells were checked by NKp46 stain to confer >90% NK cells. Cells were washed and resuspened in PBS at a concentration of 2.5 × 106 cells / ml and 200 μl was administered through tail vein injection into CS Klrk1-/- recipient mice. Mice were allowed to rest for 24 hrs before viral infection as described.
Plaque Assay
Monolayers of MDCK cells were infected with serial dilutions of lung supernatant after homogenization of frozen lung. The infected cell monolayers were incubated for 1 hr at 37°C to facilitate viral adsorption and then washed with phosphate-buffered saline. After washing, the MDCK cell monolayers were treated with minimal essential medium containing 1 mg/ml trypsin and 0.8% agarose. The infected monolayers were incubated for 72 hrs at 37°C. At 72 hrs post-infection, the agarose containing medium was gently removed and the monolayers were stained with crystal violet to visualize the influenza virus plaques.
Statistics
Significant differences among groups were identified by t-test or one-way ANOVA wherever appropriate. Significant differences in histological comparisons where determined using the Mann-Whitney Test. Differences between means were considered significant when the p-value was <0.05.
Results
Expression of NK cell cytotoxicity receptors is unchanged in mouse model of COPD
Previously, we have shown that NK cells are the predominate producers of IFNγ in response to viral ligands and that this production is enhanced after CS exposure (13). This enhanced NK cell response was independent of changes in IFN-α, IL-12, or IL-18. We explored the possibility that this altered NK cell phenotype is associated with changes in activating/inhibiting receptor expression on NK cells from mice exposed long-term to CS. We examined the levels of several key receptors including NKp46, NK1.1, NKG2D, and CD244 (2B4) on NK cells in our mouse model of COPD. The total numbers of NK cells isolated from the lung and spleen was unaltered by long term CS exposure (Fig 1A-B). The percentage of NK cells (identified as Nkp46+) (19) expressing the activating receptors NK1.1, NKG2D, and CD244 (2B4), and their receptor density were not different between exposure groups (Fig 1C-F). We also assessed CD94 expression as it heterodimerizes with NKG2A/C subunits during NK cell activation. There was no difference in the number of CD94+ cells between treatments and no significant differences were found in the expression levels of CD94-mid and -hi cells. The expression of CD49b, a common marker for NK identification, similarly exhibited no significant differences in expression between treatment groups. Thus, NK cells isolated from multiple compartments of a mouse model of COPD were phenotypically indistinguishable from the FA-exposed controls.
Figure 1. Characterization of NK cell markers in a mouse model of COPD.
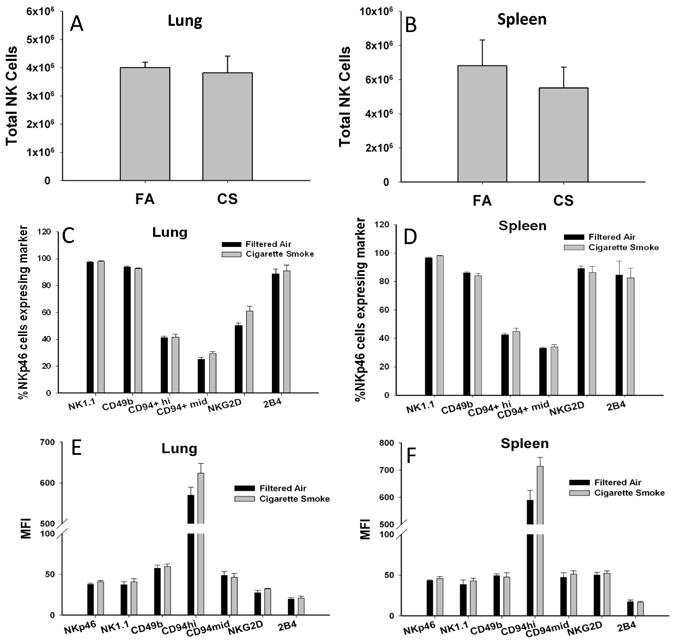
NK cells were isolated from C57BL/6 mice exposed to FA or CS for 6 months and analyzed by flow cytometry. (A-B) Total NK cells enumerated by NKp46+ expression. (C-D) The percentage of NKp46+ cells expressing the indicated NK cell markers. (E-F) The geometric mean fluorescent intensity (MFI) of NK cell receptors. Values are presented as means ± SEM. n = 5-6 per group. All data representative of two independent experiments.
NK cell cytotoxicity against MHC class I-deficient targets in a mouse model of COPD
We have previously shown that CS exposure increases CD107a expression (a marker of degranulation) (20) on NK cells (13). In the current study, we specifically examined the function of NK cell cytotoxic activity in a mouse model of COPD. Utilizing an in vivo model of NK cell cytotoxicity, splenocytes from C57BL/6 wild-type and B2m-/- mice, which lack surface MHC class I antigen, were differentially labeled with the fluorescent marker carboxyfluorescein succinimidyl ester (CFSE) and injected i.v. The ratios of peripheral blood WT cells and B2m-/- cells recovered after the assay define NK cell cytotoxic effector function (Fig 2A) (16). We found that NK cell cytotoxicity towards MHC class I-deficient cells was not different in our COPD model compared to mice exposed to FA for the same duration (Fig 2B). Notably, we did observe a similar significant decrease in cytotoxicity in mice exposed to CS or FA for 10 months, indicating an age dependent decrease in baseline NK cell function (Fig 2B).
