

Abstract
Calcium is an important signaling molecule involved in the regulation of many cellular functions. The large free energy in the Ca2+ ion membrane gradients make Ca2+ signaling inherently sensitive to the available cellular free energy, primarily in the form of ATP. In addition, Ca2+ regulates many cellular ATP consuming reactions such as muscle contraction, exocytosis, biosynthesis and neuronal signaling. Thus, Ca2+ becomes a logical candidate as a signaling molecule to modulate ATP hydrolysis and synthesis during changes in numerous forms of cellular work. Mitochondria are the primary source of aerobic energy production in mammalian cells and also maintain a large Ca2+ gradient across their inner membrane providing a signaling potential for this molecule. The demonstrated link between cytosolic and mitochondrial [Ca2+], identification of transport mechanisms as well as proximity of mitochondria to Ca2+ release sites further supports the notion that Ca2+ can be an important signaling molecule in the energy metabolism interplay of the cytosol with the mitochondria. Here we review sites within the mitochondria where Ca2+ plays a role in the regulation of ATP generation and potentially contributes to the orchestration of the cellular metabolic homeostasis. Early work on isolated enzymes pointed to several matrix dehydrogenases that are stimulated by Ca2+, which were confirmed in the intact mitochondrion as well as cellular and in vivo systems. However, studies in these intact systems suggested a more expansive influence of Ca2+ on mitochondrial energy conversion. Numerous non-invasive approaches monitoring NADH, mitochondrial membrane potential, oxygen consumption and workloads suggest significant Ca2+ effects on other elements of NADH generation as well as downstream elements of oxidative phosphorylation including the F1FO-ATPase and the cytochrome chain. These other potential elements of Ca2+ modification of mitochondrial energy conversion will be the focus of this review. Though most of specific molecular mechanisms have yet to be elucidated, it is clear that Ca2+ provides a balanced activation of mitochondrial energy metabolism which exceeds the alteration of dehydrogenases alone.
Introduction
Calcium plays a central role in cell signaling at numerous levels. The remarkably high potential energy in the gradient of Ca2+ between the cytosol, extracellular space and specialized cellular compartments (ΔGCa) approaches the ATP free energy (ΔGATP) available for cellular work(1-3) with ΔGCa and ΔGATP linked together through the Ca2+ATPase and ion transport processes. The large ΔGCa can quickly change the local [Ca2+] several fold in milliseconds with changes in membrane resistance. The cell uses this rapid and very high gain system as an important trigger in regulating many differentiated cell processes that consume ATP, such as muscle contraction, exocytosis, neuronal transmission and cellular motility. As such, linking Ca2+ concentration to the production of ATP would make a logical feed-forward or parallel control network to maintain ΔGATP during the activation of these processes, but also to support ΔGCa required for the Ca2+ signaling used to trigger these processes.
Supporting this notion, it has been appreciated for many years that that the cell has a remarkable ability to match the rate of ATP production and utilization with little or no change in metabolic intermediates including ADP and Pi (4;5). This phenomenon of constant [ATP] and ΔGATP during work transitions, especially in heart and muscle, has been termed a metabolic homeostasis (6-8) as it is implied that the metabolic regulation of the cell strives to maintain ΔGATP constant at the time it needs it most, during increases in workload. Ca2+ has been proposed as a key element in the feed-forward or parallel system for the production of mitochondrial ATP and maintenance of metabolic homeostasis in tissues due to its dual role in activating ATPase activity as well as metabolism (6;9-14). In this review, we will focus on the role of Ca2+ in modulating the mitochondrial energy conversion processes as it relates to the maintenance of cellular metabolic homeostasis.
The mitochondrion plays a critical role in cellular energy conversion of the differentiated cell by generating ATP from reduced carbon substrates. The mitochondrial membrane maintains a very large ΔGCa, however, unlike the endoplasmic reticulum or plasma membrane, this potential energy is largely supplied by the -180 to -200 mV membrane potential (ΔΨ) across the inner membrane in addition to a Ca2+ concentration gradient. The large ΔGCa across the mitochondrial inner membrane is consistent with a role for Ca2+ in signaling across this membrane. In addition, the volume of the mitochondrial matrix is very small, on the order of 1 to 15% of the cell cytosol, effectively amplifying the movement of few Ca2+ molecules into large concentration changes in the matrix space. Thus, the combination of a large ΔGCa across the inner membrane and a small volume results in a very high gain signaling potential for Ca2+ in the mitochondrial matrix.
While very controversial for many years, numerous genetically generated optical probes, as well as exogenous probes, have demonstrated that the mitochondrial Ca2+ concentration (Ca2+m) responds to rapid changes in gross or regional changes in cytosolic Ca2+ (Ca2+c), implying a dynamic import and export system for Ca2+(15-20). The contributions by Pozzan and Rizzuto using genetically coded probes for different cellular domains (18;21-23) cannot be minimized in demonstrating the role of micro-domains of Ca2+ in overcoming some of the kinetic limitations of Ca2+ signaling in mitochondria. These local regions of high Ca2+ release near mitochondria add a cellular geometry aspect to mitochondrial metabolic regulation. Finally, the transporters for Ca2+ across the mitochondrial membrane have just recently been identified (24;25) almost 50 years after the description of Ca2+ uptake by mitochondria(26). The Ca2+ uniporter system is apparently a protein complex (27) that will likely have a wide variety of regulatory mechanisms for changing its conductance. The export system in heart and brain is believed to be primarily an exchange mechanism with Na+, while in liver and kidney, Na+-independent efflux is dominant(28). However, the large discrepancy between reported influx and efflux Vmax values suggests other mechanisms may also be at play (28). For the purposes of this review, we will assume that Ca2+c influences Ca2+m, though the temporal fidelity of this relationship may vary (see (29)).
Given that Ca2+ signaling occurs across the mitochondrial membrane, we will concentrate on the identified and potential targets for Ca2+ signaling within the mitochondrion that influence the energy conversion processes.
Isolated Enzyme Studies
One of the classic systems of metabolic regulation is phosphorylation of mitochondrial pyruvate dehydrogenase (PDH) originally described by Linn et al. who, in a series of papers in 1969 using 32P labeling and activity measurements (30;31), identified a protein kinase (PDHK) and phosphatase activity (PDHP) within the PDH complex. Soon thereafter, Randle’s lab demonstrated that insulin can modulate the phosphorylation of PDH in fat cells (32;33) providing the first evidence that this was a regulated process. In very short order, Denton et al. (34) demonstrated that PDHP was Ca2+ sensitive and that physiological levels of Ca2+ can increase PDH activity by enhancing de-phosphorylation. Similar results were also found by Siess and Wieland (35). These demonstrations were the first direct linkage of Ca2+ to the activity of a mitochondrial energy conversion enzyme.
The PDH complex is a huge ~10 megadalton molecular machine that has three catalytic components, i.e. pyruvate dehydrogenase (E1), dihydrolipoyl transacetylase (E2), and dihydrolipoamide dehydrogenase (E3), a core protein E3 binding protein (E3BP), and two regulatory components, pyruvate dehydrogenase kinase (PDHK) and pyruvate dehydrogenase phosphatase (PDHP). The regulatory elements are minor constituents of the complex (~10%). There are 4 isoforms of PDHK that are tightly bound and two weakly bound isoforms of PDHP(36). The regulatory phosphorylation sites on the E1 α chain are Ser-292, Ser-299 and Ser-231(all amino acid positions are referenced to the porcine; each mammalian species has slight shifts in position) (37;38). Though there is some controversy on the relative effectiveness of these sites, the Ser-292 is apparently the most potent inhibitory site followed by Ser-299 and Ser-231. These phosphorylation sites have recently been confirmed by mass spectroscopy (39;40) while Gnad et al. (40) also identified Ser-294 and Thr-230 as sites. Though the activity of the PDHK is under numerous different controls including transcriptional control of isoforms in the tissue, attenuation by ADP, NAD+, and pyruvate, and activation by ATP, NADH and acetyl-CoA via the reduction and acetylation of the lipoyl groups of E2 (for review see(41)), there is no direct link of the PDHK isoforms activity with Ca2+. The activation of PDH via Ca2+ is apparently solely dependent on dephosphorylation of the E1 subunit by the activation of PDHP isoform 1 (PDP-1). The PDP-1 Ca2+ -binding domain enhances its association with the E2 subunit increasing phosphatase activity (42-44). PDHP-2 is the primary isoform in liver and adipose tissue (36) and is activated by polyamines but not Ca2+ {Damuni, 1984 DAMUNI1984 /id;Pezzato, 2009 PEZZATO2009 /id;Huang, 1998 HUANG1998 /id}.
McCormack and Denton screened most of the citric acid cycle enzymes of the porcine heart in 1979 (47) searching for other interactions sites of Ca2+. They found no effect of Ca2+ on aconitase, glutamate dehydrogenase, malate dehydrogenase, NADP-isocitrate dehydrogenase, succinate dehydrogenase and the non-phosphorylated form of PDH. Note that PDH was already in its active form in this screen. A modest increase in the citrate synthase affinity for oxaloacetate was detected but likely not significant in the presence of physiological Mg2+. However, a very large increase in the affinity of the α-ketoglutarate dehydrogenase complex (αKDH, also known as oxoglutarate dehydrogenase) for α–ketoglutarate with no change in Vmax was observed and confirmed by later studies in bovine kidney (48). Conversely, inhibition of αKDH by high concentrations of Ca2+ has been reported in the brain (49). The interaction of Ca2+ with ADP, ATP, NADH, and Pi levels can alter the rate of αKDH by over 100–fold, in vitro (48;50). However, while αKDH is controlled and regulated by substrates and cofactors (see review (51)), the precise mechanisms of Ca2+ modulation of αKDH activity are not known.
Calcium and ADP increase the substrate affinity for NAD+ linked isocitrate dehydrogenase (ICDH) with no change in Vmax(52). The mechanism for this process is also unknown. A sophisticated ICDH serine phosphatase/kinase system is present in Escherichia coli (see review (53)), however, no homologue in eukaryotic systems has been detected. Mitochondrial glycerol 3-phosphate dehydrogenase (G3PDH), which is highly active in fast-twitch muscle fibers and pancreatic β-cells, is also activated by Ca2+ through an increased affinity for glycerol 3-phosphate but no change in Vmax(54). Ca2+ binding to two EF-hand motifs close to the carboxy terminus of the enzyme is thought to be responsible for the Ca2+ activation of G3PDH ((55;56)).
Another important element in the interaction of Ca2+ with metabolic events is the impact on substrate transport (see review (56). Of the Ca2+ sensitive transporters, the aspartate/glutamate exchangers, citrin and aralar, are the best characterized (57-59) along with the ATP-Mg/Pi carrier (sCaMC)(60;61). These transporters belong to the carrier family with EF-hand Ca2+-binding motifs on the external membrane N-terminal domains. Through these mechanisms, Ca2+c can directly impact the transport of redox elements, the net ATP content of the matrix, as well as potentially impact cellular signaling through cytosolic amino acid levels (62;63). It has been proposed that the activation of sCaMC may be involved in the regulation of ATP synthesis with work in the heart (56), however, how an increase in ATP influx into the matrix would support ATP production is unclear other than increasing the net concentration of adenylates and perhaps improving the kinetic driving force for Complex V.
As discussed above, the actual mechanisms for Ca2+ modulation of the mitochondrial enzymes of energy conversion are not extensively illuminated as discussed over a decade ago by McCormack et al (12). The lone exception is the well characterized PDH system, where the tightly associated kinase and weakly associated Ca2+ phosphatase result in an effective Vmax regulatory mechanism, in vitro. A Vmax regulatory mechanism is much more likely to have an impact in the intact cell since it will generally not be dependent on substrate concentrations and is the result of an amplification of the Ca2+ signal through a post-translational modification (PTM) system. In contrast, the substrate affinity effects of Ca2+ on ICDH and αKDH are not well characterized, and if these effects exist in the matrix, they will require the substrate concentrations to be well below the Vmax values to have any impact. Secondly, these affinity effects are not persistent and only occur with direct association of Ca2+, again limiting the amplification of the Ca2+ signal when compared to a PTM. An excellent review on the current understanding of the mechanisms and phenomenology of Ca2+ activation of these enzymes was recently provided by Denton (64).
