

Abstract
Background
Human exome sequencing is a recently developed tool to aid in the discovery of novel coding variants. Now broadly applied, exome sequencing datasets provide a novel opportunity to evaluate the allele frequencies of previously published pathogenic rare variants.
Methods and Results
We examined the exome dataset from the NHLBI Exome Sequencing Project (ESP) and compared this dataset with a catalog of 197 previously published rare variants reported as causative of dilated cardiomyopathy (DCM) from familial and sporadic cases. Of these 197, 33 (16.8%) were also present in the ESP database, raising the question of whether they were uncommon polymorphisms. Supporting functional data has been published for 14 of the 33 (42%), suggesting they are unlikely to be false positives. The frequencies of these functional variants in the ESP dataset ranged from 0.02–1.33% (median 0.04%), which when applied as a cut-off to filter variants in a DCM pedigree identified an additional DCM candidate gene. A greater proportion of sporadic DCM cases had variants that were present in the ESP dataset vs novel variants (i.e. not in ESP; 44% vs 21%), p=0.002), suggesting some of the variants identified as disease causing in sporadic DCM are either false positives or low penetrance alleles in human populations.
Conclusions
Rare nonsynonymous variants identified in DCM subjects also present at very low frequencies in public databases are likely relevant for DCM. Allele frequencies >0.04% are of less certain pathogenicity, especially if indentified in sporadic cases, although this cut-off should be viewed as preliminary.
Keywords: cardiomyopathy, genetics, genes
One of the key challenges of medical genetics has always been to assess the pathogenic potential of rare variants in coding sequence. This is highly relevant, since assigning ‘disease causing’ to any variant may be useful for predictive testing, early disease intervention, and with further progress, specific treatment of the underlying variants or their disease pathways. The Human Genome Mutation database contains >100,000 mutations from ~4000 genes attributed to human inherited diseases, and of these, approximately 65% are single base substitutions in coding sequence or splice junctions.1
Criteria for reporting a nonsynonymous variant as pathogenic in Mendelian disease has relied upon several criteria (Table 1),2–4 including its novel identity. In earlier Mendelian studies, variants were attributed as novel based on their absence in ~200–400 unaffected chromosomes5 and more recently in larger numbers (usually 500–1000).6,7 A current method for assessing the novelty of a variant has been to determine its presence or absence in dbSNP. However, regardless of approach used, the lack of large numbers of clinically defined and ethnically matched control samples has been a confounding factor for many analyses. Further, while the recent rapid advances and application of next generation sequencing (NGS) has nearly doubled the amount of publicly available variant data in the past year alone, dbSNP is becoming increasingly “contaminated” with disease mutations that are not sufficiently annotated. Thus, variants determined as novel in earlier study designs may now be hidden in dbSNP amongst a pool of rare variants present in the general population, some of which may be benign and some pathogenic or deleterious.
Table 1.
Proposed Criteria for Evaluation of Rare Variants in Mendelian Exome Sequencing Studies
| Existing criteria | Proposed criteria / comments | |
|---|---|---|
| Rarity | Usually not identified in 300–1000 chromosomes | Disease specific iterative approach: Utilization of ESP or other large population based datasets to empirically determine frequencies of known pathogenic variants. Apply these frequencies to filtering criteria for exome data in each disease. |
| Racial/ethnic matching | Same as above - usually not identified in 300–1000 racially and ethnically matched chromosomes | Utilization of ESP and other large population based datasets will help to resolve false positives due to insufficient ethnic matching. |
| Conservation | PhastCon, GERP scoring | Conservation scores do not distinguish between ‘novel’ and ‘very rare’ pathogenic variants, but in general are high. These scores may be used for ranking or prioritization. |
| Type of variant | Missense, nonsense, splice variant | Functional classification allows for prioritization and hypothesized effect (see Functional data below). This may be impacted in the era of whole genome sequencing, when many rare non-coding variants will also be identified and conservation scores or other metrics would be needed. |
| Segregation | Present in all affected family members | No immediate change; however, with greater insight, the 'autosomal dominant' genetic paradigm may be enlarged to account for more complexity (e.g. modifiers and penetrance). New rare variant pathway analyses will highlight novel genes with rare variants in multiple sporadic cases. |
| Functional data | Pathologic change in animal or cellular model | The stringency for emphasizing relevant pathologic changes in an appropriate model will only increase. This is congruent with interest in using iPS cells to create an investigational tissue source from a subject harboring the variant that has the same genetic background. |
Our own work in a rare variant Mendelian disease has focused on idiopathic dilated cardiomyopathy (DCM). DCM is genetically heterogeneous, typically showing autosomal dominant transmission with variable age of onset and reduced penetrance.4 Rare variants in >30 previously reported DCM genes account for ~35–40% of familial cases, while the remaining genetic cause remains to be determined.4 We have contributed 95 DCM variants to the literature8–13 but unaffected individuals for these studies were limited. The Exome Sequencing project (ESP), funded by the National Heart, Lung and Blood Institute (NHLBI), recently released a searchable public database containing genetic variants identified in the human exome for >2,400 individuals. Since exome sequencing is now commonly applied in discovery of rare variants in Mendelian disease,14 we catalogued the 197 discrete variants from 235 DCM cases (Supplementary Table 1) and screened for their presence in the exome sequences of 1,351 European and 1,088 African-descent individuals. While most of these variants were not present in the ESP database, those that were present provide insight into preliminary guidance for future use of large publicly available genomic resources.