Figure 2. NK cell cytotoxicity against MHC class I-deficient and NKG2D ligand expressing targets in mouse model of COPD.
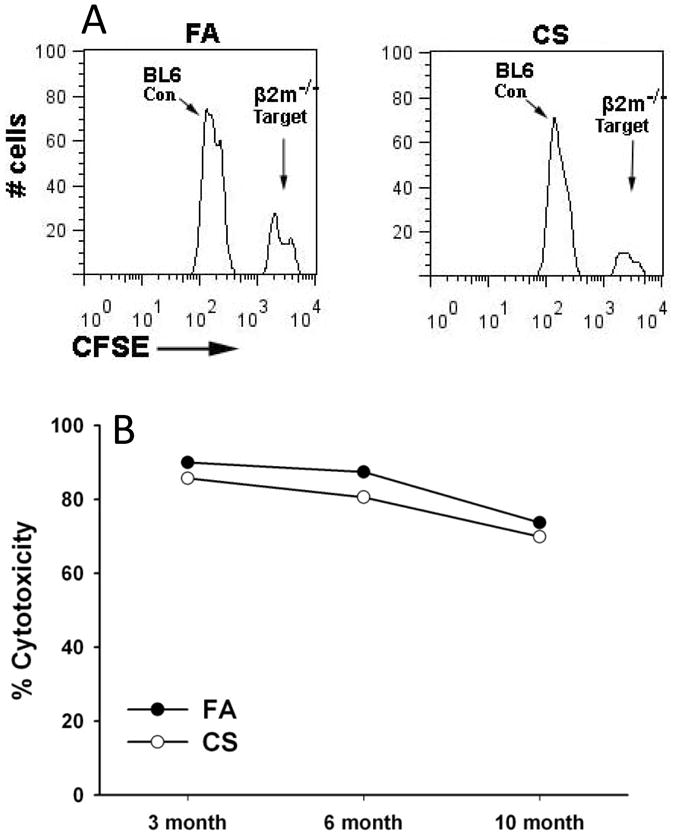
(A) Representation of flow cytometry analysis comparing in vivo clearance. CFSE labeled C57BL/6 (CFSE low) cells and C57BL/6 B2m-/- (CFSE high) cells were injected i.v .into receipt mice exposed to exposed to FA or CS. Mice were bled 16 hrs following injections and labeled target cells were analyzed by flow cytometry. (B) Time course of NK cell cytotoxicity against B2m-/- target cells, as described in (A), in mice exposed to FA or CS for 3-10 months. Data representative of results from four independent experiments.
CS exposure enhances NKG2D-mediated cytotoxicity
Engagement of the NKG2D (Klrk1) receptor is a potentially important pathway for NK cell activation in the context of COPD. Previously, we demonstrated that NKG2D ligand expression is induced on airway epithelial cells of smokers and COPD patients and the alveolar and airway epithelium of mice exposed to CS (12). However, the specific function of NKG2D in COPD has not been examined. To investigate NKG2D function on NK cells, we performed a cytotoxicity assay using an RMA mouse T lymphoma cell line transfected to express Raet1ε (21). RMA cells tend to aggregate in organs making detection in blood difficult (22). Therefore, we used an ex vivo cytotoxicity assay to measure NK activity (16). As shown in Fig. 3, NK cells from mice exposed to CS were more cytotoxic towards RMA-Raet1ε cells than mice exposed to FA. The finding that NKG2D function is enhanced in the COPD model is significant because it identifies a unique mechanism whereby CS-exposure amplifies subsequent NK cell responses towards infected cells.
Figure 3. NK cells of CS exposed mice demonstrate increased cytotoxic activity towards RAE1 expressing cells.
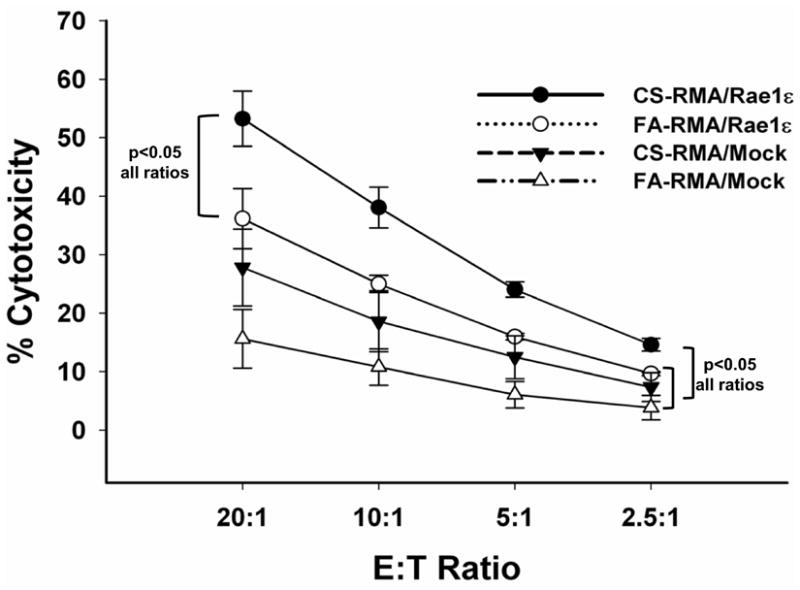
NK cells were purified from pooled splenocytes of mice exposed to FA or CS for 6 months and cytotoxicity towards RAE1ε-targets was assessed ex vivo. NK cells and CFSE-labeled RMA cells transfected to express RAE1ε (or mock transfected) were combined at increasing effector to target (E:T) ratios. Cells were incubated for 4 h, harvested and analyzed by flow cytometry as described in the methods. Figure is representative of four independent experiments each using NK cells pooled from two mice. Significant differences between groups at all E:T ratios are indicated.