The direct Ca2+ effects on PDH, NAD-IDH, αKDH and even G3PDH suggest that Ca2+ provides a large stimulus for carbohydrate oxidation. However, there is little evidence for direct Ca2+ activation of enzymes from the ketone and fatty acid oxidation pathways which supports the energy conversion of many tissues, most notably the heart. As will be seen in the discussion of intact systems, the stimulation of NAD(P)H generation in intact systems such as heart and substrate dependent effects on isolated mitochondria suggests that Ca2+ dependent mechanisms of NADH generation from fatty acid oxidation are also likely in play but not elucidated at this time.
It is interesting to consider that if the PDP-1 phosphatase had been a bit weaker associated with the PDH complex, the effect of Ca2+ on PDH in vitro would have been missed completely. In addition, the phosphorylation of PDH is persistent (65) through isolation also permitting in vitro analysis of events occurring in vivo. Due to the prevalence of protein complexes in the mitochondria matrix and the potential association of other PTM generating systems in these complexes, it is possible that many interactions have been missed due to weak associations or ill defined PTM. In addition, the matrix conformation of proteins may result in different Ca2+ interactions, in vivo, than in isolated enzymes. Thus, the evaluation of the effects of Ca2+ on intact mitochondrion might yield more interaction sites than a screen of isolated enzymes.
Isolated Mitochondria
Is PDH regulated by phosphorylation in intact mitochondria and does Ca2+ maintain its regulatory effect? For monitoring mitochondrial protein phosphorylation turnover, inorganic 32P can be added to energized mitochondria and converted into γ32P-ATP via oxidative phosphorylation as well as β32P-ADP by adenylate kinase (for methods overview see (66)). The initial 32P incorporation studies from Randle’s lab (37;67) in intact mitochondria demonstrated that the PDH E1 phosphorylation sites are dynamically modulated, responded to pyruvate and correlated with extracted PDH activity. They also demonstrated that the PDH phosphatase was under Ca2+ control in intact adipose mitochondria in 1974(68) These results have been repeated using 32P(69) and proteomic approaches(70) as well as using quantitative mass spectrometry(39) on the E1 active sites in isolated heart and liver mitochondria. In an important series of papers by McCormack and Denton (10;71;72), the observations of Ca2+ activity in isolated proteins were validated in intact heart and adipose mitochondria. These studies demonstrated that ~50 nM free Ca2+ in the extra-mitochondrial space of heart mitochondria enhanced the apparent affinity of α-ketoglutarate that was sensitive to Na+, Mg2+ and ruthenium red (RuRed), an inhibitor of Ca2+ entry across the inner membrane (73). No effect of Ca2+ was detected on the maximum coupled respiratory rate in these studies. These observations led to the notion that Ca2+ could serve as a regulator of oxidative phosphorylation independent of the classical role of ADP, ATP, Pi and NADH. This was summarized by Denton and McCormack in 1980 as “….changes in cytoplasmic and intramitochondrial Ca2+ may be a rather general means whereby stimulation of the supply of reducing equivalents for respiration can be achieved with minimum increases in the NAD+/NADH and ADP/ATP ratios.” (10). This was one of the first discussions of the role of Ca2+m signaling in the regulation of metabolic homeostasis in the cell independent of the substrates of energy conversion.
Building on this work, Moreno-Sanchez (74;75) and Moreno-Sanchez and Hansford ((76-78) determined the effects of extra-mitochondrial Ca2+ on oxidative phosphorylation using a variety of methods. Figure 1:Panel A shows the dose dependent increase in the maximum velocity of liver mitochondria oxidizing succinate despite the lack of a previously identified Ca2+-sensitive dehydrogenase linked to succinate oxidation. Indeed, the effect was not diminished in the presence of rotenone where NAD(P)H oxidation is completely. In this discussion we will refer to fluorescence studies on NADPH and NADH as NAD(P)H where the origins of the fluorescence signal has not been fully characterized (liver and most other tissues) but as NADH in the heart where the NAD(P)H contribution is believed to be minimal(79). These results were expanded by Murphy et al(80) showing that the oxidation of durohydroquinone was activated by Ca2+ suggesting an effect within Complex III. No effect on Complex IV was detected by Murphy et al. It should be noted that Johnston and Brand (81)found no effect of Ca2+ on succinate oxidation in rat liver mitochondria, however, the incubation and preparation conditions were highly varied amongst these early studies. Another series of studies follows the strategy of Koretsky and Balaban (82) in using the slope of the linear mitochondrial NADH versus respiratory rate relationship to isolate the effects of [NADH] on oxidative phosphorylation. In these studies, the effect of increasing NADH alone, via dehydrogenases, can be determined by simply titrating the concentration of carbon substrates for a given mitochondrial dehydrogenase (82). Intriguingly, Moreno-Sanchez et al. found that the [NADH] to respiration relationship had a steeper slope and was shifted to the left (i.e. higher rates at lower [NADH]) with the addition of Ca2+ (Figure 1:Panle B) (83). These data implied that the effect of Ca2+ cannot be ascribed to changes in NADH from alterations in the original Ca2+ dehydrogenases alone and other elements should be considered.
Figure 1.
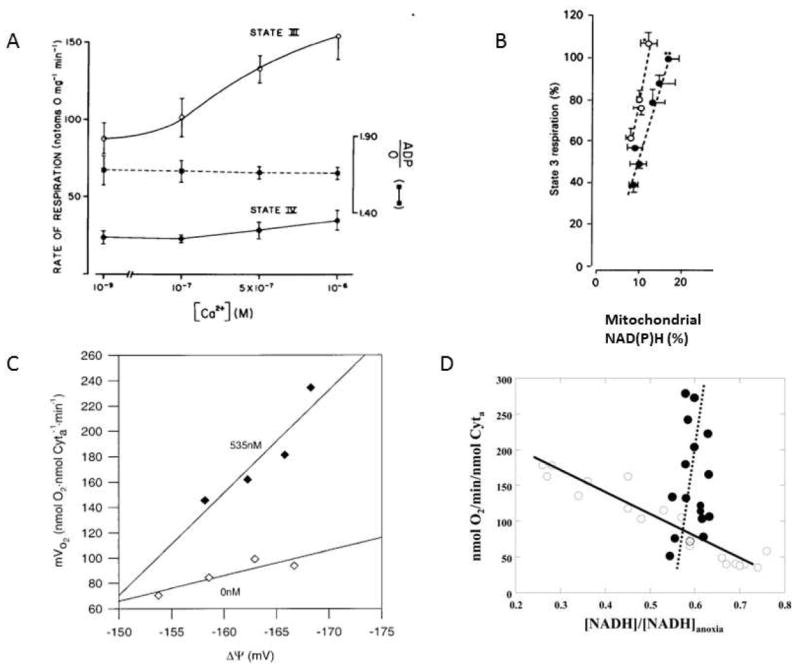
A) Effect of extra-mitochondrial Ca2+ on the rate of respiration in isolated rat liver mitochondria respiring on 5 mM succinate. A 5 minute pre-incubation with Ca2+ was used before driving State 3 respiration with 250 μM ADP(74). Reproduced with permission. B) Effect of extramitochondrial Ca2+ on the relationship between [NAD(P)H] and respiration in isolated liver mitochondria. All experiments were conducted with 5 mM malate. Solid points represent the control study with effectively no Ca2+ and mitochondrial [NAD(P)] increased with increasing glutamate (0.25, 0.5, 1, 2, 5 and 10 mM). The open circles are with fixed substrates 1 mM glutamate and 5 mM malate with the extramitochondrial Ca2+ varied from 5 nM (initial value), 66, 130, 225 and 400 nM (adapted from(83) reproduced with permission) C) Mitochondrial membrane potential versus oxygen consumption of State-3 respiration with variable substrates in the presence and absence of Ca2+in porcine heart mitochondria. In all experiments only the glutamate /malate total concentration was varied from 0.5-5.0 mM to generate a difference in driving force from the citric acid cycle. The open symbols are in the nominal absence of Ca2+. The filled symbols are in the presence of the previously determined optimal Ca2+ concentration of 535 nM. Data from (87), reproduced with permission. D) Relationship between [NADH] and oxygen consumption with apyrase or a sarcoplasmic reticulum(SR)-mitochondria reconstitution system Open symbols are data collected by varying [Apyrase]. Increasing [Apyrase] increased respiration but decreased [NADH]. Closed Signals are fixed [SR] and [mitochondria] (ratio of SR to mitochondria 0.5) with varying [Ca2+] over low physiological levels of 0 to 492 nM (calculated free concentration). Increasing [Ca2+] increased respiration over 5 fold with a slight increase in [NADH]. Lines are the linear regression of the data points. Data from (92), reproduced with permission.
These initial results and subsequent reports suggested that Ca2+ might be doing much more in the regulation of oxidative phosphorylation than just modifying dehydrogenase activity including the alteration of the adenine nucleotide translocase (ANT)(75), the F1-F0-ATPase (Complex V)(84), the cytochrome chain (80)or other intermediary metabolism enzymes. It is also important to note that the concentration range of these studies represents the range we believe that normal physiological Ca2+ signaling occurs (0.01 to 1 μM). Many earlier studies on isolated mitochondria focused on concentrations exceeding 50 μM such as the convincing demonstration of the inhibition of Complex V ATPase activity (85) as well as the early demonstrations of an activation of ANT by external Ca2+ that was shown not to occur at physiological [Ca2+](86). Many of these early studies with high μM Ca concentrations may provide important information on Ca2+ effects during patho-physiological conditions but likely play little role under normal physiological conditions.
Territo et al (87) expanded the approach of titrating substrates and Ca2+ (82;83) and applied it to both [NADH] as well as the mitochondrial membrane potential (ΔΨ) in porcine heart mitochondria. ΔΨ is of primary interest since it is the major driving force for ATP production by Complex V and the relationship between the free energy from NADH (ΔGNADH) and ΔΨ reveals information on the efficiency of the cytochrome chain in converting ΔGNADH to ΔΨ. An example from this study is presented in Figure 1 Panel C for a glutamate + malate dose response curve in the presence and absence of Ca2+. What is shown here is a current (i.e. oxygen consumption) versus voltage (ΔΨ) plot of oxidative phosphorylation. The slope of this relationship is proportional to the resistance of the system for converting a given voltage into a current flux down the cytochrome chain. In the absence of a considerable leak pathway, this must also reflect the flux of protons across Complex V, as this is the major source of proton re-entry into mitochondria under State 3 conditions. As seen in Figure 1 Panel C, the effective resistance of Complex V in the membrane was remarkably dependent on [Ca2+]. Under low Ca2+ conditions, mitochondria were capable of generating very high State 3 ΔΨ, however; ΔΨ could not be used to generate ATP. These data suggest that dehydrogenase reducing equivalent supply to the cytochrome chain was more than adequate to generate ΔΨ; only this energy could not be used by Complex V in the absence of Ca2+. Similar observations were made with all substrates used, including succinate where NADH is not utilized. Thus, Ca2+ activated oxidative phosphorylation independent of dehydrogenase activity as suggested by the earlier studies. Data with arsenate as a substrate to isolate ANT(75) suggested that the majority of this non-dehydrogenase effect in heart mitochondria was due to an activation of Complex V by Ca2+ (87), and, as previously demonstrated by Beis and Newshome (86), there was no Ca2+ effect on ANT.
Another important finding from this study was that the linear dependence of respiration on ΔΨ had a maximum slope (in the presence of Ca2+) of only ~9 nmoles O2 /min/nmole cyto a/mV ΔΨ. This is an interesting number since it provides the relationship of ΔΨ versus ATP production by Complex V at Vmax (maximum [ADP] and [Pi]). Note that without calcium (87) or with non-saturating ADP(88), this number is even lower. Taking the maximum oxygen consumption per nmole cyto a in the heart approaching 700 nmoles O2/nmole cyto a/min (89), the normalized rate of respiration only changes ~1.3% /mV ΔΨ. Thus, ΔΨ alone is not a powerful modulator of ATP production rate and will be discussed further below.