Methods
Data collection
We collated single nucleotide substitutions or small insertion/deletion allelic variants reported in the literature as ‘mutations’ or ‘rare variants’ in 30 autosomal DCM genes (LMNA, MYH6, MYPN, TNNT2, SCN5A, MYBPC3, RBM20, TMPO, LAMA4, VCL, LDB3, TCAP, PSEN1, PSEN2, ACTN2, CRYAB, TPM1, ABCC9, ACTC, PDLIM3, ILK, TNNC1, TNNI3, PLN, DES, SGCD, CSRP3, TTN, ANKRD1, BAG3) as previously published in a review4 and subsequent publications.8,15
Genomic positions (Hg19) and the reference and alternate allele on the forward strand were determined for each variant. Conservation for single base variants and prediction of functional effects was assessed with PhastCons,16 GERP17 and Grantham scores,18 using SeattleSeq SNP annotation (http://snp.gs.washington.edu/SeattleSeqAnnotation131/) and PolyPhen2.19
Search for published DCM variants in Exome Sequencing project data
Details of the ESP dataset are described in supplemental methods. Exome sequencing was performed at the Broad Institute or the University of Washington. Single base variant data for these analyses are available on the ESP Exome Variant Server (EVS- snp.gs.washington.edu/EVS/).
Genomic positions of DCM single base variants were searched against exome data for 2,439 individuals, of which 1,351 were of European and 1,088 were of African descent respectively. For small insertion deletions, genomic positions 15 bases either side of the reported variant were searched against VCF files for 1,200 ESP controls of which 1,028 were of European and 167 were of African descent and 5 were of unknown ancestry, respectively. Of 1,200 ESP individuals, ~600 overlapped with those used for single base analysis.
Exome sequencing of DCM pedigree
The exomes of three related individuals with DCM were sequenced as previously described.8
Statistical analysis
Wilcoxon two-sample tests to determine statistical difference of Grantham, PhastCon and GERP scores between DCM variants present and absent in the ESP dataset were performed in SAS v. 9.2.
Results
ESP data across DCM genes and variant positions
Only one of 30 autosomal DCM genes, RBM20, was not captured in this version of the ESP dataset, eliminating its nine known DCM variants from consideration.11,20 The remaining 29 genes were included in the ESP target sequence with an average sequence coverage exceeding 60x and with a minimum of >80% of the target sequence having coverage exceeding 20x (Supplementary Table 2).
DCM allelic variation
Allelic variants from 29 genes reported as either ‘mutations’ or ‘rare variants’ causative of DCM, and in one case as a ‘disease modifier,’ from 53 published studies are summarized (Figure 1). A total of 235 DCM cases with variants attributed as causative of disease were reported, of which 197 variants were discrete. Of these 197, 87 were reported in our studies 8–13 and eleven were reported by more than one study. Most variants (88%) were ‘private’ to a single family with the remaining occurring in two to four DCM probands; 89% were single base substitutions (93% missense, 5% nonsense and 2% occurring in consensus splice sites). The remaining 11% of variants were small insertion/deletions (indels) ranging from one to four bases in size, of which 86% resulted in frameshifts within the coding regions. Most variants (73.6%) occurred in familial dilated cardiomyopathy, 24.4% occurred in sporadic cases, and 2% were observed in both familial and sporadic DCM cases.
Figure 1.
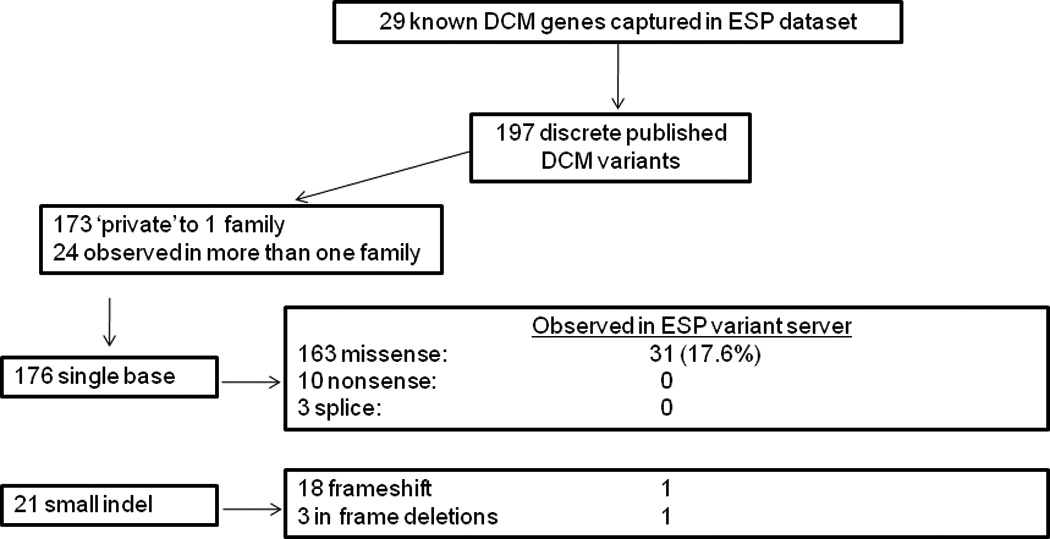
DCM variant data collected from 53 publications. ESP, Exome Sequencing Project.
Known DCM allelic variants present in dbSNP build 131
52 of 176 single base substitution variants were present in dbSNP, of which eighteen have no reported population frequency data, 20 have reported frequency of <0.1%, two have frequencies between 0.1 and 1% and seven have frequencies > 1% in at least one population. Only 22 of 52 (42%) of known DCM variants present in dbSNP were tagged as ‘clinically associated.’ Eight of 21 small indels were also present in dbSNP and seven of eight were flagged as ‘clinically associated’. Seven of eight indels had no population frequency data and one (TNNT2 Lys210del) was not identified in 243 individuals from European, Asian, Hispanic and Yoruban populations.