NKG2D deletion abolishes NK cell hyperresponsiveness in mouse model of COPD
Previously, we reported NK cell hyperresponsiveness in the context of CS exposure and in an inducible, lung-specific Raet1 expressing transgenic mouse model (13). To investigate the role of NKG2D in mediating CS-induced NK cell hyperresponsiveness, we utilized mice deficient in NKG2D (Klrk1-/-). NK cells were purified (∼99%) from FA and CS-exposed mice and stimulated with IL-18 or IL-12/IL-18. Consistent with previous data (13), CS exposure enhances NK cell IFNγ production after stimulation with cytokines (Fig 4). The increased IFNγ production occurred absent significant differences in IL-12 or IL-18 receptor expression on NK cells between FA- and CS-exposed mice (data not shown). In contrast, NKG2D-deficient mice failed to exhibit increased cytokine responsiveness in a mouse model of COPD. Together with the current results demonstrating enhanced NKG2D function in our CS model of COPD (Fig 3), our findings suggest that a hyperresponsive NK cell phenotype may be achieved through chronic, lung-specific NKG2D stimulation.
Figure 4. Klrk1-/- mice do not develop enhanced cellular responses in mouse model of COPD.
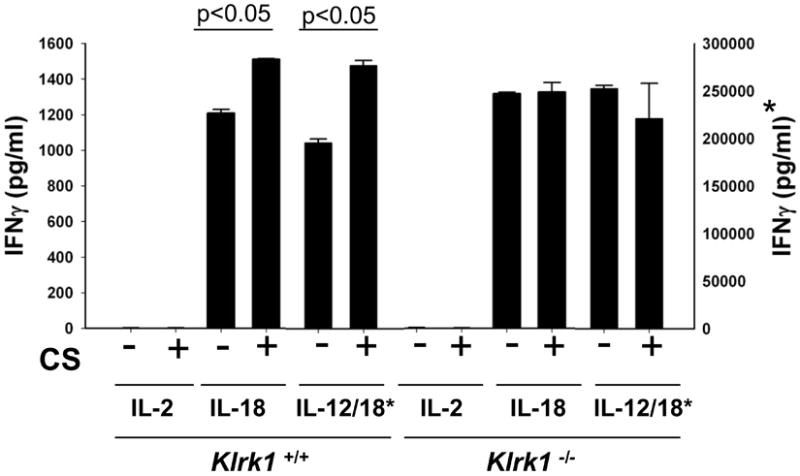
NK cells from the pooled spleens of 5 mice were highly purified (>99% NKp46+) and stimulated overnight with cytokines as labeled. IFNγ was quantified by ELISA. The marked (*) axis is the scale representing levels of IFNγ released by IL-12/18 stimulation. Values are presented as means ± SEM . Relevant significant differences between groups are highlighted. Figure is representative of three independent experiments.
NKG2D on NK cells is necessary for enhanced pulmonary inflammation and airway injury following influenza infection in COPD
Viral infections contribute significantly to the pathogenesis of COPD. The above findings that NKG2D is required for NK cell hyperresponsiveness led us to examine the role of NKG2D in influenza-induced exacerbations of COPD. As we were interested in the role of NK cells, we chose to examine the early effects (i.e. 4 days post infection) of influenza infection in our COPD model. Compared to influenza-infected mice exposed to FA, infection of mice exposed to CS augmented inflammation and airway epithelial injury that included both loss of epithelial integrity and differentiated epithelial cell morphology (Fig 5A, C). Furthermore, we found that these exaggerated pathologies were reduced in mice lacking the NKG2D receptor (Fig 5B, D).
Figure 5. CS-exposed Klrk1-/- mice lack enhanced cellular responses associated with influenza infection.

Mice were infected with 2 × 103 pfu influenza virus and endpoints were measured 4 days after infection. (A-B) H&E stained lung sections representing changes in lung inflammation and airways obstruction between groups. (C-D) Clara cell secretory protein (CCSP) immunohistochemistry staining of lung sections showing epithelial damage of large airways and small airways. (E-F) Total RNA was isolated from lung homogenates of FA- or CS-exposed Klrk1+/+ and Klrk1-/- mice with and without influenza infection. Raet1 and Mult1 transcripts were assayed by quantitative RT-PCR and normalized to Rpl32. (G) H&E stained lung sections of influenza-infected CS-exposed Klrk1-/- mice which received NK cells from FA or CS-exposed Klrk1+/+ mice. (H) Semi-quantitative assessment of inflammation severity and distribution. (I) Quantitation of the percentage of intrapulmonary airways exhibiting any degree of airways obstruction. (n = 4-8 mice per group)
By staining with antibodies against Clara cell secretory protein (CCSP), a specific marker for airway epithelial cells, we assessed the extent of damage to the airway epithelium. CCSP staining demonstrated increased disruption of epithelial integrity within the large airways of CS-exposed Klrk1+/+ mice and that surviving epithelial cells had reduced expression of CCSP as a marker of epithelial differentiation relative to FA-infected Klrk1+/+ mice (Fig 5C). In contrast, the damage to the airway epithelium of Klrk1-/- mice was similar to FA-exposed Klrk+/+ mice regardless of exposure (Fig 5D). Similarly, influenza infection caused blockage of many of the small airways of CS-exposed Klrk1+/+ mice by large aggregates of cells that appeared to be a mix of denuded epithelial cells and leukocytes (Fig 5A, C). These clumps of cells were infrequently observed in the FA-exposed Klrk1+/+ mice. Such mixed aggregates of cells/cellular debris were infrequently seen in the influenza infected Klrk1-/- mice regardless of exposure (Fig 5B, D).