Panov and Scaduto (90) also observed an increase in NADH, ΔΨ and respiration rate by adding Ca2+ during the titration of ADP in the presence of saturating carbon substrate. Similar results were also obtained using ATPase additions by Territo et al. (91) in the presence and absence of Ca2+, where the activation of oxidative phosphorylation by Ca2+ was associated with an increase in NADH but a decrease in ΔΨ polarization consistent with an increase in both NADH generation and Complex V activity. Panov and Scaduto also confirmed that Ca2+ increased the Vmax of respiration for the many fuels tested, while the apparent affinity for ADP was unchanged.
Another issue that could be tested in isolated mitochondria is whether the affinity of Ca2+ to stimulate ATPase activity matches the affinity for the generalized activation of oxidative phosphorylation discussed above. This was tested by Balaban et al. (92) for the limited case of a reconstituted system of the Ca2+ ATPase of the sarcoplasmic reticulum (SERCA) and cardiac mitochondria. By reintroducing the isolated sarcoplasmic reticulum (SR) to a suspension of isolated mitochondria of the porcine heart, the effects of Ca2+ on SERCA and mitochondrial oxidative phosphorylation simultaneously can be determined. An example from this study is shown in Figure 1 Panel D with the SR reconstituted system and a control experiment with a non-Ca2+ dependent ATPase. In Figure 1 Panel D, the resistance of the system is examined by plotting the rate of respiration versus the normalized change in [NADH]. Regrettably, mitochondrial ΔΨ could not be measured due the interference from the membrane potential in the active SR vesicles in the system. As seen with the addition of apyrase, a Ca2+ independent ATPase, an increase in respiratory rate was associated with a net decrease in NADH as the rate of utilization increased beyond the capacity for NADH production. However, simply adding Ca2+ to the SR-mitochondria reconstituted system resulted in a larger increase in respiration, >5 fold, with a slight increase in [NADH]. Thus, the affinity for Ca2+ of SERCA and mitochondrial respiration is appropriate to increase respiration over 5 fold with little or no change in driving force evaluated as [NADH]. These data offer strong support that Ca2+ could provide the balanced activation of the mitochondria and ATPase activity in the cell to result in metabolic homeostasis, at least at the level of NADH, observed during increases in cardiac workload.
What is the alteration in Complex V resulting in the change in vitro ATPase activity and increased Vmax of ATP synthesis with Ca2+ in the intact mitochondria? Hopper et al. (70) found a Ca2+ sensitive de-phosphorylation of the γ subunit of Complex V using a phosphoprotein sensitive dye. In Saris’ lab, evidence was found for a proportional increase in the phosphorylation of the c subunit with Ca2+ stimulation of liver mitochondria (93). However, there are no clear mechanisms linking Ca2+ effects in the intact mitochondria and subsequent in vitro activity. What is clear is that, with regard to protein phosphorylation of Complex V, there are many more potential sites that could influence enzymatic activity (39;94-100)however, none have been shown to be Ca2+ sensitive as of yet. It is interesting to note that many of these sites are not detected in different mass spectrometry studies, suggesting tissue, species, tissue preparation, or technology driven differences in the detection. Also possible is that the mole fraction of protein that is phosphorylated is very small making the detection difficult and very dependent on the peptide enhancement schemes (100;101). Due to the variability in detection and continuing growth of the discovered sites, it is unlikely that the full phosphorylation scheme of Complex V, as well as other mitochondrial proteins, is fully described. As such, the precise role of Ca2+ in Complex V phosphorylation, or for other PTMs, is still to be determined.
Cellular Studies
To study the interplay of mitochondrial energy metabolism and Ca2+ it would be ideal to monitor the mitochondrial metabolic status and the Ca2+ concentrations in the matrix and adjacent cytosol during cellular work transitions. Using the enhanced mitochondrial NADH fluorescence (79;102;103) (NADHm), likely originating from binding in Complex I(103), fluorescent probes of mitochondrial membrane potential (104-106) as well as Ca2+c and Ca2+m using various fluorescent indicators (107-109), this ideal situation is now nearly realized and has provided many new insights. Measurement limitations in most microscopy studies include the direct determination of ΔGATP or, more importantly, free ADP and Pi, and the net energetic “current”, oxygen consumption, as these are usually limited to larger samples using 31P NMR and oxygen electrodes, respectively. Both of these parameters are critical in the analysis of energy metabolic state. Another limitation of cellular systems is the lack of true work when compared to in vivo tissues or isolated organ systems, especially muscle cells, which cannot be easily mechanically loaded in culture or isolated cell systems. Indeed, most cells in established or primary culture are likely working at least an order of magnitude lower than in vivo. Thus, interpretation of these experiments needs to be tempered by these limitations especially when dealing with the regulation of energy conversion in the mitochondria which may be working close to resting conditions in many preparations. Exceptions to this work level limitation likely includes neuronal preparations where the metabolic work associated with ion transport in re-polarization may be appropriately simulated or even over-stimulated (see (15)) as well as secretory systems like β cell preparations.
With many cellular systems the addition of Ca2+, via a wide variety of methods, has convincingly demonstrated an activation of NAD(P)H generation by the entry of Ca2+ into the mitochondrial matrix (12). One of the earliest demonstrations of a Ca2+ activation of NAD(P)H generation in suspensions of rat liver cells (110) demonstrated that the respiratory stimulation by glucagon, vasoactive intestinal peptide and antidiuretic hormone is proportional to the increase in NAD(P)H fluorescence and was dependent on extracellular Ca2+ (Figure 2: Panel A). Since respiration, or NAD(P)H oxidation, increased with these hormones, there is no question that the increase in [NAD(P)H] was the result of increased generation of NAD(P)H and not a decrease in oxidation rate. Using a much more sophisticated and integrative approach, Robb-Gaspers et al. (111) measured Ca2+c, Ca2+m, NAD(P)H, ΔΨ, and PDH activity and were able establish some of the cellular mechanisms involved in this phenomenon. The hormone induced increase in [NAD(P)H] was associated with an increase in Ca2+c, but also Ca2+m. Also unique to this study was the demonstration that a small increase in ΔΨ was dependent upon Ca2+m (Figure 2: Panel B). These studies demonstrated that the hormone induced increase in respiration, and ATP hydrolysis is supported, in part, by an increase in ΔΨ as a result of net increase in NAD(P)H, implying that, in liver, the potential energy for performing work was actually increased with these hormones. This group also showed that the activation of PDH alone did not result in the changes in NAD(P)H, suggesting other mechanisms must be in play. The lack of PDH involvement in liver could be due to the dominance of the non-Ca2+ sensitive PDP-2 isoform in liver (36) reducing the sensitivity of PDH activity to Ca2+. Related to this point, it is important to point out that the overall dynamic range of ATP production in the liver is very low, that is only 50% (112) while the heart, skeletal muscle, kidney and brain could have alterations in ATP production of 10 fold or more. Thus, the liver may not have the same dynamic modulation of energy metabolism as other tissues possibly explaining the presence of the Ca2+ insensitive PDP.
Figure 2.
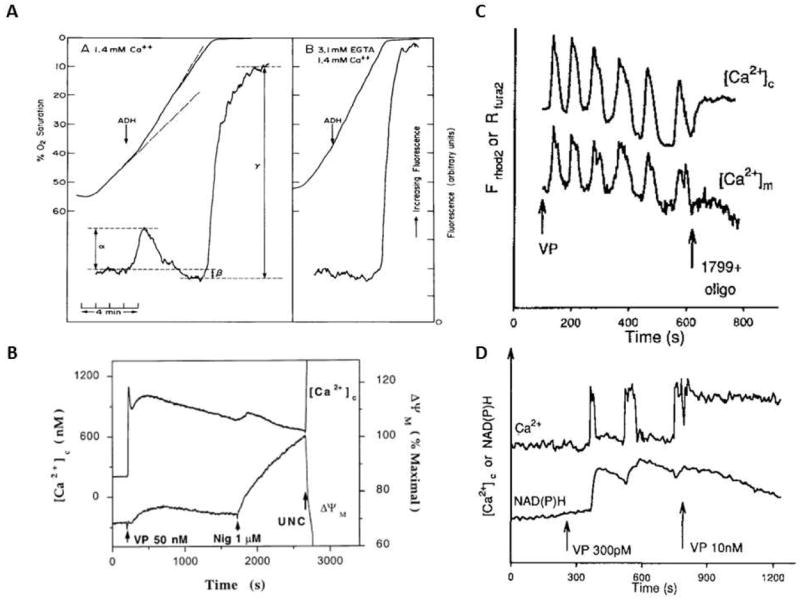
A) Effect of ADH on NADH and oxygen consumption of isolated rat hepatocytes. In both panels 100 nM ADH was added to the chambers at the arrow in the absence (A) and presence (B) of 3.1 mM EGTA. Data from (110), reproduced with permission. B) Measurements of Ca2+c and ΔΨ in cultured hepatocytes with vasopressin (50 nM). [Ca2+]c was measured using fura-2 and ΔΨ was monitored with TMREE. Data from (111), reproduced with permission. C) Temporal correlation of Ca2+c and Ca2+m during hormonal stimulation of hepatocytes. [Ca2+]m was monitored in hepatocytes loaded with dihydro-Rhod 2-AM, and [Ca2+]c was measured in cells loaded with Fura 2-AM. Data from (114), reproduced with permission. D) 4. Relationship between the frequency of Ca2+c and NAD(P)Hm fluorescence. NAD(P)H and [Ca2+]c were measured simultaneously during the addition of vasopressin (VP). Data from (114), reproduced with permission
Pralong et al. (113) and Hajnóczky et al. (114)made important observations that the frequency of changes in [Ca2+c] may encode information for activating metabolism in liver and pancreatic β cells. Hajnóczky et al. showed remarkable temporal fidelity between Ca2+c and Ca2+m during the transient increases in Ca2+ observed upon exposure to hormones (Figure 2: Panel C). However, the NAD(P)H responses were clearly initiated by Ca2+ but had a much longer time constant of recovery when compared to Ca2+c or Ca2+m (Figure 2:Panel D). Is the persistent increase in NAD(P)H observed due to the slow metabolic rate to dissipate a transient increase in NAD(P)H production level, or is it due to a persistent increase in NAD(P)H production after the Ca2+ pulse? We can estimate the NAD(P)H turnover from the initial rate of reduction of NAD(P)H with anoxia or with a metabolic inhibitor. That is, the initial rate of reduction of NAD(P)H should reflect the rate of oxidation during the steady state when the production and oxidation are matched. In these series of experiments, the rate of NAD(P)H reduction at anoxia or with inhibitors in these cells far exceeds the delay in NAD(P)H oxidation after Ca2+ stimulation suggesting that the NAD(P)H turnover is very high in these tissues. Thus, the high turnover of NAD(P)H predict a rapid oxidation of NAD(P)H when Ca2+m is reduced if its production tracked Ca2+m. Clearly, this does not occur and the NAD(P)H levels remain elevated for a long times after the Ca2+m pulse. These data imply that some-type of persistent modification of the NAD(P)H generation system was occurring that could not be explained by the rapid reversible binding of Ca2+ to ICDH and αKDH. This persistent effect suggests that a persistent PTM was generated by Ca2+m in the NAD(P)H generation system. Since liver PDH was ruled out in previous studies, it is likely that other, un-described, persistent enzymatic perturbation associated with NAD(P)H generation are modulated by Ca2+m. One of these PTM in liver could be mitochondrial matrix volume which has been shown to be modified by Ca2+ and alter several aspects of energy metabolism (115). However the source of this persistent NAD(P)H production stimulation effect in the liver is unknown.
As discussed above, numerous single cell studies have demonstrated similar Ca2+ dependent activation of NAD(P)H generation (29;116). Duchen (117) showed that Ca2+ influx during a depolarization in neuronal cells increased NAD(P)Hm and was blocked with RuRed (Figure 3: Panel A). This result was replicated in cardiac cells by Liu and O’Rourke (118) in a comprehensive study where the previously discussed metabolic homeostasis was demonstrated for NADHm in single paced myocytes (Figure 3: Panel B). ATP hydrolysis was increased by 4 Hz pacing that increased Ca2+m but was associated with no change in NADHm. This result suggests that the delivery of reducing equivalents was matched to consumption despite maintaining ΔGNADH implying a matched increase in NADHm generating capacity during the work transition. This effect was dependent on Ca2+m since RuRed disrupted the NADHm homeostasis with pacing. These data confirm the notion that the activation of NADHm generation by Ca2+m is required for the maintenance of metabolic homeostasis.