Known DCM allelic variants present in >2000 ESP exomes
A total of 31 single base substitutions (17.6%) reported in the literature as ‘mutations’ or ’rare variants’, and in one case, (LDB3 Lys204Arg),21 as a ‘disease modifier’ were also present in the ESP dataset (Table 2). These variants occurred in seventeen of the 29 DCM genes captured in the ESP data set (Table 3). Frequencies in ESP individuals ranged from 0 to 0.59 % in Europeans, and from 0 to 4.64% in Africans. Two variants occurred at frequency >1% in ESP individuals of African ancestry: LDB3 Lys204Arg (4.64%) and LDB3 Asp117Asn (1.33%). These variants were highly conserved with PhastCons scores of 1 and 0.997 and GERP scores of 5.55 and 3.48, respectively, although Grantham scores for these variants were not suggestive of structural impact, (26 and 23 respectively, where a score >50 is suggestive of impact). Lys204Arg was predicted to be ‘probably-damaging’ by PolyPhen2, while the discovery study observed this variant in 6 DNAs from African American control individuals and suggested it was a disease modifier or polymorphism.21 Asp117Asn was predicted to be benign by PolyPhen2, but despite these predictions, cells transfected with this particular variant showed cytoskeleton disarray.21
Table 2.
DCM Rare Variants Present in the ESP Data Set
| Gene | Variant | Type* | rs Number | PhastCon | GERP | Clinical Assoc, dbSNP* |
N, DCM families |
%, all ESP* |
% European ESP* |
% African ESP* |
DCM Functional Data |
Refs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ABCC9 | Leu1524fs | fs | NA | NA | 1 | 0.04 | 0.05 | 0 | yes | 32 | ||
| ACTN2 | Gln9Arg | m | 1 | 3.17 | 1 | 0.04 | 0.08 | 0 | yes | 26 | ||
| ANKRD1 | Thr116Met | m | 1 | 5.47 | 1 | 0.02 | 0 | 0.05 | 40 | |||
| ANKRD1 | Arg66Gln | m | 1 | 5.71 | 1 | 0.1 | 0.19 | 0 | 40 | |||
| CRYAB | Arg157His | m | 1 | NA | 1 | 0.02 | 0.04 | 0 | yes | 24 | ||
| CSRP3 | Trp4Arg | m | rs45550635 | 1 | 5.67 | yes | 2 | 0.35 | 0.59 | 0.05 | yes | 10, 25–26, 35 |
| CSRP3 | Gly72Arg | m | rs45552933 | 1 | 5.29 | 1 | 0.02 | 0.04 | 0 | 10 | ||
| DES | Asp312Asn | m | rs34337334 | 0.241 | 4.44 | yes | 1 | 0.06 | 0 | 0.14 | yes | 30 |
| LDB3 | Ser196Leu | m | rs45487699 | 0.002 | 2.48 | 1 | 0.02 | 0.04 | 0 | 21 | ||
| LDB3 | Asp117Asn | m | 0.997 | 3.48 | 2 | 0.73 | 0.37 | 1.33 | yes | 21 | ||
| LDB3 | Lys204Arg | m | rs34423165 | 1 | 5.55 | 1 | 2.15 | 0.15 | 4.64 | 21 | ||
| LDB3 | Ala698Thr | m | rs45577134 | 0.992 | 5.07 | 2 | 0.04 | 0.07 | 0 | 10 | ||
| MYBPC3 | Glu619Lys | m | 0.961 | 3.2 | 1 | 0.06 | 0.1 | 0 | 41 | |||
| MYBPC3 | Arg326Gln | m | rs34580776 | 1 | 3.48 | 2 | 0.15 | 0.23 | 0 | 41 | ||
| MYBPC3 | Asp605Gly | m | 1 | 4.14 | 1 | 0.03 | 0.05 | 0 | 9 | |||
| MYBPC3 | Gly5Arg | m | 0.964 | 4.13 | 1 | 0.06 | 0.09 | 0 | 9 | |||
| MYBPC3 | Gly490Arg | m | 0.954 | 4.28 | 1 | 0.09 | 0.14 | 0 | 9 | |||
| MYBPC3 | Ala833Thr | m | 0.873 | 4.16 | 3 | 0.17 | 0.28 | 0 | 9 | |||
| MYH6 | Ala1004Ser | m | 1 | 4.84 | 4 | 0.06 | 0.11 | 0 | 9, 42 | |||
| MYH6 | Ala1440Pro | m | 0.847 | 4.51 | 1 | 0.02 | 0.04 | 0 | 9 | |||
| MYH6 | Arg568Cys | m | 0.878 | 4.44 | 1 | 0.04 | 0.04 | 0.05 | 9 | |||
| MYH7 | Val964Leu | m | rs45496496 | 1 | 4.86 | 1 | 0.04 | 0.07 | 0 | 10 | ||
| MYPN | Val1195Met | m | rs71534280 | 0.924 | 4.87 | 1 | 0.06 | 0 | 0.14 | yes | 22 | |
| MYPN | Pro1112Leu | m | rs71534278 | 1 | 5.03 | 1 | 0.14 | 0.19 | 0.09 | yes | 22 | |
| PSEN2 | Ser130Leu | m | rs63750197 | 0.021 | 3.26 | yes | 1 | 0.02 | 0.04 | 0 | yes | 12 |
| SCN5A | Thr220Ile | m | rs45620037 | 1 | 3.95 | 1 | 0.03 | 0.05 | 0 | yes | 28, 43 | |
| SCN5A | Ser216Leu | m | rs41276525 | 1 | 3.95 | 2 | 0.06 | 0.09 | 0 | 10 | ||
| SCN5A | Pro2005Ala | m | rs45489199 | 0.133 | −9.85 | 1 | 0.12 | 0.18 | 0 | 10 | ||
| SCN5A | Ile1835Thr | m | rs45563942 | 1 | 4.27 | 1 | 0.2 | 0 | 0.54 | yes | 10, 31 | |
| SGCD | Lys238 del | In fr del | 1 | 0.3 | 0 | 1.8 | 44 | |||||
| TMPO | Arg690Cys | m | rs17028450 | 0.956 | 0.893 | 1 | 0.03 | 0.04 | 0 | yes | 29 | |
| TNNT2 | Glu244Asp | m | rs45466197 | 0.996 | 2.28 | 1 | 0.02 | 0 | 0.05 | yes | 23, 45 | |
| VCL | Ala934Val | m | rs16931179 | 0.998 | 4.74 | 1 | 0.04 | 0.04 | 0.05 | yes | 27 |
fs, frameshift; m, missense; in fr del, in-frame deletion; Clin Assoc dbSNP, clinical association denoted in the dbSNP database; % all ESP, % European or African ESP, all reflect the frequency expressed in percent against the 2,439 exomes of the entire dataset, the 1,351 European or 1,088 African samples in the ESP. Refs; references
Table 3.