To verify that the lack of the exaggerated response to influenza in the CS-Klrk1-/- was attributable to receptor function and not the lack of NKG2D ligand induction, we assessed the affects of CS and influenza infection on transcript levels of multiple NKG2D ligands. As previously reported by our group, CS exposure induces RAE1 expression in lung tissue (12). Here, we demonstrate that CS exposure and influenza infections similarly upregulate Raet1 expression. An analogous pattern of Raet1 expression occurred in CS-exposed and influenza-infected Klrk1+/+ and Klrk1-/- mice (Fig 5E). Another NKG2D ligand, Mult1, demonstrated similar expression patterns to CS and influenza in both Klrk1+/+ and Klrk1-/- mice (Fig 5F).
We next sought to define the role of NKG2D specifically on NK cells in the exaggerated pathologies associated with influenza infection and COPD. We transferred purified lung NK cells from FA and CS-exposed Klrk1+/+ mice into CS-exposed Klrk1-/- recipients which were then infected with influenza. Histopathological assessments were conducted to calculate inflammation severity and distribution as well as calculate the extent of airway obstruction. Influenza-infected Klrk1+/+ mice exposed to FA for 6 months and influenza-infected Klrk1-/- mice exposed to FA or CS for 6 months exhibited significantly less inflammation than Klrk1+/+ CS-exposed mice (Fig 5H, I). Transfer of NK cells from CS-exposed Klrk1+/+ donors into CS-exposed Klrk1-/- recipients nearly recapitulated the phenotype of influenza infected CS-exposed Klrk1+/+ mice (compare Fig 5G and 5A). The pathologies elicited with the transfer of NK cells from CS-exposed donors was also significantly greater than the pathologies associated with the transfer of NK cells from FA-exposed donors into the CS-exposed Klrk1-/- recipients (Fig 5H, I). These experiments demonstrate that NKG2D+ NK cells from CS-exposed mice play a major role in inflammation and airway obstruction in the mouse model of COPD viral exacerbation.
In addition to the inflammatory and histopathologic assays, we assessed the viral titers in the lungs of influenza-infected mice (Fig 6). There were no significant differences between FA- and CS-exposed Klrk1+/+ or Klrk1-/- mice or CS-exposed Klrk1-/- mice that received Klrk1+/+ FA or CS NK cells. These studies demonstrate that NKG2D is necessary for the enhanced pulmonary inflammation and airway injury after influenza infection in COPD independent of the ability of the immune system to eliminate the virus.
Figure 6. Long-term CS exposure does not affect viral clearance in Klrk1+/+ or Klrk1-/- mice.
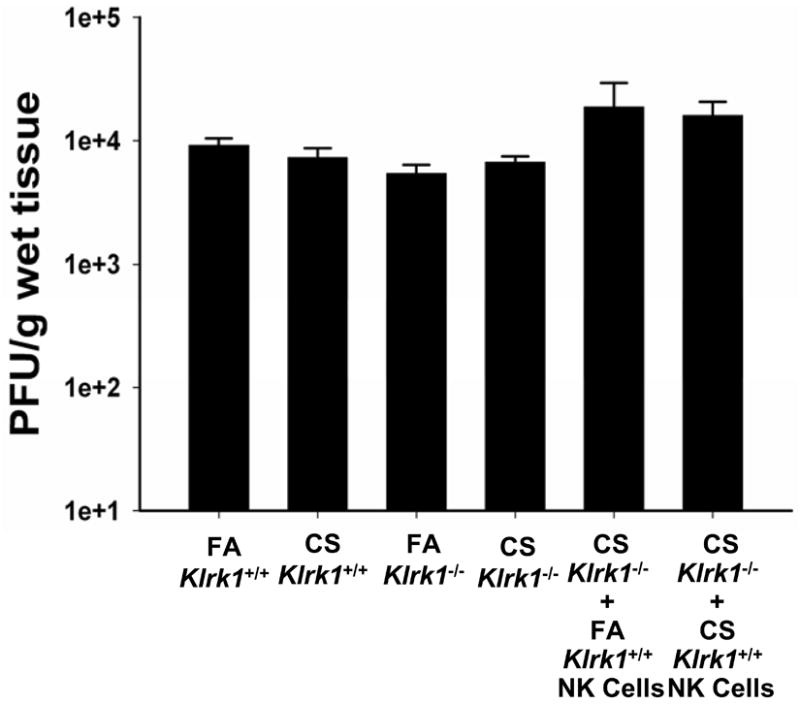
Influenza titers in individual mice were determined by plaque assay as described in Methods. Data are presented as plaque-forming units (PFU) per gram of wet lung tissue. All data representative of two independent experiments (n = 4-8 mice per group).