Figure 3.
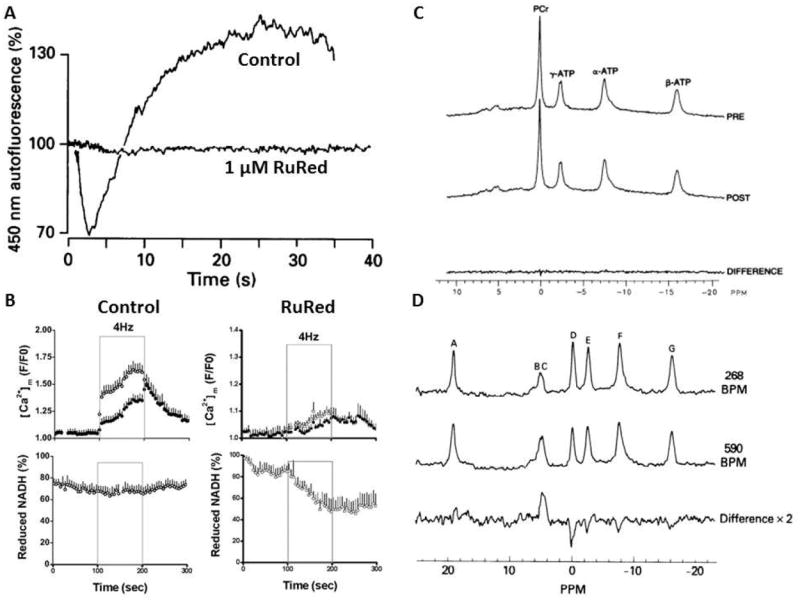
A) Effect of patch clamp depolarization (from -70 mV holding potential to 0 mV) of freshly isolated neurons NAD(P)H fluorescence in the absence and presence of RuRed. RuRed did not block the cellular membrane Ca2+ current associated with the depolarization but blocked the NAD(P)H fluorescence response. Data adapted from(117), reproduced with permission. B) Effect of 4 Hz pacing on isolated cardiac myocytes NADH fluorescence and Ca2+m in the absence (control) and presence RuRed. The pacing of the myocytes occurred at the times indicated. Data from (118) reproduced with permission. C) Effect of pacing work on the 31P NMR spectrum of the intact canine heart. Top spectrum represents the control spectrum while the post spectrum represents the average 31P NMR spectrum during pacing the heart to maximum pacing induced coronary blood flow and oxygen consumption. The bottom trace is the difference of the control and maximum pacing protocol. Data from(141), reproduced with permission. D) Effect of RuRed on the 31P NMR spectrum of the perfused rat heart undergoing an increase in pacing rate. The top spectrum is during the control pacing rate, middle spectrum was collected during the increase in pacing rate to 580 beats/min, bottom spectrum represents the difference between these two conditions with a large increase in Pi and decrease in CrP. The peak assignments are A: external reference, B-C: Pi, D: CrP, E: γP-ATP, F: αP-ATP/NAD and G: βP-ATP. Data from (146), reproduced with permission.
Das and Harris in a series of papers (119-121) found that the extracted ATPase activity of cardiac Complex V was increased with pacing and adrenergic stimulation suggesting that work may also impact the downstream elements in oxidative phosphorylation and not just NADH generation. Similar observations have also been made in the canine heart with dobutamine, in vivo (122). These studies were accomplished with a rapid extraction of sub-mitochondrial particles to assay Complex V ATPase activity; that is Complex V working in “reverse” hydrolyzing ATP. Our own experience confirms the necessity to perform these studies immediately after collecting tissue or after perturbations in mitochondria (122). It is also important to note that these in vitro Complex V assays are done in the presence of high EGTA concentrations making the direct contribution of Ca2+ on the Complex V ATPase activity unlikely(84), despite the fact that a Ca2+ binding site has been found in the Complex V β subunit (123). Indeed, we have found no effect of added Ca2+ on the isolated Complex V ATPase activity in blue native gel assays. Thus, the effect on Complex V by Ca2+ is apparently occurring from indirect effects within the tissue or mitochondria. These studies demonstrate the persistent Ca2+ effect on in vitro ATPase activity, much like what has been observed for PDH activity with increases in workload in heart and muscle (124-126), consistent with a PTM alteration of the F1-ATPase interfering with the normal substrate and product binding, regulatory protein interactions or possible rotation of the gamma subunit occurring in the ATPase reaction, in vitro (84;122). The extrapolation of these ATPase activity measures to the ATP synthetic reaction in the intact mitochondria, in vitro or in vivo, is also tenuous, but clearly reflects persistent changes in the enzyme that are responsive to tissue workload. If Complex V maximum activity is altered by Ca2+ as suggested by these extraction studies and in intact mitochondria, this would provide a direct mechanism for altering ATP production with little or no change in its reactants, ADP and Pi, due to a matched increase in the Vmax of ATP production capacity.
Recently, evidence has been generated that suggest the effect of Ca2+ on NADH generation and mitochondrial function may also be important in the steady state and not just during workload transitions. In several cell lines, Cardenas et al showed that inability to release Ca2+ from the endoplasmic reticulum via inositol trisphosphate receptor Ca2+ release channel revealed a phenotype characteristic of an overall compromised mitochondrial function(14). These novel data are consistent with the notion that Ca2+ modulation of mitochondrial function might be critically important in the maintenance of mitochondrial function in the steady state.
Intact tissues and in vivo measures
The gold standard in establishing a signaling network associated with physiological events is a demonstration of its presence in vivo or in intact tissues performing normal functions. Indeed, it was critical to demonstrate early on that PDH activation occurs in tissues during changes in workload or increases in Ca2+c. This was demonstrated in several intact systems in the early 1980’s including heart (125;127), skeletal muscle (128), brain (129) and liver (130). The activation of PDH with exercise in human muscle has also been well established (131). Again, the effects in liver might be due to parameters other than Ca2+c due to the lack of a Ca2+ sensitive pyruvate dehydrogenase phosphatase isozyme (36) and low energy conversion dynamic range. These early observations were consistent with the activation of PDH in maintenance of energy metabolic homeostasis in these tissues, matching the rate of NADH generation with demand.
The heart has been the major model for these studies due to its large metabolic dynamic range, the ease of measuring work both in intact systems in vitro and in vivo, and its ability to maintain metabolic homeostasis in the face of large alterations in ATP hydrolysis rates. This has been illustrated in numerous studies over the last 50 years with the non-destructive 31P NMR studies (132-135), including novel human data (136;137), providing important confirmation of several decades earlier tissue extractions (138-140). An example from one of these studies is presented in Figure 3, Panel C where the steady state 31P NMR detected ATP metabolites were monitored in a canine heart in vivo using a directly applied surface coil(141). As seen in this example, large increases in ATP turnover with work transitions were not, even transiently(141), associated with a change in the ATP hydrolysis products. Portman et al. (142) demonstrated that the capacity to maintain cardiac metabolic homeostasis may not be fully developed in neonatal hearts, while the hypertrophied heart (143) or post-ischemic heart may also have a disrupted metabolic response to work (144).
The best evidence that the metabolic homeostasis is partially dependent on Ca2+m signaling is, again the effect of RuRed on this process. RuRed disrupts the ability of the perfused heart to maintain ΔGATP with work transitions (145;146). An example is shown in Figure 3, Panel D where the heart is shown not to maintain [CrP] and [Pi] during a pacing experiment in the presence of RuRed. Thus, as for the cellular studies shown in Figure 2 for NADHm levels, RuRed disrupts the ability to a metabolic homeostasis in a perfused heart work transition. As discussed earlier, McCormack et al. (124) demonstrated that RuRed also blocks the activation of PDH during hormonal stimulation of intact rat hearts, again consistent with a role of Ca2+ entry in dehydrogenase activation with work. We have attempted to use RuRed on the heart in vivo using a local infusion into the coronary arteries (for method see (147)) however, we found that the heart could not tolerate the normal doses of RuRed when directly infused in vivo. Thus, a definitive study demonstrating a role for Ca2+ in regulating the metabolic homeostasis in vivo is still lacking.
These studies suggested that metabolic homeostasis exists within the cytosolic ΔGATP with changes in work load, but what is occurring with regard to the free energy within the mitochondria? Several studies have demonstrated that with physiological increases in workload that NADHm remains near nearly constant (140;148;149) as demonstrated in cells under some conditions. In a series of studies, Brandes and Bers (150-153) evaluated the role of Ca2+c and Ca2+m in the regulation of NADHm isolated intact trabeculae during pacing transitions. An excellent temporal correlation between Ca2+m and NADHm was observed (153). However, a clear initial mismatch of NADH generation and production was observed during a work transition both immediately after a pacing increase or decrease. Thus, in this preparation the temporal fidelity of workload and metabolic compensation is poor but the metabolic compensation observed was dependent on alterations in Ca2+m.
It is important to note that if the cardiac workload is extended over normal physiological ranges, the metabolic homeostasis breaks down. Examples include observation of a net NADH oxidation as the ATPase activity is increased from moving an unloaded vented heart to a normal loaded perfused heart in vitro (154) and a net reduction in NADH in the over-paced working heart due to demand ischemia(148). In addition to NADH levels, the cytosolic ΔGATP is also not maintained in these extremes, increasing with KCl arrest (155;156) and decreasing as maximum workloads (133;135). Thus, not surprisingly, metabolic homeostasis only operates effectively over physiological workloads. It is also important to point out that Arai et al. (157) also demonstrated that the redox state of cytochrome c was unchanged in vivo with dobutamine infusions using reflection absorption spectrophotometry consistent with the notion that the redox state of the respiratory chain is also maintained nearly constant, in vivo. Regrettably, fluorescence interference from the visceral pericardium makes NADHm fluorescence measurements from large animal hearts, in vivo, difficult to perform (158).
The measurement of mitochondrial ΔΨ in working intact tissues is difficult to accomplish. However, several investigators have made estimates using a variety of approaches. Wan et al.(159) in the perfused rat heart using 3H tetraphenylphosphonium to estimate ΔΨ found a slight depolarization with increasing workload with unchanged ADP, Pi and NADHm. The authors suggested that the only way to accomplish this is modification of Complex V kinetics since ΔΨ depolarized and its substrates remained constant. This observation and conclusion is similar to the isolated mitochondria studies with Ca2+ and ATPase additions by Territo et al (91). Kauppinen and Hassinen(160) using safranin and Kauppinen(161) using 3H triphenylmethylphosphonium also found small depolarization with pacing in the perfused heart. Piwnica-Worms et al (162) demonstrated that the retention of Tc sestamibi in heart cells is critically dependent on the ΔΨ to the extent that ΔΨ can be estimated using this distribution. Nuclear imaging of Tc-sestamibi has been used extensively in clinical studies to determine cardiac perfusion and viability. In these studies, it is common to collect resting and maximum exercise Tc-sestamibi images. In general, the clinician is looking for the same signal distribution in both conditions, implying that in the normal heart, ΔΨ only slightly decreases with exercise (for example see Verzijlbergen et al. (163) ) Thus, the best evidence available suggests that the mitochondrial ΔΨ depolarizes modestly during increases in workload and associated increases in net Ca2+c and no evidence exists in the heart that ΔΨ is hyperpolarizing and could drive ATP production higher during increases in workload in the heat.