DCM variants by gene
| Gene | Number reported DCM variants (%) |
Number variants in ESP |
Freq ESP % | Number observations in ESP |
|---|---|---|---|---|
| LMNA | 43 (20.57) | 0 | 0 | 0 |
| MYH7 | 28 (13.40) | 1 | 0.07 | 2 |
| MYBPC3 | 16 (7.66) | 6 | 0.05–0.28 | 1–6 |
| BAG3 | 13 (6.25) | 0 | 0 | 0 |
| TNNT2 | 12 (5.74) | 1 | 0.05 | 1 |
| SCN5A | 10 (4.78) | 4 | 0.05–0.54 | 1–7 |
| TPM1 | 9 (4.31) | 0 | 0 | 0 |
| MYH6 | 9 (4.31) | 3 | 0.05–0.11 | 1–3 |
| RBM20* | 9 (4.31) | NA | NA | NA |
| LDB3 | 7 (3.35) | 4 | 0.04–4.64 | 1–101 |
| DES | 6 (2.87) | 1 | 0.14 | 3 |
| TNNC1 | 5 (2.39) | 0 | 0 | 0 |
| ANKRD1 | 5 (2.39) | 2 | 0.05–0.19 | 1–5 |
| TNNI3 | 4 (1.91) | 0 | 0 | 0 |
| MYPN | 4 (1.91) | 2 | 0.14–0.19 | 3–5 |
| PLN | 3 (1.44) | 0 | 0 | 0 |
| TCAP | 3 (1.44) | 0 | 0 | 0 |
| VCL | 3 (1.44) | 1 | 0.05 | 1 |
| CSRP3 | 3 (1.44) | 2 | 0.04–0.59 | 1–16 |
| ACTC1 | 2 (0.96) | 0 | 0 | 0 |
| TTN | 2 (0.96) | 0 | 0 | 0 |
| ABCC9 | 2 (0.96) | 1 | 0.05 | 1 |
| SGCD | 2 (0.96) | 1 | 1.8 | 6 |
| ILK | 1 (0.48) | 0 | 0 | 0 |
| LAMA4 | 1 (0.48) | 0 | 0 | 0 |
| PSEN1 | 1 (0.48) | 0 | 0 | 0 |
| ACTN2 | 1 (0.48) | 1 | 0.08 | 2 |
| CRYAB | 1 (0.48) | 1 | 0.04 | 1 |
| TMPO | 1 (0.48) | 1 | 0.04 | 1 |
| PSEN2 | 1 (0.48) | 1 | 0.04 | 1 |
The number of variants per DCM gene that also occurred in the ESP dataset are shown with the highest allele frequency and highest number of observations in a single ESP population. Where more than one variant per gene is observed in the ESP population, the range of frequency and occurrence is shown.
RBM20 was not captured in the ESP dataset, but is included here to demonstrate its frequency in the DCM in context to other known genes.
Eighteen of 31 single base substitutions present in the ESP dataset were also present in dbSNP (three of which were flagged as clinically associated). Thirteen variants not present in dbSNP but present in ESP individuals had frequencies ranging from 0.04 to 1.33% in at least one population. Published functional data was available for fifteen of these 31 variants, with thirteen of fifteen showing some functional deficit12, 21–30 while two of fifteen showed no significant functional effect. In the case of SCN5A Ile1835Thr, observed in the ESP individuals of African origin at a frequency of 0.54% (absent in those of European ancestry), sodium current was significantly altered but only in the presence of a constitutively expressed SCN5A transcript variant, Q1077del.31
Among the 21 DCM indels, an in-frame deletion of a single amino acid (SGCD Lys238del) was detected in the ESP individuals, occurring in six of 134 chromosomes at this position (1.8%) from individuals of African descent but not present in any individuals of European descent. A second DCM indel was identified in a single heterozygous individual of European descent within the ESP data as insertion A at chr12: 21958185. The DCM variant at this locus was reported as a complex deletion of ATT and an insertion of AAAT resulting in a frameshift in ABCC9, with a functional deficit supported by cell based assays showing an abnormal ATP-sensitive potassium channel phenotype32 (chr12:21958186–21958188).
Of the DCM variants present in ESP individuals, five were present only in samples of European origin, whilst none were unique to samples of African American origin. This is likely a reflection of the initial DCM discovery samples which were predominantly of European origin. The exact composition of the DCM discovery samples is not reported in several of the published manuscripts but for our own published cohort8–13 which contributed 44% of variants in the study, patients is 93% European, 5% African American, and 2% other origin.
Lack of discriminating features of DCM variants present versus those absent in the ESP dataset
For single base substitutions, we compared DCM variants present in the ESP dataset against those that were absent (novel), under the assumption that DCM variants identified in the general population may be less conserved, and thus less pathogenic, than novel variants unique to DCM. However, conservation scores at the nucleotide (GERP) or amino acid level (PhastCons and Grantham scores) and PolyPhen2 classification did not discriminate between DCM variants present in the ESP dataset and those that were absent. Of those present in the ESP dataset, seven were classed as ‘benign’, sixteen were classified as ‘probably damaging’ and eight as ‘possibly damaging’. Wilcoxon two-sample tests of Grantham and GERP scores for variants present in the ESP dataset verses those that were novel were not significantly different, p-values 0.26 and 0.20 (1-tailed) respectively. PhastCons scores between the two groups showed weak evidence for statistical difference, p=0.049 (1-tailed), average 0.951, compared with 0.858 in DCM variants that were present in the ESP dataset, although this was not significant after correction for multiple testing.