Discussion
A prevailing assumption is that CS is a toxic irritant that broadly inhibits leukocyte function (23, 24). However, a growing body of literature suggests that this perspective is too narrow and that chronic CS exposure can augment specific innate and adaptive immune functions in COPD patients (7, 25-27). In the course of infection, NK cells play an important dual role, the early release of cytokines and directed cytotoxicity. Previously, we have shown that NK cells from CS-exposed mice have enhanced cytokine production in response to stimulating cytokines or to TLR ligands that mimic pathogen associated molecular patterns (PAMPS) (13). In the current study, we show that this enhanced function is dependent on the presence of NKG2D. NK cells from CS-exposed, NKG2D-deficient do not exhibit enhanced IFNγ production when stimulated with IL-12 and/or IL-18. Excessive NK cell activation after infection could worsen the outcomes associated with CS exposure. In the context of COPD, the presence of NK cells hyperresponsive to viral provocation could increase the inflammatory response and contribute to the progression of COPD.
Although there have been a number of studies in humans investigating the effect of CS exposure on cytotoxic lymphocyte effector functions, primarily cytotoxicity, the outcomes of these studies are equivocal. Several studies reported increased cytotoxic activity (28-32) while others show decreased activity in smokers with and without COPD (27, 33-36). Analyses of NK cell function in mice exposed to CS show a similar variability in results (13, 37). The recent reports from Urbanowicz et al. indicate that compartmental differences in cytotoxic lymphocyte function may account for these discordant results. Those investigators found that NK cells collected from the peripheral blood of long term smokers showed reduced cytotoxicity (27), whereas NK cells collected from the sputum of similar patient populations showed increased cytotoxicity (32). Ultimately, the reasons for the disparities are unclear but the inconsistencies highlight the need for mechanistic studies to understand the effects of CS exposure on NK cell function and the role of NK cells in COPD pathogenesis.
NK cells interact with cells through an extensive array of stimulatory and inhibitory receptor-ligand interactions including NKG2D (8, 38). In this study, we report increased NKG2D-mediated cytotoxicity in a mouse model of COPD. Previously, we demonstrated that inducible expression of RAET1 on mouse pulmonary epithelial cells, and subsequent activation of NKG2D, causes an emphysematous phenotype that was associated with increased epithelial cell apoptosis (12). Moreover, these mice exhibited enhanced cytokine release in response to TLR stimulation (13) and enhanced clearance of pulmonary bacterial infection (39). Our findings appear contradictory to reports that repeated activation of NKG2D in transgenic mice caused receptor downregulation and loss of function (8, 40-42). However, these studies examined the effects of constitutive expression of NKG2D ligands. In contrast, our mouse model conditionally expresses Raet1α in pulmonary epithelial cells, an exposure more comparable to the CS model of COPD, and did not exhibit downregulation of NKG2D expression (12). Together with the current results demonstrating enhanced NKG2D function in our CS model of COPD, our findings suggest that a hyperresponsive NK cell phenotype is achieved through chronic, lung-specific NKG2D stimulation.
Cigarette smoke exposure increases pulmonary inflammation and lung remodeling in response to viral infection. Earlier studies did not identify specific mechanisms that may contribute to CS-induced viral exacerbations (43-46), whereas Kang et al. identified multiple soluble mediators (IL-18, IFNγ) and signaling pathways (e.g., TLR3) that contribute to viral-induced pathologies in CS-exposed mice (47). However, the specific cell populations and pathways affected by CS exposure that, in turn contribute to the heightened response to influenza, have not been identified. This distinction is important because neither cytokine nor receptor pathways were altered by CS exposure in the absence of infection in the previous studies (43-47). The integrity of these pathways may reflect their importance for responses to pulmonary viral pathogens with established tobacco smoke induced injury. Here, we have identified significant cell-specific alterations in response to CS exposure that directly contribute to the increased pathology after viral infection. In the context of influenza infection of CS-exposed mice, we demonstrate the requirement for NKG2D+ NK cells in the exacerbation of COPD pathologies. The CS-exposed NKG2D- deficient mice could not mount the prototypic injury response of CS exposed wild-type mice that included the influx of inflammatory cells, the loss/destruction of an intact respiratory epithelium and diminished expression of the CCSP, a phenotypic marker of the protective Clara cell. However, reconstitution with NKG2D+ NK cells from CS-exposed wild-type mice in the NKG2D-deficient mice reestablishes the prototypic injury response. Of note, there were no differences in viral load between any groups of influenza-infected mice. This finding is important because it suggests that the exaggerated immune response to virus and the subsequent increased pathology in COPD, which is dependent on NKG2D, is not a function of immunosuppression and increased susceptibility to infection. Rather, the reported increased incidence and severity of symptoms in smokers is likely due to the increased inflammation and airway obstruction as a consequence of enhanced NK cell function in response to the viral infection.
Although not directly examined in this study, it is tempting to speculate that the diminished pathologies in CS-exposed, influenza-infected, NKG2D-deficient mice was due to the failure of these NK cells to develop a hyperresponsive phenotype in the COPD model. An alternative explanation for these observations is that there is a fundamental requirement for NKG2D in response to pulmonary influenza infection. If so, the FA-exposed Klrk1+/+ or Klrk1-/- mice would reveal this requirement for homeostatic responses to influenza infection in terms of inflammation, injury and clearance. However, the lack of effect of NKG2D deficiency on these basal responses strongly supports the concept that NKG2D-dependent, CS-induced, NK cell hyperresponsiveness is an important mechanism in disease pathogenesis.