So how is cardiac ATP synthesis balanced with ATP hydrolysis during these dynamic changes in cardiac workload without significant changes in [ADP] and [Pi], the substrates for ATP synthesis? As discussed above, the current consensus is that Ca2+ dependent activation of NADH generation via known and likely unknown, activation of NADH generation pathways is important. Indeed, the demonstrated activation of PDH with workload is consistent with this notion (125;127). However, if NADH alone was driving ATP production higher, then the driving force for Complex V, ΔΨ, would have to be increased sufficiently to drive ATP synthesis at a fixed concentration of ADP, Pi and ΔGATP. How big a change in ΔΨ would have to occur to increase ATP production 3 to 4 fold under these conditions? As discussed above, the relationship between ΔΨ and ATP production is linear over the physiological rates with a slope ~1.3% change in ATP production/mV ΔΨ (87). This suggests that increasing the heart rate by 3 fold would require an additional ~150mV over the resting value of 180 mV taking ΔΨ to an unreasonable value >300 mV. Indeed, most of the current evidence suggests that the ΔΨ decreases with increasing workload, decreasing the driving force for ATP production, not increasing it. Thus, a model that just uses ΔΨ or an activation of the dehydrogenases, to attain a metabolic homeostasis would not work if this linear observation between ΔΨ and ATP production in isolated mitochondria is correct. Clearly the activation of NADH generation by Ca2+ must be coupled to other “downstream” events in oxidative phosphorylation to be effective in maintaining the metabolic homeostasis.
Alternatively, activation of the Complex V Vmax by mitochondrial Ca2+ would permit a higher rate of ATP production at a fixed or reduced ΔΨ and a constant ΔGATP as suggested by several investigators. This is consistent with isolated mitochondria data where Ca2+ increases the maximum rate of ATP production of Complex V and the observation that the maximum velocity of Complex V ATPase activity is nearly proportional to the workload of the hearts it was extracted from(122). Therefore, it is reasonable to propose that activation of the maximum velocity of Complex V for ATP synthesis is part of the Ca2+ activation scheme toward maintenance of metabolic homeostasis. Although this activation of Complex V ATP synthetic activity has been demonstrated in isolated mitochondria with appropriate sensitivity to Ca2+ and has been demonstrated to be persistent in the isolated Complex V, implying a PTM, the precise mechanism of this modification remains unknown. Phillips et al. has suggested this might be due the labile nature of many PTM’s in the bacterial system that might be still in play in the mitochondrial matrix relying on autophosphorylation mechanisms(164).
An activation of NADH generation and ATP synthetic capacity at Complex V by Ca2+ provides a balanced activation of mitochondrial metabolism at the entry of reducing equivalents to the respiratory chain with the re-entry of protons via actual ATP production at Complex V. Both of these effects are generated by apparent changes in the actual kinetics of these reaction systems. However, this simple model still leaves some important gaps in how the respiratory rate is matched to the ATP hydrolysis rate. The classical mechanism for the regulation of the rate of oxygen consumption in the mitochondria is the redox state of Complex IV or cytochrome a, a3 (cyto a). Though still somewhat controversial, the general consensus is that the redox state of cyto a must become more reduced for an increase in respiration to occur and that this process is roughly linear. Simply viewed, the only reactants in the terminal reduction of oxygen are reduced cyto a and oxygen. With saturating oxygen, the only parameter that can regulate the rate of reduction of oxygen, or oxygen consumption, is the redox state of Complex IV or a modification of its kinetics. It has been appreciated since the earliest studies that with increased ATP production in isolated mitochondria via the activation of Complex V, or even increased substrate delivery, Complex IV becomes more reduced, permitting the higher rate of oxygen reduction and removal of reducing equivalents(165;166). Since cyto a is believed to be highly oxidized (~98%) under resting conditions, even the linear relationship between redox state and oxygen consumption results in the very large dynamic range required in many tissues. With cyto a becoming just 10% reduced, a 5-fold or greater increase in respiration can be realized. Thus, as stated above, the fact that respiration increases with workload implies that the redox elements in Complex IV are becoming more reduced or the kinetics of the reaction are modified. Currently, the best hypothesis for how the respiratory rate is modulated is through a net small depolarization of ΔΨ, as observed in most studies to date, increasing the net delivery of reducing equivalents to Complex IV. Recent evidence is suggesting that the kinetics of Complex IV can be altered by PTM as well as different allosteric regulators (167-171), and even though a Ca2+ binding site has been identified (172), no direct evidence is available that Ca2+ alters the kinetics of this enzyme between its redox state and oxygen. Bender and Kadenbach (167) reported that Ca2+ attenuated the inhibition of Complex IV caused by incubating mitochondria with cAMP, though Ca2+ had no effect on Complex IV when added without cAMP or to the isolated enzyme. Due to the importance of Complex IV in the determination of oxidative phosphorylation rate as discussed above, the further study of both direct and indirect effects of Ca2+on the kinetics of this reaction, as well as the relationship between ΔΨ and oxidative phosphorylation in intact systems are warranted.
Another possibility for the observed metabolic homeostasis in intact tissues is the compartmentation of metabolic intermediates in the cytosol much like that demonstrated for Ca2+ (173-175). The basic concept is that regional changes in ADP, Pi and creatine in the regions around the mitochondria are major factors in driving mitochondrial ATP production. While the maintenance of ΔGNADH with workloads is inconsistent with a local delivery of ADP and Pi(8), the slight depolarization of ΔΨ however could be reflecting local increases in these metabolites activating Complex V directly. Currently, direct imaging of the cellular distribution of these metabolites, so important in the analysis of Ca2+ compartmentation, is not feasible. Hopefully, the development of imaging tools capable of determining the local metabolite concentrations within working cells will contribute to a better understanding of the role of cellular metabolite compartmentation in this process.
General Summary
The strong link between ΔGCa and ΔGATP makes Ca2+ a primary candidate as the molecular signal in the maintenance of the cellular metabolic homeostasis. Isolated studies identified several dehydrogenases activated by Ca2+ along with substrate transport mechanisms. Studies on intact mitochondria and cells suggest that Ca2+ stimulates the production of reducing equivalents through these mechanisms as well as other processes in addition to these of these classical sites. The activation of metabolism by Ca2+ in cells is apparently distributed over many elements of oxidative phosphorylation as witnessed by near homeostatic regulation of NADH, ΔΨ and ΔGATP in active tissues that is disrupted by altering mitochondrial Ca2+ transport. We have summarized these processes in a schematic diagram (Figure 4) to emphasize the distribution of Ca2+ regulated sites of oxidative phosphorylation. The best evidence for Ca2+ activation downstream of the dehydrogenases is a Vmax activation of Complex V, but the specific mechanisms remain elusive. Thus, it is evident that Ca2+ is playing a much more integrative role in the regulation of mitochondrial energy metabolism that exceeds a simple activation of dehydrogenases and may include a systemic activation of the metabolic network contributing to the overall metabolic homeostasis of the cell. Fortunately, continuing advances in the proteomics field have already begun to provide specific information on how Ca2+ may alter several types of PTM in oxidative phosphorylation Complexes. It is anticipated that the molecular mechanisms of Ca2+ interaction with these distributed sites in mitochondrial energy metabolism will be forthcoming in the very near future.
Figure 4.
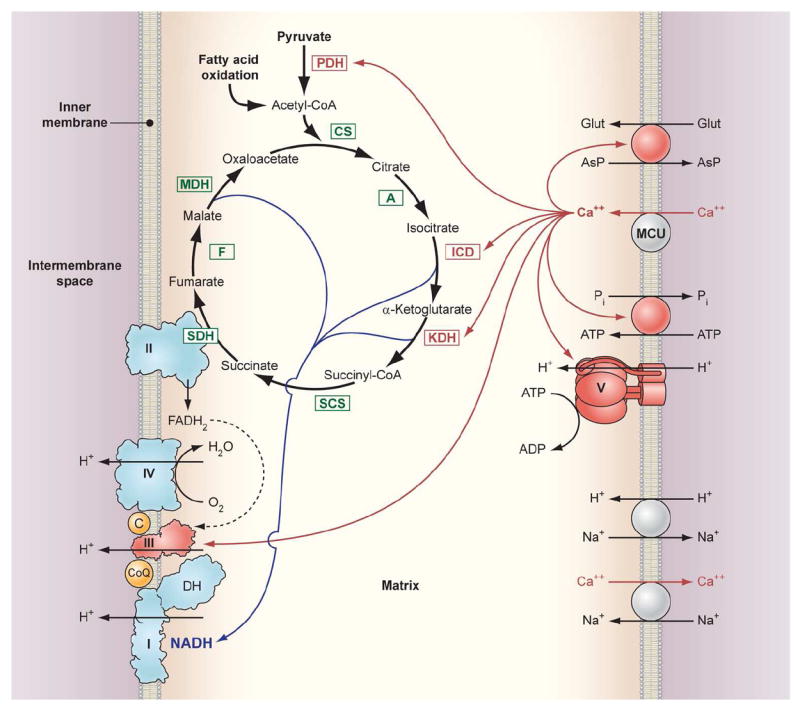
Schematic diagram of the interaction of matrix Ca2+ with processes involved in oxidative phosphorylation. Enzyme abbreviations: PDH: Pyruvate dehydrogenase, CS: Citrate synthase, A: Aconitase, ICD: Isocitrate dehydrogenase, KDH: α ketoglutarate dehydrogenase, SCS: Succinyl CoA synthetase, SDH: Succinate dehydrogenase (also Complex II), F: Fumarase, MDH: Malate dehydrogenase, MCU: Mitochondria calcium uniporter. The Complexes of oxidative phosphorylation are labeled as roman numerals from I-V. The DH on Complex 1 refers to the intrinsic and possibly associated NADH dehydrogenase activity. The red arrows from Ca2+ to the different interaction sites imply either a direct or indirection modulation of the transport or enzymatic activity.