We further stratified those DCM variants present in the ESP against those that were novel by whether they occurred in familial or sporadic DCM cases and the amount of supporting functional data. Forty-four percent of DCM variants present in the ESP dataset occurred in sporadic cases compared to 21% of the novel variants, p=0.002 (2-tailed). One possibility for the excess of sporadic DCM cases with variants that were also observed in the ESP dataset is that these variants were false positives for disease causation. An alternate possibility is that these rare variants were low penetrance or susceptibility alleles that could be resolved to some extent by functional assays. Six of fifteen variants present in both sporadic DCM cases and the ESP dataset showed some functional deficit, suggesting that they were low penetrance alleles. Furthermore, no significant difference was observed between the amount of supporting functional data in those variants present and those that were absent in the ESP data, p=0.43 (2-tailed).
The true picture may become clearer once functional assays are available for many more variants. Based on the assumption that DCM variants with functional data that have also been observed in the ESP dataset are indeed pathogenic but with reduced penetrance, we propose an empirically derived frequency cut-off to aid in determining pathogenicity. In this dataset there were fourteen DCM variants with supporting functional data that were also observed in the 2,439 ESP individuals. In the ESP dataset the median frequency of these variants was 0.04% (2/4878 alleles), which we suggest may be a useful cut point for lower penetrance alleles when filtering exome data.
Application of a 0.04% cut point to exome sequencing data in a multiply affected DCM pedigree
To further illustrate the potential utility of the ESP dataset and evaluate our proposed cut point of allele frequency of 0.04%, we performed exome sequence analysis on three DCM cases within the same pedigree, known to be point mutation negative for sixteen known DCM genes, 8–13 (Figure 2, Table 4). After filtering against dbSNP and 1000 Genomes Project, an average of 368 nonsynonymous or splice site variants remained per individual (Table 4). We then screened the remaining variants against the ESP data and categorized these into two discrete categories: novel variants (absent from ESP data) and very rare variants (present one and two times in the ESP dataset, a frequency of 0.04%. This reduced the number of novel variants to ~100 per individual, and sharing across first cousins, reduced this number to eighteen. Sharing across all three affected individuals reduced the number of novel variants to eight genes (OTOP1, FSIP1, LOC100130086, FAM115C, CCL3L3, FRG1, CDC27 and EPB41L3). Ten additional rare variants were shared between first cousins that also occurred between one and two times in the ESP data, and this was reduced to two variants (COLQ at chr3:15507925 and OR11H12 at chr14:19378189) when sharing across all three affected members. Upon further examination of DCM transcriptome data, previously described8 only FRG1, EPB41L3 and COLQ were expressed in heart tissue. FRG1 (Facioscapulohumeral muscular dystrophy Region Gene 1) is a valid candidate, given that other DCM genes such as DMD1, LMNA and BAG3 are also causative of muscular dystrophy.33–34 However, this variant resulted from paralagous sequence (unpublished data). The remaining variants occurred in EPB41L3, (erthyrocyte protein band 4.1) and COLQ (Collagenic tail of endplate acetylcholinesterase), the former being a novel variant (chr18:5423409 T>C) and the latter (chr3:15507925 C>G) occurring in 2/2702 ESP alleles of European ancestry and in 0/2174 ESP alleles of African ancestry. On the basis of our findings with known DCM variants occurring at very low frequency in the ESP dataset, we consider COLQ to be an equally valid candidate as EPB41L3. Further investigation is underway.
Figure 2.
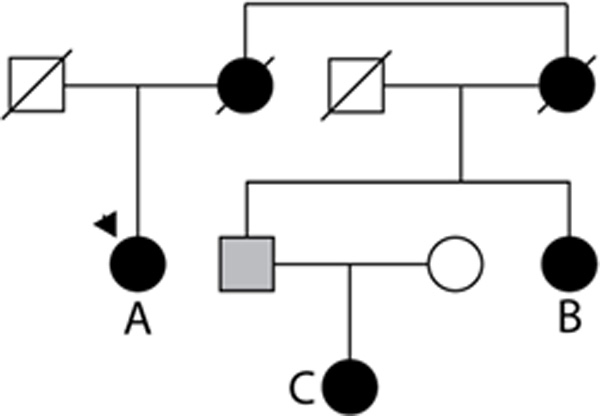
DCM family with exome sequencing. The number of variants in affected individuals A, B and C. AB, AC, BC, and ABC are shown in Table 3 and reflect the variants in common between these combinations of patients. Relationships are AB, first cousins; BC aunt/niece, and AC are first cousin/first cousin once removed.
Table 4.
Exome sequence variants in a DCM pedigree (see Figure 2), mutation negative for 16 known DCM genes
| A | B | C | AB | AC | BC | ABC | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total Variants: Nonsense, Missense, or Splice Site |
6878 | 6887 | 6851 | 4524 | 4429 | 4799 | 3708 | |||||||
| Above, not in dbSNP, 1000 genomes, other local databases |
357 | 379 | 369 | 98 | 61 | 137 | 48 | |||||||
| novel | very rare |
novel | very rare |
novel | very rare |
novel | very rare |
novel | very rare |
novel | very rare |
novel | very rare |
|
| Above, compared to ESP: | 101 | 36 | 98 | 54 | 101 | 56 | 18 | 10 | 11 | 2 | 26 | 19 | 8 | 2 |
| Expressed in heart | 39 | 15 | 40 | 24 | 43 | 19 | 6 | 7 | 2 | 2 | 11 | 6 | 2 | 1 |
Novel means not identified in the ESP dataset; very rare means identified only 1 or 2 times.
Discussion
Exome sequencing is a recently developed approach with the potential to uncover pathogenic mutations for Mendelian diseases.14 Because of its comprehensive nature, the ESP exome dataset (ESP2500) provides a new coding reference database (http://snp.gs.washington.edu/EVS/). Using this resource, we analyzed DCM, a rare variant Mendelian adult onset disease, for the number of reported DCM variants present among the ESP dataset and dbSNP. Our results raise several issues.