Together with our previous report demonstrating CS-induced Raet1 expression in the lung (12), our new findings suggest a model whereby CS induces the chronic local expression of NKG2D ligands on resident pulmonary cells which primes NK cells to independent stimulation by cytokines, TLR ligands, and NKG2D ligands. These NK activating mediators are known to increase during viral infection and lead to enhanced inflammation and cytotoxicity. Important to the exacerbation of COPD, the enhanced NK cell activation leads to increased pulmonary injury and airways obstruction. Although we demonstrate a clear role for NKG2D in the priming of NK cell function in the context of long-term CS exposure, the mechanisms whereby NKG2D mediates NK cell priming and the subsequent response to influenza remain to be fully elucidated.
In the current study, we present evidence that NK cells regulate inflammation in a model of viral infection of COPD and that NKG2D mediates enhanced NK cell effector function and influenza-induced pathologies in a mouse model of COPD. These findings indicate that alterations in NK cell function will have significant consequences in COPD patients infected with virus. Although NK cells are by far the predominant cell type expressing NKG2D in mice, NKG2D is also expressed almost ubiquitously on human CD8 T cells (8). However, studies suggest that NKG2D plays a co-stimulatory role based on the relative inability to stimulate T cell responses with NKG2D ligands alone (48-50). This difference in NKG2D expression and function between mice and humans coupled with evidence for a role of CD8 T cells in COPD pathogenesis (51-53) necessitates further examination of the contribution of NKG2D-bearing lymphocyte subpopulations in COPD pathogenesis.
Acknowledgments
The authors acknowledge the generosity of Dr. Kevin Harrod of the Lovelace Respiratory Research Institute for providing Influenza A virus and Dr. Lewis Lanier for providing the RAET1ε expressing RMA cells. We would also like to thank the Cincinnati Children's Hospital Research Flow Cytometry Core for expert assistance with cell sorting.
This work was supported by National Institutes of Health research Grants R01 ES015036 (to M.T.B.), T32 ES016646 (to B.W.W.), the University of Cincinnati Center for Environmental Genetics P30 ES006096, a Flight Attendant Medical Research Institute grant (to M.T.B.), NIH HL050046 (SWG) and American Lung Association RG-122427-N (APS).
References
- 1.Pauwels R. Global initiative for chronic obstructive lung diseases (GOLD): time to act. Eur Respir J. 2001;18:901–902. [PubMed] [Google Scholar]
- 2.Rennard SI, Vestbo J. COPD: the dangerous underestimate of 15% Lancet. 2006;367:1216–1219. doi: 10.1016/S0140-6736(06)68516-4. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez-Roisin R. Toward a consensus definition for COPD exacerbations. Chest. 2000;117:398S–401S. doi: 10.1378/chest.117.5_suppl_2.398s. [DOI] [PubMed] [Google Scholar]
- 4.Anzueto A. Impact of exacerbations on COPD. Eur Respir Rev. 2010;19:113–118. doi: 10.1183/09059180.00002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wedzicha JA. Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1:115–120. doi: 10.1513/pats.2306030. [DOI] [PubMed] [Google Scholar]
- 6.Wesseling G. Occasional review: influenza in COPD: pathogenesis, prevention, and treatment. Int J Chron Obstruct Pulmon Dis. 2007;2:5–10. doi: 10.2147/copd.2007.2.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freeman CM, Han MK, Martinez FJ, Murray S, Liu LX, Chensue SW, Polak TJ, Sonstein J, Todt JC, Ames TM, Arenberg DA, Meldrum CA, Getty C, McCloskey L, Curtis JL. Cytotoxic potential of lung CD8(+) T cells increases with chronic obstructive pulmonary disease severity and with in vitro stimulation by IL-18 or IL-15. J Immunol. 2010;184:6504–6513. doi: 10.4049/jimmunol.1000006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Champsaur M, Lanier LL. Effect of NKG2D ligand expression on host immune responses. Immunol Rev. 2010;235:267–285. doi: 10.1111/j.0105-2896.2010.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gasser S, Raulet DH. The DNA damage response arouses the immune system. Cancer Res. 2006;66:3959–3962. doi: 10.1158/0008-5472.CAN-05-4603. [DOI] [PubMed] [Google Scholar]
- 10.Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol. 2011;89:216–224. doi: 10.1038/icb.2010.78. [DOI] [PubMed] [Google Scholar]
- 11.Borchers MT, Harris NL, Wesselkamper SC, Zhang S, Chen Y, Young L, Lau GW. The NKG2D-activating receptor mediates pulmonary clearance of Pseudomonas aeruginosa. Infect Immun. 2006;74:2578–2586. doi: 10.1128/IAI.74.5.2578-2586.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borchers MT, Wesselkamper SC, Curull V, Ramirez-Sarmiento A, Sanchez-Font A, Garcia-Aymerich J, Coronell C, Lloreta J, Agusti AG, Gea J, Howington JA, Reed MF, Starnes SL, Harris NL, Vitucci M, Eppert BL, Motz GT, Fogel K, McGraw DW, Tichelaar JW, Orozco-Levi M. Sustained CTL activation by murine pulmonary epithelial cells promotes the development of COPD-like disease. J Clin Invest. 2009;119:636–649. doi: 10.1172/JCI34462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motz GT, Eppert BL, Wortham BW, Amos-Kroohs RM, Flury JL, Wesselkamper SC, Borchers MT. Chronic cigarette smoke exposure primes NK cell activation in a mouse model of chronic obstructive pulmonary disease. J Immunol. 2010;184:4460–4469. doi: 10.4049/jimmunol.0903654. [DOI] [PubMed] [Google Scholar]