References
- 1.Hasselbach W, Oetliker H. Energetics and electrogenicity of the sarcoplasmic reticulum calcium pump. Annu Rev Physiol. 1983;45:325–339. doi: 10.1146/annurev.ph.45.030183.001545. [DOI] [PubMed] [Google Scholar]
- 2.Chen W, London R, Murphy E, Steenbergen C. Regulation of the Ca2+ gradient across the sarcoplasmic reticulum in perfused rabbit heart. A 19F nuclear magnetic resonance study. Circ Res. 1998;83:898–907. doi: 10.1161/01.res.83.9.898. [DOI] [PubMed] [Google Scholar]
- 3.Tian R, Halow JM, Meyer M, Dillmann WH, Figueredo VM, Ingwall JS, Camacho SA. Thermodynamic limitation for Ca2+ handling contributes to decreased contractile reserve in rat hearts. Am J Physiol. 1998;275:H2064–H2071. doi: 10.1152/ajpheart.1998.275.6.H2064. [DOI] [PubMed] [Google Scholar]
- 4.Hill AV. A challange to biochemists. Biochim Biophys Acta. 1950;4:4–11. doi: 10.1016/0006-3002(50)90003-5. [DOI] [PubMed] [Google Scholar]
- 5.Devin A, Rigoulet M. Mechanisms of mitochondrial response to variations in energy demand in eukaryotic cells. Am J Physiol Cell Physiol. 2007;292:C52–C58. doi: 10.1152/ajpcell.00208.2006. [DOI] [PubMed] [Google Scholar]
- 6.Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol. 2002;34:1259–1271. doi: 10.1006/jmcc.2002.2082. [DOI] [PubMed] [Google Scholar]
- 7.Hochachka PW, McClelland GB. Cellular metabolic homeostasis during large-scale change in ATP turnover rates in muscles. J Exp Biol. 1997;200:381–386. doi: 10.1242/jeb.200.2.381. [DOI] [PubMed] [Google Scholar]
- 8.Balaban RS. Maintenance of the metabolic homeostasis of the heart: developing a systems analysis approach. Ann N Y Acad Sci. 2006;1080:140–153. doi: 10.1196/annals.1380.013. [DOI] [PubMed] [Google Scholar]
- 9.Denton RM, McCormack JG. On the role of the calcium transport cycle in heart and other mammalian imtochondria. FEBS Lett. 1980;119:1–8. doi: 10.1016/0014-5793(80)80986-0. [DOI] [PubMed] [Google Scholar]
- 10.Denton RM, McCormack JG, Edgell NJ. Role of calcium ions in the regulation of intramitochondrial metabolism. Effects of Na+, Mg2+ and ruthenium red on the Ca2+-stimulated oxidation of oxoglutarate and on pyruvate dehydrogenase activity in intact rat heart mitochondria. Biochem J. 1980;190:107–117. doi: 10.1042/bj1900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balaban RS. The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta. 2009;1787:1334–1341. doi: 10.1016/j.bbabio.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 13.Hansford RG. Role of calcium in respiratory control. Med Sci Sports Exerc. 1994;26:44–51. [PubMed] [Google Scholar]
- 14.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duchen MR. Mitochondria and Ca(2+)in cell physiology and pathophysiology. Cell Calcium. 2000;28:339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- 16.Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+ homeostasis in intact cells. J Cell Biol. 1994;126:1183–1194. doi: 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 18.Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, Pozzan T. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 2001;20:4998–5007. doi: 10.1093/emboj/20.17.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trollinger DR, Cascio WE, Lemasters JJ. Mitochondrial calcium transients in adult rabbit cardiac myocytes: inhibition by ruthenium red and artifacts caused by lysosomal loading of Ca(2+)-indicating fluorophores. Biophys J. 2000;79:39–50. doi: 10.1016/S0006-3495(00)76272-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ricken S, Leipziger J, Greger R, Nitschke R. Simultaneous Measurements of Cytosolic and Mitochondrial Ca2+ Transients in HT29 Cells. Journal of Biological Chemistry. 1998;273:34961–34969. doi: 10.1074/jbc.273.52.34961. [DOI] [PubMed] [Google Scholar]
- 21.Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- 22.Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE. 2004;2004:re1. doi: 10.1126/stke.2152004re1. [DOI] [PubMed] [Google Scholar]
- 23.Rutter GA, Burnett P, Rizzuto R, Brini M, Murgia M, Pozzan T, Tavare JM, Denton RM. Subcellular imaging of intramitochondrial Ca2+ with recombinant targeted aequorin: significance for the regulation of pyruvate dehydrogenase activity. Proc Natl Acad Sci U S A. 1996;93:5489–5494. doi: 10.1073/pnas.93.11.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeStefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci USA. 1961;47:1744–1750. doi: 10.1073/pnas.47.11.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim Biophys Acta. 2009;1787:1291–1308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529(Pt 1):57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linn TC, Pettit FH, Reed LJ. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc Natl Acad Sci U S A. 1969;62:234–241. doi: 10.1073/pnas.62.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linn TC, Pettit FH, Hucho F, Reed LJ. Alpha-keto acid dehydrogenase complexes. XI. Comparative studies of regulatory properties of the pyruvate dehydrogenase complexes from kidney, heart, and liver mitochondria. Proc Natl Acad Sci U S A. 1969;64:227–234. doi: 10.1073/pnas.64.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coore HG, Denton RM, Martin BR, Randle PJ. Regulation of adipose tissue pyruvate dehydrogenase by insulin and other hormones. Biochem J. 1971;125:115–127. doi: 10.1042/bj1250115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denton RM, Coore HG, Martin BR, Randle PJ. Insulin activates pyruvate dehydrogenase in rat epididymal adipose tissue. Nat New Biol. 1971;231:115–116. doi: 10.1038/newbio231115a0. [DOI] [PubMed] [Google Scholar]
- 34.Denton RM, Randle P, Martin BR. Stimulation by Ca2+ of pyruvate dehydrogenase phosphate phosphatase. Biochem J. 1972;128:161–163. doi: 10.1042/bj1280161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siess EA, Wieland OH. Purification and characterization of pyruvate-dehydrogenase phosphatase from pig-heart muscle. Eur J Biochem. 1972;26:96–105. doi: 10.1111/j.1432-1033.1972.tb01744.x. [DOI] [PubMed] [Google Scholar]
- 36.Huang B, Gudi R, Wu P, Harris RA, Hamilton J, Popov KM. Isoenzymes of Pyruvate Dehydrogenase Phosphatase. Journal of Biological Chemistry. 1998;273:17680–17688. doi: 10.1074/jbc.273.28.17680. [DOI] [PubMed] [Google Scholar]
- 37.Sale GJ, Randle PJ. Analysis of site occupancies in [32P]phosphorylated pyruvate dehydrogenase complexes by aspartyl-prolyl cleavage of tryptic phosphopeptides. Eur J Biochem. 1981;120:535–540. doi: 10.1111/j.1432-1033.1981.tb05733.x. [DOI] [PubMed] [Google Scholar]
- 38.Yeaman SJ, Hutcheson ET, Roche TE, Pettit FH, Brown JR, Reed LJ, Watson DC, Dixon GH. Sites of phosphorylation on pyruvate dehydrogenase from bovine kidney and heart. Biochemistry. 1978;17:2364–2370. doi: 10.1021/bi00605a017. [DOI] [PubMed] [Google Scholar]
- 39.Boja ES, Phillips D, French SA, Harris RA, Balaban RS. Quantitative Mitochondrial Phosphoproteomics Using iTRAQ on an LTQ-Orbitrap with High Energy Collision Dissociation. J Proteome Res. 2009;8:4665–4675. doi: 10.1021/pr900387b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gnad F, Forner F, Zielinska DF, Birney E, Gunawardena J, Mann M. Evolutionary constraints of phosphorylation in eukaryotes, prokaryotes, and mitochondria. Mol Cell Proteomics. 2010;9:2642–2653. doi: 10.1074/mcp.M110.001594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. American Journal of Physiology - Endocrinology And Metabolism. 2003;284:E855–E862. doi: 10.1152/ajpendo.00526.2002. [DOI] [PubMed] [Google Scholar]
- 42.Pettit FH, Roche TE, Reed LJ. Function of calcium ions in pyruvate dehydrogenase phosphatase activity. Biochem Biophys Res Commun. 1972;49:563–571. doi: 10.1016/0006-291x(72)90448-2. [DOI] [PubMed] [Google Scholar]
- 43.Turkan A, Gong X, Peng T, Roche TE. Structural Requirements within the Lipoyl Domain for the Ca2+-dependent Binding and Activation of Pyruvate Dehydrogenase Phosphatase Isoform 1 or Its Catalytic Subunit. Journal of Biological Chemistry. 2002;277:14976–14985. doi: 10.1074/jbc.M108434200. [DOI] [PubMed] [Google Scholar]
- 44.Vassylyev DG, Symersky J. Crystal structure of pyruvate dehydrogenase phosphatase 1 and its functional implications. J Mol Biol. 2007;370:417–426. doi: 10.1016/j.jmb.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Damuni Z, Humphreys JS, Reed LJ. Stimulation of pyruvate dehydrogenase phosphatase activity by polyamines. Biochemical and Biophysical Research Communications. 1984;124:95–99. doi: 10.1016/0006-291x(84)90921-5. [DOI] [PubMed] [Google Scholar]
- 46.Pezzato E, Battaglia V, Brunati A, Agostinelli E, Toninello A. Ca;-independent effects of spermine on pyruvate dehydrogenase complex activity in energized rat liver mitochondria incubated in the absence of exogenous Ca and Mg. Amino Acids. 2009;36:449–456. doi: 10.1007/s00726-008-0099-5. [DOI] [PubMed] [Google Scholar]
- 47.McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J. 1979;180:533–544. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawlis VB, Roche TE. Regulation of bovine kidney .alpha.-ketoglutarate dehydrogenase complex by calcium ion and adenine nucleotides. Effects on S0.5 for .alpha.-ketoglutarate. Biochemistry. 1981;20:2512–2518. doi: 10.1021/bi00512a023. [DOI] [PubMed] [Google Scholar]
- 49.Lai JCK, Cooper AJL. Brain +¦-Ketoglutarate Dehydrogenase Complex: Kinetic Properties, Regional Distribution, and Effects of Inhibitors. Journal of Neurochemistry. 1986;47:1376–1386. doi: 10.1111/j.1471-4159.1986.tb00768.x. [DOI] [PubMed] [Google Scholar]
- 50.Lawlis VB, Roche TE. Inhibition of bovine kidney .alpha.-ketoglutarate dehydrogenase complex by reduced nicotinamide adenine dinucleotide in the presence or absence of calcium ion and effect of adenosine 5’-diphosphate on reduced nicotinamide adenine dinucleotide inhibition. Biochemistry. 1981;20:2519–2524. doi: 10.1021/bi00512a024. [DOI] [PubMed] [Google Scholar]
- 51.Strumilo S. Short-term regulation of the +¦-ketoglutarate dehydrogenase complex by energy-linked and some other effectors. Biochemistry (Moscow) 2005;70:726–729. doi: 10.1007/s10541-005-0177-1. [DOI] [PubMed] [Google Scholar]
- 52.Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J. 1978;176:899–906. doi: 10.1042/bj1760899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.LaPorte DC. The isocitrate dehydrogenase phosphorylation cycle: regulation and enzymology. J Cell Biochem. 1993;51:14–18. doi: 10.1002/jcb.240510104. [DOI] [PubMed] [Google Scholar]
- 54.Rutter GA, Pralong WF, Wollheim CB. Regulation of mitochondrial glycerol-phosphate dehydrogenase by Ca2+ within electropermeabilized insulin-secreting cells (INS-1) Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1992;1175:107–113. doi: 10.1016/0167-4889(92)90016-5. [DOI] [PubMed] [Google Scholar]
- 55.MacDonald MJ, Brown LJ. Calcium activation of mitochondrial glycerol phosphate dehydrogenase restudied. Arch Biochem Biophys. 1996;326:79–84. doi: 10.1006/abbi.1996.0049. [DOI] [PubMed] [Google Scholar]
- 56.Satrustegui J, Pardo B, del AA. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol Rev. 2007;87:29–67. doi: 10.1152/physrev.00005.2006. [DOI] [PubMed] [Google Scholar]
- 57.Palmieri L, Pardo B, Lasorsa FM, del AA, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrustegui J, Palmieri F. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001;20:5060–5069. doi: 10.1093/emboj/20.18.5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, Satrustegui J. Ca2+ Activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J Biol Chem. 2007;282:7098–7106. doi: 10.1074/jbc.M610491200. [DOI] [PubMed] [Google Scholar]