A filtering approach based on removing variants found in exome data from unaffected individuals and variants in dbSNP/1000 Genomes Project has proved effective in many Mendelian diseases,14 but these likely represent extremely rare diseases from highly penetrant mutations. When this approach is applied to other disease models with variable penetrance and very rare allele frequency (0.1%) it will likely be less effective in discerning causative alleles. The development of NGS technology has rapidly increased the amount of publicly available sequence variant data. As we report herein for DCM, 16.8% of variants previously reported as disease causative are present in the sequenced individuals within the 2,439 ESP samples. It is possible that a fraction of these rare variants were false positives, although the supporting functional data suggested they may actually be lower penetrance disease-causing alleles. Importantly, prior to large scale datasets such as the ESP it has been nearly impossible to accurately assess the level of penetrance due to the lack of large population-based estimates of variant allele frequencies relative to their disease frequencies.
This raises a second issue - how to distinguish pathogenic variants present at very low frequency in the general population from benign rare variants. Understandably, an oversimplified filtering approach could result in discarding variants that are truly pathogenic. Fourteen of the 33 (42%) DCM variants that were also present in ESP individuals had functional effects in either heterologous cell-based assays or in animal models. In an effort to derive guidance from this previously published data, we suggest an allele frequency of 0.04% as a conservative initial cut point, which is the median allele frequency of variants present in the ESP but also with supporting functional data. In our own example of exome sequencing of a DCM pedigree (table 4), this analysis yielded an additional candidate gene.
Ideally, functional assays would be performed for all variants, as the use of any cut-point has caveats. CSRP3 Trp4Arg is a pertinent example, as it has been observed in multiple DCM families from independent studies,10, 25–26 and was observed within the ESP individuals at a frequency of 0.35% in individuals of European and 0.05% in individuals of African descent, respectively. The CSRP3 Trp4Arg variant specifically affects the ability of Csrp3 to interact with T-cap,35 and in a knock-in mouse model resulted in an age- and gene dosage-dependent hypertrophic cardiomyopathy and heart failure phenotype.25
Further discussion is deserved for the SCN5A variant, Ile1835Thr, also present in dbSNP (rs45563942) with no clinical association. This variant was observed at a frequency of 0.54% (7/1292 alleles) in the ESP individuals of African ancestry and absent in those of European origin. In a cell based assay, Ile1835Thr significantly reduced peak sodium current but the effect was dependent on genetic background in the context of transcriptional variation and combination with a common polymorphism, SCN5A H558R (rs1805124).31 Two constitutively expressed SCN5A transcript variants (not allelic variants) exist at frequencies of 65% and 35% expression in heart.36 These transcripts are identical with the exception that the more highly expressed transcript has a deletion of a single amino acid, Q1077. The functional change in sodium current due to the rare DCM variant, Ile1835Thr was specifically observed in cDNA constructs for the Q1077deletion transcript in combination with the common H558R variant and no significant change was observed on the Q1077 background.
These levels of complexity are likely just one example of genetic interactions, and DCM is but one paradigm that the ESP data can illuminate. Supporting evidence for our preliminary cut-point is derived from the example of the long-QT syndrome (LQTS). Although as with DCM, inheritance is predominantly autosomal dominant, a study of 147 discrete variants from two LQTS genes in 753 families suggested that long-QT alleles are more frequently transmitted to daughters than to sons.37 In this example, careful matching of unaffected individuals by gender could also be important. However, only one variant, KCNQ1 Pro320His, is present in the ESP dataset, frequency 0.037% in individuals of European origin, falling within our suggested cutpoint.
Traditionally, Mendelian disease has been viewed as caused by one unique or very rare variant in an individual or pedigree. However, this is likely an over simplification, as other genetic disease paradigms are possible.38 We note that DCM has been shown to result principally from autosomal dominant transmission, commonly understood to result in gain of function mutations, even though loss of function, at least for calcium handling contractile proteins in DCM, also commonly occurs.39 Much greater understanding of disease mechanisms may be relevant, including the possibility of other more complex genetic mechanisms such as di- or multi-genic ‘recessive’ mechanisms where multiple dysfunctional protein cascades interact, all of which leads to the final phenotype of DCM.38
Limitations
Use of unaffected individuals from the same ethnicity and geographical region is highly important. The ESP dataset consists of European and African American ancestries, and a small proportion of the DCM cases are of other origins. This is a potential bias in our study, most strongly highlighted by the failure to observe the LMNA Leu85Arg variant in the ESP data. This variant is present in dbSNP, has a reported frequency >1% in Asian and Sub-Saharan Africans and >20% in Maasai.
Our proposed allele frequency cut point of 0.04% is the median of a small number of observations, and thus with larger sample sizes and more functional data will likely need to be revised. This is the first empiric evidence derived from an exome dataset that provides any guidance, although it may differ for other phenotypes.
Summary
In the exome era, traditional boundaries between Mendelian and more complex diseases will be tested, as illustrated in this example of DCM. This will contribute a degree of uncertainty in clinical interpretation of Mendelian gene variants. The importance of careful annotation of variants cannot be underestimated and through this we predict the level of evidence to assign pathogenicity will be enhanced.
Supplementary Material
Acknowledgements
We thank the National Heart, Lung, and Blood Institute GO Exome Sequencing Project and its ongoing studies that produced and provided exome variant calls for comparison: the Lung Cohorts Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Heart Cohorts Sequencing Project (HL-103010), the Broad Institute Sequencing Project (HL-102925), the Northwest Genomics Center Sequencing Project (HL-102926; D.A.N., M.J.R.), and the Family Studies Project Team.