- 14.Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, Knoblaugh S, Cado D, Greenberg NM, Raulet DH. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. 2008;28:571–580. doi: 10.1016/j.immuni.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Motz GT, Eppert BL, Sun G, Wesselkamper SC, Linke MJ, Deka R, Borchers MT. Persistence of lung CD8 T cell oligoclonal expansions upon smoking cessation in a mouse model of cigarette smoke-induced emphysema. J Immunol. 2008;181:8036–8043. doi: 10.4049/jimmunol.181.11.8036. [DOI] [PubMed] [Google Scholar]
- 16.Barnes MJ, Aksoylar H, Krebs P, Bourdeau T, Arnold CN, Xia Y, Khovananth K, Engel I, Sovath S, Lampe K, Laws E, Saunders A, Butcher GW, Kronenberg M, Steinbrecher K, Hildeman D, Grimes HL, Beutler B, Hoebe K. Loss of T cell and B cell quiescence precedes the onset of microbial flora-dependent wasting disease and intestinal inflammation in Gimap5-deficient mice. J Immunol. 2010;184:3743–3754. doi: 10.4049/jimmunol.0903164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baez M, Palese P, Kilbourne ED. Gene Composition of High-Yielding Influenza Vaccine Strains Obtained by Recombination. J Infect Dis. 1980;141:362–365. doi: 10.1093/infdis/141.3.362. [DOI] [PubMed] [Google Scholar]
- 18.Glasser SW, Witt TL, Senft AP, Baatz JE, Folger D, Maxfield MD, Akinbi HT, Newton DA, Prows DR, Korfhagen TR. Surfactant protein C-deficient mice are susceptible to respiratory syncytial virus infection. Am J Physiol Lung Cell Mol Physiol. 2009;297:L64–72. doi: 10.1152/ajplung.90640.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walzer T, Blery M, Chaix J, Fuseri N, Chasson L, Robbins SH, Jaeger S, Andre P, Gauthier L, Daniel L, Chemin K, Morel Y, Dalod M, Imbert J, Pierres M, Moretta A, Romagne F, Vivier E. Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Natl Acad Sci U S A. 2007;104:3384–3389. doi: 10.1073/pnas.0609692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 21.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci U S A. 2001;98:11521–11526. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoglund P, Glas R, Ohlen C, Ljunggren HG, Karre K. Alteration of the natural killer repertoire in H-2 transgenic mice: specificity of rapid lymphoma cell clearance determined by the H-2 phenotype of the target. J Exp Med. 1991;174:327–334. doi: 10.1084/jem.174.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sopori M. Effects of cigarette smoke on the immune system. Nat Rev Immunol. 2002;2:372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- 24.Stampfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat Rev Immunol. 2009;9:377–384. doi: 10.1038/nri2530. [DOI] [PubMed] [Google Scholar]
- 25.Feghali-Bostwick CA, Gadgil AS, Otterbein LE, Pilewski JM, Stoner MW, Csizmadia E, Zhang Y, Sciurba FC, Duncan SR. Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:156–163. doi: 10.1164/rccm.200701-014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SH, Goswami S, Grudo A, Song LZ, Bandi V, Goodnight-White S, Green L, Hacken-Bitar J, Huh J, Bakaeen F, Coxson HO, Cogswell S, Storness-Bliss C, Corry DB, Kheradmand F. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13:567–569. doi: 10.1038/nm1583. [DOI] [PubMed] [Google Scholar]
- 27.Urbanowicz RA, Lamb JR, Todd I, Corne JM, Fairclough LC. Altered effector function of peripheral cytotoxic cells in COPD. Respir Res. 2009;10:53. doi: 10.1186/1465-9921-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chrysofakis G, Tzanakis N, Kyriakoy D, Tsoumakidou M, Tsiligianni I, Klimathianaki M, Siafakas NM. Perforin expression and cytotoxic activity of sputum CD8+ lymphocytes in patients with COPD. Chest. 2004;125:71–76. doi: 10.1378/chest.125.1.71. [DOI] [PubMed] [Google Scholar]
- 29.Kratikanont P, deShazo RD, Banks DE, Chapman Y. Cytotoxic cell function in bronchogenic carcinoma. Chest. 1987;92:90–94. doi: 10.1378/chest.92.1.90. [DOI] [PubMed] [Google Scholar]
- 30.Lethbridge MW, Kemeny DM, Ratoff JC, O'Connor BJ, Hawrylowicz CM, Corrigan CJ. A novel technique to explore the functions of bronchial mucosal T cells in chronic obstructive pulmonary disease: application to cytotoxicity and cytokine immunoreactivity. Clin Exp Immunol. 2010;161:560–569. doi: 10.1111/j.1365-2249.2010.04198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newman LS, Kreiss K, Campbell PA. Natural killer cell tumoricidal activity in cigarette smokers and in silicotics. Clin Immunol Immunopathol. 1991;60:399–411. doi: 10.1016/0090-1229(91)90096-s. [DOI] [PubMed] [Google Scholar]