- 59.Marmol P, Pardo B, Wiederkehr A, del AA, Wollheim CB, Satrustegui J. Requirement for aralar and its Ca2+-binding sites in Ca2+ signal transduction in mitochondria from INS-1 clonal beta-cells. J Biol Chem. 2009;284:515–524. doi: 10.1074/jbc.M806729200. [DOI] [PubMed] [Google Scholar]
- 60.delArco A, Satrustegui J. Identification of a novel human subfamily of mitochondrial carriers with calcium-binding domains. J Biol Chem. 2004;279:24701–24713. doi: 10.1074/jbc.M401417200. [DOI] [PubMed] [Google Scholar]
- 61.Nosek MT, Dransfield DT, Aprille JR. Calcium stimulates ATP-Mg/Pi carrier activity in rat liver mitochondria. J Biol Chem. 1990;265:8444–8450. [PubMed] [Google Scholar]
- 62.Maechler P, Wollheim CB. Mitochondrial signals in glucose-stimulated insulin secretion in the beta cell. J Physiol. 2000;529(Pt 1):49–56. doi: 10.1111/j.1469-7793.2000.00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Satrustegui J, Contreras L, Ramos M, Marmol P, del AA, Saheki T, Pardo B. Role of aralar, the mitochondrial transporter of aspartate-glutamate, in brain N-acetylaspartate formation and Ca(2+) signaling in neuronal mitochondria. J Neurosci Res. 2007;85:3359–3366. doi: 10.1002/jnr.21299. [DOI] [PubMed] [Google Scholar]
- 64.Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787:1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 65.Denton RM, McCormack JG, Marshall SE. Persistence of the effect of insulin on pyruvate dehydrogenase activity in rat white and brown adipose tissue during the preparation and subsequent incubation of mitochondria. Biochem J. 1984;217:441–452. doi: 10.1042/bj2170441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aponte AM, Phillips D, Harris RA, Blinova K, French S, Johnson DT, Balaban RS. 32P labeling of protein phosphorylation and metabolite association in the mitochondria matrix. Methods Enzymol. 2009;457:63–80. doi: 10.1016/S0076-6879(09)05004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sale GJ, Randle PJ. Incorporation of [32P]phosphate into the pyruvate dehydrogenase complex in rat heart mitochondria. Biochem J. 1980;188:409–421. doi: 10.1042/bj1880409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Randle PJ, Denton RM, Pask HT, Severson DL. Calcium ions and the regulation of pyruvate dehydrogenase. Biochem Soc Symp. 1974;39:75–87. [PubMed] [Google Scholar]
- 69.Aponte AM, Phillips D, Hopper RK, Johnson DT, Harris RA, Blinova K, Boja ES, French S, Balaban RS. Use of (32)P to study dynamics of the mitochondrial phosphoproteome. J Proteome Res. 2009;8:2679–2695. doi: 10.1021/pr800913j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, Witzmann FA, Harris RA, Balaban RS. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry. 2006;45:2524–2536. doi: 10.1021/bi052475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McCormack JG, Denton RM. The activation of isocitrate dehydrogenase (NAD+) by Ca2+ within intact uncoupled rat brown adipose tissue mitochondria incubated in the presence and absence of albumin [proceedings] Biochem Soc Trans. 1980;8:339. doi: 10.1042/bst0080339. [DOI] [PubMed] [Google Scholar]
- 72.McCormack JG, Denton RM. Role of calcium ions in the regulation of intramitochondrial metabolism. Properties of the Ca2+-sensitive dehydrogenases within intact uncoupled mitochondria from the white and brown adipose tissue of the rat. Biochem J. 1980;190:95–105. doi: 10.1042/bj1900095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rossi CS, Vasington FD, Carafoli E. The effect of ruthenium red on the uptake and release of Ca2+ by mitochondria. Biochemical and Biophysical Research Communications. 1973;50:846–852. doi: 10.1016/0006-291x(73)91322-3. [DOI] [PubMed] [Google Scholar]
- 74.Moreno-Sanchez R. Regulation of oxidative phosphorylation in mitochondria by external free Ca2+ concentrations. J Biol Chem. 1985;260:4028–4034. [PubMed] [Google Scholar]
- 75.Moreno-Sanchez R. Inhibition of oxidative phosphorylation by a Ca2+-induced diminution of the adenine nucleotide translocator. Biochim Biophys Acta. 1983;724:278–285. doi: 10.1016/0005-2728(83)90146-9. [DOI] [PubMed] [Google Scholar]
- 76.Moreno-Sanchez R, Hansford RG. Dependence of cardiac mitochondrial pyruvate dehydrogenase activity on intramitochondrial free Ca2+ concentration. Biochem J. 1988;256:403–412. doi: 10.1042/bj2560403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hansford RG, Moreno-Sanchez R, Lewartowski B. Activation of pyruvate dehydrogenase complex by Ca2+ in intact heart, cardiac myocytes, and cardiac mitochondria. Ann N Y Acad Sci. 1989;573:240–53. 240–253. doi: 10.1111/j.1749-6632.1989.tb15001.x. [DOI] [PubMed] [Google Scholar]
- 78.Moreno-Sanchez R, Devars S, Lopez-Gomez F, Uribe A, Corona N. Distribution of control of oxidative phosphorylation in mitochondria oxidizing NAD-linked substrates. Biochim Biophys Acta. 1991;1060:284–292. doi: 10.1016/s0005-2728(05)80318-4. [DOI] [PubMed] [Google Scholar]
- 79.Eng J, Lynch RM, Balaban RS. NADH fluorescence spectroscopy and imaging of isolated cardiac myocytes. Biophys J. 1989;55:621–630. doi: 10.1016/S0006-3495(89)82859-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Murphy AN, Kelleher JK, Fiskum G. Submicromolar Ca2+ regulates phosphorylating respiration by normal rat liver and AS-30D hepatoma mitochondria by different mechanisms. J Biol Chem. 1990;265:10527–10534. [PubMed] [Google Scholar]
- 81.Johnston JD, Brand MD. Stimulation of the respiration rate of rat liver mitochondria by sub-micromolar concentrations of extramitochondrial Ca2+ Biochem J. 1987;245:217–222. doi: 10.1042/bj2450217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Koretsky AP, Balaban RS. Changes in pyridine nucleotide levels alter oxygen consumption and extramitochondrial phosphates in isolated mitochondria: A 31P NMR and fluorescence study. Biochim Biophys. 1987;893:398–408. doi: 10.1016/0005-2728(87)90092-2. [DOI] [PubMed] [Google Scholar]
- 83.Moreno-Sanchez R, Hogue BA, Hansford RG. Influence of NAD-linked dehydrogenase activity on flux through oxidative phosphorylation. Biochem J. 1990;268:421–428. doi: 10.1042/bj2680421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harris DA, Das AM. Control of mitochondrial ATP synthesis in the heart. Biochem J. 1991;280:561–573. doi: 10.1042/bj2800561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gomez-Puyou A, de Gomez-Puyou MT, Ernster L. Inactive to active transitions of the mitochondrial ATPase complex as controlled by the ATPase inhibitor. Biocim Biophys Acta. 1979;547:252–257. doi: 10.1016/0005-2728(79)90008-2. [DOI] [PubMed] [Google Scholar]
- 86.Beis I, Newsholme EA. Effects of calcium ions on adenine nucleotide translocase from cardiac muscle. Journal of Molecular and Cellular Cardiology. 1976;8:863–876. doi: 10.1016/0022-2828(76)90069-9. [DOI] [PubMed] [Google Scholar]
- 87.Territo PR, Mootha VK, French SA, Balaban RS. Ca(2+) activation of heart mitochondrial oxidative phosphorylation:role of F0/F1ATPase. Am J Physiol. 2000;278:c423–c435. doi: 10.1152/ajpcell.2000.278.2.C423. [DOI] [PubMed] [Google Scholar]
- 88.Borutaite V, Mildaziene V, Brown GC, Brand MD. Control and kinetic analysis of ischemia-damaged heart mitochondria: which parts of the oxidative phosphorylation system are affected by ischemia? Biochim Biophys Acta. 1995;1272:154–158. doi: 10.1016/0925-4439(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 89.Mootha VK, Arai AE, Balaban RS. Maximum oxidative phosphorylation capacity of the mammalian heart. Am J Physiol. 1997;272:H769–H775. doi: 10.1152/ajpheart.1997.272.2.H769. [DOI] [PubMed] [Google Scholar]
- 90.Panov AV, Scaduto RC., Jr Substrate specific effects of calcium on metabolism of rat heart mitochondria. Am J Physiol. 1996;270:H1398–H1406. doi: 10.1152/ajpheart.1996.270.4.H1398. [DOI] [PubMed] [Google Scholar]
- 91.Territo PR, French SA, Balaban RS. Simulation of cardiac work transitions, in vitro: effects of simultaneous Ca(2+)and ATPase additions on isolated porcine heart mitochondria. Cell Calcium. 2001;30:19–27. doi: 10.1054/ceca.2001.0211. [DOI] [PubMed] [Google Scholar]
- 92.Balaban RS, Bose S, French SA, Territo PR. Role of calcium in metabolic signaling between cardiac sarcoplasmic reticulum and mitochondria in vitro. American Journal of Physiology-Cell Physiology. 2003;284:C285–C293. doi: 10.1152/ajpcell.00129.2002. [DOI] [PubMed] [Google Scholar]
- 93.Evtodienko YV, Azarashvili TS, Teplova VV, Odinokova IV, Saris N. Regulation of oxidative phosphorylation in the inner membrane of rat liver mitochondria by calcium ions. Biochemistry (Mosc) 2000;65:1023–1026. [PubMed] [Google Scholar]
- 94.Hojlund K, Wrzesinski K, Larsen PM, Fey SJ, Roepstorff P, Handberg A, Dela F, Vinten J, McCormack JG, Reynet C, Beck-Nielsen H. Proteome analysis reveals phosphorylation of ATP synthase beta -subunit in human skeletal muscle and proteins with potential roles in type 2 diabetes. J Biol Chem. 2003;278:10436–10442. doi: 10.1074/jbc.M212881200. [DOI] [PubMed] [Google Scholar]
- 95.Feng J, Zhu M, Schaub MC, Gehrig P, Roschitzki B, Lucchinetti E, Zaugg M. Phosphoproteome analysis of isoflurane-protected heart mitochondria: phosphorylation of adenine nucleotide translocator-1 on Tyr194 regulates mitochondrial function. Cardiovascular Research. 2008;80:20–29. doi: 10.1093/cvr/cvn161. [DOI] [PubMed] [Google Scholar]
- 96.Kane LA, Van Eyk JE. Post-translational modifications of ATP synthase in the heart: biology and function. J Bioenerg Biomembr. 2009;41:145–150. doi: 10.1007/s10863-009-9218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Arrell DK, Elliott ST, Kane LA, Guo Y, Ko YH, Pedersen PL, Robinson J, Murata M, Murphy AM, Marbán E, Van Eyk JE. Proteomic Analysis of Pharmacological Preconditioning. Circ Res. 2006;99:706–714. doi: 10.1161/01.RES.0000243995.74395.f8. [DOI] [PubMed] [Google Scholar]
- 98.Deng N, Zhang J, Zong C, Wang Y, Lu H, Yang P, Wang W, Young GW, Wang Y, Korge P, Lotz C, Doran P, Liem DA, Apweiler R, Weiss JN, Duan H, Ping P. Phosphoproteome analysis reveals regulatory sites in major pathways of cardiac mitochondria. Molecular & Cellular Proteomics. 2010;10(2):1–14. doi: 10.1074/mcp.M110.000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao X, Len IR, Bak S, Mogensen M, Wrzesinski K, Hjlund K, Jensen ON. Phosphoproteome Analysis of Functional Mitochondria Isolated from Resting Human Muscle Reveals Extensive Phosphorylation of Inner Membrane Protein Complexes and Enzymes. Molecular & Cellular Proteomics. 2011;10(1):1–14. doi: 10.1074/mcp.M110.000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee J, Xu Y, Chen Y, Sprung R, Kim SC, Xie S, Zhao Y. Mitochondrial Phosphoproteome Revealed by an Improved IMAC Method and MS/MS/MS. Molecular & Cellular Proteomics. 2007;6:669–676. doi: 10.1074/mcp.M600218-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Villen J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc. 2008;3:1630–1638. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chance B, THORELL B. Localization and kinetics of reduced pyridine nucleotide in living cells by microfluorometry. J Biol Chem. 1959;234:3044–3050. [PubMed] [Google Scholar]
- 103.Blinova K, Levine RL, Boja ES, Griffiths GL, Shi ZD, Ruddy B, Balaban RS. Mitochondrial NADH fluorescence is enhanced by complex I binding. Biochemistry. 2008;47:9636–9645. doi: 10.1021/bi800307y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dykens JA, Stout AK. Assessment of mitochondrial membrane potential in situ using single potentiometric dyes and a novel fluorescence resonance energy transfer technique. Methods Cell Biol. 2001;65:285–309. doi: 10.1016/s0091-679x(01)65018-0. [DOI] [PubMed] [Google Scholar]