Funding sources. This work was supported by National Institute of Health awards HL58626 (R.E.H.), 5U54NS065712-02 and 1R01NS072248-01 (S.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures. None
References
- 1.Chen JM, Ferec C, Cooper DN. Revealing the human mutome. Clin Genet. 2010;78:310–320. doi: 10.1111/j.1399-0004.2010.01474.x. [DOI] [PubMed] [Google Scholar]
- 2.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 3.Ho CY, MacRae CA. Defining the pathogenicity of DNA sequence variation. Circ Cardiovasc Genet. 2009;2:95–97. doi: 10.1161/CIRCGENETICS.109.864793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hershberger RE, Siegfried JD. Update 2011: Clinical and Genetic Issues in Familial Dilated Cardiomyopathy. J American College of Cardiology. 2011;57:1641–1649. doi: 10.1016/j.jacc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr., Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 6.Zuchner S, Dallman J, Wen R, Beecham G, Naj A, Farooq A, Kohli MA, Whitehead PL, Hulme W, Konidari I, Edwards YJ, Cai G, Peter I, Seo D, Buxbaum JD, Haines JL, Blanton S, Young J, Alfonso E, Vance JM, Lam BL, Pericak-Vance MA. Whole-Exome Sequencing Links a Variant in DHDDS to Retinitis Pigmentosa. Am J Hum Genet. 2011;88:201–206. doi: 10.1016/j.ajhg.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Regalado ES, Guo DC, Villamizar C, Avidan N, Gilchrist D, McGillivray B, Clarke L, Bernier F, Santos-Cortez RL, Leal SM, Bertoli-Avella AM, Shendure J, Rieder MJ, Nickerson DA, Milewicz DM. Exome Sequencing Identifies SMAD3 Mutations as a Cause of Familial Thoracic Aortic Aneurysm and Dissection With Intracranial and Other Arterial Aneurysms. Circ Res. 2011;109:680–686. doi: 10.1161/CIRCRESAHA.111.248161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, Mangos S, Gonzalez-Quintana J, Wang L, McGee S, Reiser J, Martin E, Nickerson DA, Hershberger RE. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as cause for dilated cardiomyopathy. American J Human Genet. 2011;88:273–282. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hershberger RE, Norton N, Morales A, Li D, Siegfried JD, Gonzalez-Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:155–161. doi: 10.1161/CIRCGENETICS.109.912345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J, Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1:21–26. doi: 10.1111/j.1752-8062.2008.00017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li D, Morales A, Gonzalez-Quintana J, Norton N, Siegfried JD, Hofmeyer M, Hershberger RE. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci. 2010;3:90–97. doi: 10.1111/j.1752-8062.2010.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li D, Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Partain J, Nixon RR, Allen CN, Irwin RP, Jakobs PM, Litt M, Hershberger RE. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet. 2006;79:1030–1039. doi: 10.1086/509900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008;156:161–169. doi: 10.1016/j.ahj.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ku CS, Naidoo N, Pawitan Y. Revisiting Mendelian disorders through exome sequencing. Hum Genet. 2011;129:351–370. doi: 10.1007/s00439-011-0964-2. [DOI] [PubMed] [Google Scholar]
- 15.Villard E, Perret C, Gary F, Proust C, Dilanian G, Hengstenberg C, Ruppert V, Arbustini E, Wichter T, Germain M, Dubourg O, Tavazzi L, Aumont MC, DeGroote P, Fauchier L, Trochu JN, Gibelin P, Aupetit JF, Stark K, Erdmann J, Hetzer R, Roberts AM, Barton PJ, Regitz-Zagrosek V, Aslam U, Duboscq-Bidot L, Meyborg M, Maisch B, Madeira H, Waldenstrom A, Galve E, Cleland JG, Dorent R, Roizes G, Zeller T, Blankenberg S, Goodall AH, Cook S, Tregouet DA, Tiret L, Isnard R, Komajda M, Charron P, Cambien F. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur Heart J. 2011;32:1065–1076. doi: 10.1093/eurheartj/ehr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, Kent WJ, Miller W, Haussler D. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper GM, Goode DL, Ng SB, Sidow A, Bamshad MJ, Shendure J, Nickerson DA. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat Methods. 2010;7:250–251. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 19.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol. 2009;54:930–941. doi: 10.1016/j.jacc.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, Sinagra G, Lin JH, Vu TM, Zhou Q, Bowles KR, Di Lenarda A, Schimmenti L, Fox M, Chrisco MA, Murphy RT, McKenna W, Elliott P, Bowles NE, Chen J, Valle G, Towbin JA. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42:2014–2027. doi: 10.1016/j.jacc.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 22.Duboscq-Bidot L, Xu P, Charron P, Neyroud N, Dilanian G, Millaire A, Bors V, Komajda M, Villard E. Mutations in the Z-band protein myopalladin gene and idiopathic dilated cardiomyopathy. Cardiovasc Res. 2008;77:118–125. doi: 10.1093/cvr/cvm015. [DOI] [PubMed] [Google Scholar]
- 23.Hershberger RE, Pinto JR, Parks SB, Kushner JD, Li D, Ludwigsen S, Cowan J, Morales A, Parvatiyar MS, Potter JD. Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ Cardiovasc Genet. 2009;2:306–313. doi: 10.1161/CIRCGENETICS.108.846733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inagaki N, Hayashi T, Arimura T, Koga Y, Takahashi M, Shibata H, Teraoka K, Chikamori T, Yamashina A, Kimura A. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem Biophys Res Commun. 2006;342:379–386. doi: 10.1016/j.bbrc.2006.01.154. [DOI] [PubMed] [Google Scholar]
- 25.Knoll R, Kostin S, Klede S, Savvatis K, Klinge L, Stehle I, Gunkel S, Kotter S, Babicz K, Sohns M, Miocic S, Didie M, Knoll G, Zimmermann WH, Thelen P, Bickeboller H, Maier LS, Schaper W, Schaper J, Kraft T, Tschope C, Linke WA, Chien KR. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res. 2010;106:695–704. doi: 10.1161/CIRCRESAHA.109.206243. [DOI] [PubMed] [Google Scholar]