- 32.Urbanowicz RA, Lamb JR, Todd I, Corne JM, Fairclough LC. Enhanced effector function of cytotoxic cells in the induced sputum of COPD patients. Respir Res. 2010;11:76. doi: 10.1186/1465-9921-11-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hughes DA, Haslam PL, Townsend PJ, Turner-Warwick M. Numerical and functional alterations in circulatory lymphocytes in cigarette smokers. Clin Exp Immunol. 1985;61:459–466. [PMC free article] [PubMed] [Google Scholar]
- 34.Mian MF, Lauzon NM, Stampfli MR, Mossman KL, Ashkar AA. Impairment of human NK cell cytotoxic activity and cytokine release by cigarette smoke. J Leukoc Biol. 2008;83:774–784. doi: 10.1189/jlb.0707481. [DOI] [PubMed] [Google Scholar]
- 35.O'Shea D, Cawood TJ, O'Farrelly C, Lynch L. Natural killer cells in obesity: impaired function and increased susceptibility to the effects of cigarette smoke. PLoS One. 2010;5:e8660. doi: 10.1371/journal.pone.0008660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillips B, Marshall ME, Brown S, Thompson JS. Effect of smoking on human natural killer cell activity. Cancer. 1985;56:2789–2792. doi: 10.1002/1097-0142(19851215)56:12<2789::aid-cncr2820561213>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Lu LM, Zavitz CC, Chen B, Kianpour S, Wan Y, Stampfli MR. Cigarette smoke impairs NK cell-dependent tumor immune surveillance. J Immunol. 2007;178:936–943. doi: 10.4049/jimmunol.178.2.936. [DOI] [PubMed] [Google Scholar]
- 38.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wesselkamper SC, Eppert BL, Motz GT, Lau GW, Hassett DJ, Borchers MT. NKG2D is critical for NK cell activation in host defense against Pseudomonas aeruginosa respiratory infection. J Immunol. 2008;181:5481–5489. doi: 10.4049/jimmunol.181.8.5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coudert JD, Scarpellino L, Gros F, Vivier E, Held W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood. 2008;111:3571–3578. doi: 10.1182/blood-2007-07-100057. [DOI] [PubMed] [Google Scholar]
- 41.Oppenheim DE, Roberts SJ, Clarke SL, Filler R, Lewis JM, Tigelaar RE, Girardi M, Hayday AC. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat Immunol. 2005;6:928–937. doi: 10.1038/ni1239. [DOI] [PubMed] [Google Scholar]
- 42.Wiemann K, Mittrucker HW, Feger U, Welte SA, Yokoyama WM, Spies T, Rammensee HG, Steinle A. Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J Immunol. 2005;175:720–729. doi: 10.4049/jimmunol.175.2.720. [DOI] [PubMed] [Google Scholar]
- 43.Bauer CM, Zavitz CC, Botelho FM, Lambert KN, Brown EG, Mossman KL, Taylor JD, Stampfli MR. Treating viral exacerbations of chronic obstructive pulmonary disease: insights from a mouse model of cigarette smoke and H1N1 influenza infection. PLoS ONE. 2010;5:e13251. doi: 10.1371/journal.pone.0013251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gualano RC, Hansen MJ, Vlahos R, Jones JE, Park-Jones RA, Deliyannis G, Turner SJ, Duca KA, Anderson GP. Cigarette smoke worsens lung inflammation and impairs resolution of influenza infection in mice. Respir Res. 2008;9:53. doi: 10.1186/1465-9921-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mackenzie JS, Flower RLP. Effect of Long-Term Exposure to Cigarette-Smoke on the Height and Specificity of the Secondary Immune-Response to Influenza-Virus in a Murine Model System. J Hyg-Cambridge. 1979;83:135–141. doi: 10.1017/s0022172400025900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robbins CS, Bauer CM, Vujicic N, Gaschler GJ, Lichty BD, Brown EG, Stampfli MR. Cigarette smoke impacts immune inflammatory responses to influenza in mice. Am J Respir Crit Care Med. 2006;174:1342–1351. doi: 10.1164/rccm.200604-561OC. [DOI] [PubMed] [Google Scholar]
- 47.Kang MJ, Lee CG, Lee JY, Dela Cruz CS, Chen ZJ, Enelow R, Elias JA. Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J Clin Invest. 2008;118:2771–2784. doi: 10.1172/JCI32709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol. 2001;2:255–260. doi: 10.1038/85321. [DOI] [PubMed] [Google Scholar]
- 49.Maasho K, Opoku-Anane J, Marusina AI, Coligan JE, Borrego F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J Immunol. 2005;174:4480–4484. doi: 10.4049/jimmunol.174.8.4480. [DOI] [PubMed] [Google Scholar]
- 50.Roberts AI, Lee L, Schwarz E, Groh V, Spies T, Ebert EC, Jabri B. NKG2D receptors induced by IL-15 costimulate CD28-negative effector CTL in the tissue microenvironment. J Immunol. 2001;167:5527–5530. doi: 10.4049/jimmunol.167.10.5527. [DOI] [PubMed] [Google Scholar]
- 51.O'Shaughnessy TC, Ansari TW, Barnes NC, Jeffery PK. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med. 1997;155:852–857. doi: 10.1164/ajrccm.155.3.9117016. [DOI] [PubMed] [Google Scholar]
- 52.Saetta M, Di Stefano A, Turato G, Facchini FM, Corbino L, Mapp CE, Maestrelli P, Ciaccia A, Fabbri LM. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157:822–826. doi: 10.1164/ajrccm.157.3.9709027. [DOI] [PubMed] [Google Scholar]
- 53.Zhu X, Gadgil AS, Givelber R, George MP, Stoner MW, Sciurba FC, Duncan SR. Peripheral T cell functions correlate with the severity of chronic obstructive pulmonary disease. J Immunol. 2009;182:3270–3277. doi: 10.4049/jimmunol.0802622. [DOI] [PubMed] [Google Scholar]