- 26.Mohapatra B, Jimenez S, Lin JH, Bowles KR, Coveler KJ, Marx JG, Chrisco MA, Murphy RT, Lurie PR, Schwartz RJ, Elliott PM, Vatta M, McKenna W, Towbin JA, Bowles NE. Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol Genet Metab. 2003;80:207–215. doi: 10.1016/s1096-7192(03)00142-2. [DOI] [PubMed] [Google Scholar]
- 27.Olson TM, Illenberger S, Kishimoto NY, Huttelmaier S, Keating MT, Jockusch BM. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation. 2002;105:431–437. doi: 10.1161/hc0402.102930. [DOI] [PubMed] [Google Scholar]
- 28.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor MR, Slavov D, Gajewski A, Vlcek S, Ku L, Fain PR, Carniel E, Di Lenarda A, Sinagra G, Boucek MM, Cavanaugh J, Graw SL, Ruegg P, Feiger J, Zhu X, Ferguson DA, Bristow MR, Gotzmann J, Foisner R, Mestroni L. Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat. 2005;26:566–574. doi: 10.1002/humu.20250. [DOI] [PubMed] [Google Scholar]
- 30.Taylor MR, Slavov D, Ku L, Di Lenarda A, Sinagra G, Carniel E, Haubold K, Boucek MM, Ferguson D, Graw SL, Zhu X, Cavanaugh J, Sucharov CC, Long CS, Bristow MR, Lavori P, Mestroni L. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation. 2007;115:1244–1251. doi: 10.1161/CIRCULATIONAHA.106.646778. [DOI] [PubMed] [Google Scholar]
- 31.Cheng J, Morales A, Siegfried JD, Li D, Norton N, Song J, Gonzalez-Quintana J, Makielski JC, Hershberger RE. SCN5A rare variants in familial dilated cardiomyopathy decrease peak sodium current depending on the common polymorphism H558R and common splice variant Q1077del. Clin Transl Sci. 2010;3:287–294. doi: 10.1111/j.1752-8062.2010.00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O'Cochlain F, Gao F, Karger AB, Ballew JD, Hodgson DM, Zingman LV, Pang YP, Alekseev AE, Terzic A. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36:382–387. doi: 10.1038/ng1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta P, Bilinska ZT, Sylvius N, Boudreau E, Veinot JP, Labib S, Bolongo PM, Hamza A, Jackson T, Ploski R, Walski M, Grzybowski J, Walczak E, Religa G, Fidzianska A, Tesson F. Genetic and ultrastructural studies in dilated cardiomyopathy patients: a large deletion in the lamin A/C gene is associated with cardiomyocyte nuclear envelope disruption. Basic Res Cardiol. 2010;105:365–377. doi: 10.1007/s00395-010-0085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Selcen D, Muntoni F, Burton BK, Pegoraro E, Sewry C, Bite AV, Engel AG. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009;65:83–89. doi: 10.1002/ana.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knoll R, Hoshijima M, Hoffman HM, Person V, Lorenzen-Schmidt I, Bang ML, Hayashi T, Shiga N, Yasukawa H, Schaper W, McKenna W, Yokoyama M, Schork NJ, Omens JH, McCulloch AD, Kimura A, Gregorio CC, Poller W, Schaper J, Schultheiss HP, Chien KR. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111:943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 36.Makielski JC, Ye B, Valdivia CR, Pagel MD, Pu J, Tester DJ, Ackerman MJ. A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ Res. 2003;93:821–828. doi: 10.1161/01.RES.0000096652.14509.96. [DOI] [PubMed] [Google Scholar]
- 37.Imboden M, Swan H, Denjoy I, Van Langen IM, Latinen-Forsblom PJ, Napolitano C, Fressart V, Breithardt G, Berthet M, Priori S, Hainque B, Wilde AA, Schulze-Bahr E, Feingold J, Guicheney P. N Engl J Med. 2006;355:2744–2751. doi: 10.1056/NEJMoa042786. [DOI] [PubMed] [Google Scholar]
- 38.Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010;12:655–667. doi: 10.1097/GIM.0b013e3181f2481f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Venkatraman G, Harada K, Gomes AV, Kerrick WG, Potter JD. Different functional properties of troponin T mutants that cause dilated cardiomyopathy. J Biol Chem. 2003;278:41670–41676. doi: 10.1074/jbc.M302148200. [DOI] [PubMed] [Google Scholar]
- 40.Duboscq-Bidot L, Charron P, Ruppert V, Fauchier L, Richter A, Tavazzi L, Arbustini E, Wichter T, Maisch B, Komajda M, Isnard R, Villard E. Mutations in the ANKRD1 gene encoding CARP are responsible for human dilated cardiomyopathy. Eur Heart J. 2009;17:2128–2136. doi: 10.1093/eurheartj/ehp225. [DOI] [PubMed] [Google Scholar]
- 41.Moller DV, Andersen PS, Hedley P, Ersboll MK, Bundgaard H, Moolman-Smook J, Christiansen M, Kober L. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur J Hum Genet. 2009;17:1241–1249. doi: 10.1038/ejhg.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carniel E, Taylor MR, Sinagra G, Di Lenarda A, Ku L, Fain PR, Boucek MM, Cavanaugh J, Miocic S, Slavov D, Graw SL, Feiger J, Zhu XZ, Dao D, Ferguson DA, Bristow MR, Mestroni L. Alpha-myosin heavy chain: a sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation. 2005;112:54–59. doi: 10.1161/CIRCULATIONAHA.104.507699. [DOI] [PubMed] [Google Scholar]
- 43.Gui J, Wang T, Jones RP, Trump D, Zimmer T, Lei M. Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS One. 2010;5:e10985. doi: 10.1371/journal.pone.0010985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsubata S, Bowles KR, Vatta M, Zintz C, Titus J, Muhonen L, Bowles NE, Towbin JA. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest. 2000;106:655–662. doi: 10.1172/JCI9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harada K, Potter JD. Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin T result in different effects on the pH and Ca2+ sensitivity of cardiac muscle contraction. J Biol Chem. 2004;279:14488–14495. doi: 10.1074/jbc.M309355200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.