- 105.Lemasters JJ, Chacon E, Ohata H, Harper IS, Nieminen AL, Tesfai SA, Herman B. Measurement of electrical potential, pH, and free calcium ion concentration in mitochondria of living cells by laser scanning confocal microscopy. Methods Enzymol. 1995;260:428–44. 428–444. doi: 10.1016/0076-6879(95)60156-2. [DOI] [PubMed] [Google Scholar]
- 106.Freedman JC, Novak TS. Optical measurement of membrane potential in cells, organelles, and vesicles. Methods Enzymol. 1989;172:102–122. doi: 10.1016/s0076-6879(89)72011-5. [DOI] [PubMed] [Google Scholar]
- 107.Tsien RY, Pozzan T, Rink TJ. Calcium homeostasis in intact lymphocytes: cytoplasmic free calcium monitored with a new, intracellularly trapped fluorescent indicator. J Cell Biol. 1982;94:325–334. doi: 10.1083/jcb.94.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brini M, De GF, Murgia M, Marsault R, Massimino ML, Cantini M, Rizzuto R, Pozzan T. Subcellular analysis of Ca2+ homeostasis in primary cultures of skeletal muscle myotubes. Mol Biol Cell. 1997;8:129–143. doi: 10.1091/mbc.8.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- 110.Balaban RS, Blum JJ. Hormone-induced changes in NADH fluorescence and O2 consumption of rat hepatocytes. Am J Physiol. 1982;242:C172–C177. doi: 10.1152/ajpcell.1982.242.3.C172. [DOI] [PubMed] [Google Scholar]
- 111.Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998;17:4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Soboll S. Regulation of energy metabolism in liver. J Bioenerg Biomembr. 1995;27:571–582. doi: 10.1007/BF02111655. [DOI] [PubMed] [Google Scholar]
- 113.Pralong WF, Spat A, Wollheim CB. Dynamic pacing of cell metabolism by intracellular Ca2+ transients. Journal of Biological Chemistry. 1994;269:27310–27314. [PubMed] [Google Scholar]
- 114.Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- 115.Halestrap AP. Regulation of mitochondrial metabolism through changes in matrix volume. Biochem Soc Trans. 1994;22:522–529. doi: 10.1042/bst0220522. [DOI] [PubMed] [Google Scholar]
- 116.Voronina S, Sukhomlin T, Johnson PR, Erdemli G, Petersen OH, Tepikin A. Correlation of NADH and Ca2+ signals in mouse pancreatic acinar cells. J Physiol. 2002;539:41–52. doi: 10.1113/jphysiol.2001.013134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Duchen MR. Ca(2+)-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J. 1992;283(Pt 1):41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Das AM, Harris DA. Reversible modulation of the mitochondrial ATP synthase with energy demand in cultured rat cardiomyocytes. FEBS Lett. 1989;256:97–100. doi: 10.1016/0014-5793(89)81725-9. [DOI] [PubMed] [Google Scholar]
- 120.Das AM, Harris DA. Control of mitochondrial ATP synthase in heart cells: inactive to active transitions caused by beating or postive inotrophic agents. Cardiovasc Res. 1990;24:411–417. doi: 10.1093/cvr/24.5.411. [DOI] [PubMed] [Google Scholar]
- 121.Das AM. Regulation of mitochondrial ATP synthase activity in human myocardium. Clin Sci (Lond) 1998;94:499–504. doi: 10.1042/cs0940499. [DOI] [PubMed] [Google Scholar]
- 122.Scholz TD, Balaban RS. Mitochondrial F1-ATPase activity of canine myocardium: effects of hypoxia and stimulation. Am J Physiol. 1994;266:H2396–H2403. doi: 10.1152/ajpheart.1994.266.6.H2396. [DOI] [PubMed] [Google Scholar]
- 123.Hubbard MJ, McHugh NJ. Mitochondrial ATP synthase F1-beta-subunit is a calciumbinding protein. FEBS Lett. 1996;391:323–329. doi: 10.1016/0014-5793(96)00767-3. [DOI] [PubMed] [Google Scholar]
- 124.McCormack JG, England PJ. Ruthenium Red inhibits the action of pyruvate dehydrogenase caused by positive inotropic agents in the perfused rat heart. Biochem J. 1983;214:581–585. doi: 10.1042/bj2140581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kobayashi K, Neely JR. Mechanism of pyruvate dehydrogenase activation by increased cardiac work. J Mol Cell Cardiol. 1983;15:369–382. doi: 10.1016/0022-2828(83)90321-8. [DOI] [PubMed] [Google Scholar]
- 126.Parolin ML, Chesley A, Matsos MP, Spriet LL, Jones NL, Heigenhauser GJ. Regulation of skeletal muscle glycogen phosphorylase and PDH during maximal intermittent exercise. Am J Physiol. 1999;277:E890–E900. doi: 10.1152/ajpendo.1999.277.5.E890. [DOI] [PubMed] [Google Scholar]
- 127.McCormack JG, Denton RM. The activation of pyruvate dehydrogenase in the perfused rat heart by adrenaline and other inotropic agents. Biochem J. 1981;194:639–643. doi: 10.1042/bj1940639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hennig G, Loffler G, Wieland OH. Active and inactive forms of pyruvatedehydrogenase in skeletal muscle as related to the metabolic and functional state of the muscle cell. FEBS Lett. 1975;59:142–145. doi: 10.1016/0014-5793(75)80361-9. [DOI] [PubMed] [Google Scholar]
- 129.Morgan DG, Routtenberg A. Brain pyruvate dehydrogenase: phosphorylation and enzyme activity altered by a training experience. Science. 1981;214:470–471. doi: 10.1126/science.7291989. [DOI] [PubMed] [Google Scholar]
- 130.James G. Evidence that adrenaline activates key oxidative enzymes in rat liver by increasing intramitochondrial [Ca2+] FEBS Letters. 1985;180:259–264. doi: 10.1016/0014-5793(85)81082-6. [DOI] [PubMed] [Google Scholar]
- 131.ConstantinTeodosiu D, Carlin J, Cederblad G, Harris R, Hultman Acetyl group accumulation and pyruvate dehydrogenase activity in human muscle during incremental exercise. Acta Physiologica Scandinavica. 1991;143:367–372. doi: 10.1111/j.1748-1716.1991.tb09247.x. [DOI] [PubMed] [Google Scholar]
- 132.Balaban RS, Kantor HL, Katz LA, Briggs RW. Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science. 1986;232:1121–1123. doi: 10.1126/science.3704638. [DOI] [PubMed] [Google Scholar]
- 133.Katz LA, Swain JA, Portman MA, Balaban RS. Relation between phosphate metabolites and oxygen consumption of heart in vivo. Am J Physiol. 1989;256:H265–H274. doi: 10.1152/ajpheart.1989.256.1.H265. [DOI] [PubMed] [Google Scholar]
- 134.Arthur HL, Petein MA, Michurski SP, Zimmer SD, Ugurbil K. 31P-NMR studies of respiratory regulation in the intact myocardium. FEBS LET. 1986;206:257–261. doi: 10.1016/0014-5793(86)80992-9. [DOI] [PubMed] [Google Scholar]
- 135.Zhang J, Duncker DJ, Xu Y, Zhang Y, Path G, Merkle H, Hendrich K, From AHL, Bache RJ, Ugurbil K. Transmural bioenergetic responses of normal myocardium to high workstates. Am J Physiol. 1995;268:H1891–H1911. doi: 10.1152/ajpheart.1995.268.5.H1891. [DOI] [PubMed] [Google Scholar]
- 136.Schaefer S, Schwartz GG, Steinman SK, Meyerhoff DJ, Massie BM, Weiner MW. Metabolic response of the human heart to inotropic stimulation: in vivo phosphorus-31 studies of normal and cardiomyopathic myocardium. Mag Res Med. 1992;25:260–272. doi: 10.1002/mrm.1910250205. [DOI] [PubMed] [Google Scholar]
- 137.Weiss RG, Bottomley PA, Hardy CJ, Gerstenblith G. Regional myocardial metabolism of high-energy phosphates during isometric exercise in patients with coronary artery disease. N Engl J Med. 1990;323:1593–1600. doi: 10.1056/NEJM199012063232304. [DOI] [PubMed] [Google Scholar]
- 138.Wollenberger A. Relation between work and labile phosphate content in the isolated dog heart. Circ Res. 1957;5:175–178. doi: 10.1161/01.res.5.2.175. [DOI] [PubMed] [Google Scholar]
- 139.Boerth RC, Covell JW, Seagren SC, Pool PE. High -energy phosphate concentrations in dog myocardium during stress. Am J Physiol. 1969;216:1103–1106. doi: 10.1152/ajplegacy.1969.216.5.1103. [DOI] [PubMed] [Google Scholar]
- 140.Neely JR, Denton RM, England PJ, Randle PJ. The effects of increased heart work on the tricarboxylate cycle and its interactions with glycolysis in the perfused rat heart. Biochem J. 1972;128:147–159. doi: 10.1042/bj1280147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Heineman FW, Balaban RS. Phosphorus-31 nuclear magnetic resonance analysis of transient changes of canine myocardial metabolism in vivo. J Clin Invest. 1990;85:843–852. doi: 10.1172/JCI114511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Portman MA, Heineman FW, Balaban RS. Developmental changes in the relation between phosphate metabolites and oxygen consumption in the sheep heart in vivo. J Clin Invest. 1989;83:456–464. doi: 10.1172/JCI113904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 144.Zimmer SD, Ugurbil K, Michurski SP, Mohanakrishnan P, Ulstad VK, Foker JE, From AHL. Alterations in oxidative function and respiratory regulation in the postischemic myocardium. J Biol Chem. 1989;264:12402–12411. [PubMed] [Google Scholar]
- 145.Unitt JF, McCormack JG, Reid D, Maclachlan LK. Direct evidence for a role of intramitochondrial Ca+2 in the regulation of oxidative phosphorylation in the stimulated rat heart. Biochem J. 1989;262:293–301. doi: 10.1042/bj2620293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Katz LA, Koretsky AP, Balaban RS. Activation of dehydrogenase activity and cardiac respiration: a 31P-NMR study. Amer J Physiol. 1988;255:185–188. doi: 10.1152/ajpheart.1988.255.1.H185. [DOI] [PubMed] [Google Scholar]
- 147.Samaha FF, Heineman FW, Ince C, Fleming J, Balaban RS. ATP-sensitive potassium channel is essential to maintain basal coronary vascular tone in vivo. Am J Physiol. 1992;262:C1220–C1227. doi: 10.1152/ajpcell.1992.262.5.C1220. [DOI] [PubMed] [Google Scholar]
- 148.Heineman FW, Balaban RS. Effects of afterload and heart rate on NAD(P)H redox state in the isolated rabbit heart. Am J Physiol. 1993;264:H433–H440. doi: 10.1152/ajpheart.1993.264.2.H433. [DOI] [PubMed] [Google Scholar]
- 149.Vuorinen K, ALARAMI A, YAN Y, INGMAN P, Hassinen IE. Respiratory control in heart muscle during fatty acid oxidation: Energy-state or substrate-level regulation by Ca2+ J Mol Cell Cardiol. 1995;27:1581–1591. doi: 10.1016/s0022-2828(95)90458-1. [DOI] [PubMed] [Google Scholar]
- 150.Brandes R, Bers DM. Intracellular Ca2+ increases the mitochondrial NADH concentration during elevated work in intact cardiac muscle. Circ Res. 1997;80:82–87. doi: 10.1161/01.res.80.1.82. [DOI] [PubMed] [Google Scholar]
- 151.Brandes R, Bers DM. Analysis of the mechanisms of mitochondrial NADH regulation in cardiac trabeculae. Biophys J. 1999;77:1666–1682. doi: 10.1016/S0006-3495(99)77014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Brandes R, Maier LS, Bers DM. Regulation of mitochondrial [NADH] by cytosolic [Ca2+] and work in trabeculae from hypertrophic and normal rat hearts. Circ Res. 1998;82:1189–1198. doi: 10.1161/01.res.82.11.1189. [DOI] [PubMed] [Google Scholar]
- 153.Brandes R, Bers DM. Simultaneous measurements of mitochondrial NADH and Ca(2+) during increased work in intact rat heart trabeculae. Biophys J. 2002;83:587–604. doi: 10.1016/S0006-3495(02)75194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ashruf JF, Coremans JMCC, Bruining HA, Ince C. Increase of cardiac work is associated with decrease of mitochondrial NADH Am. J Physiol. 1995;269:H856–H862. doi: 10.1152/ajpheart.1995.269.3.H856. [DOI] [PubMed] [Google Scholar]
- 155.Heineman FW, Kupriyanov VV, Marshall R, Fralix TA, Balaban RS. Myocardial oxygenation in the isolated working rabbit heart as a function of work. Am J Physiol. 1992;262:H255–H267. doi: 10.1152/ajpheart.1992.262.1.H255. [DOI] [PubMed] [Google Scholar]
- 156.Van Dorsten FA, Nederhoff MGJ, Nicolay K, Van Echteld CJA. 31P NMR studies of creatine kinase flux in M-creatine kinase-deficient mouse heart. American Journal of Physiology - Heart and Circulatory Physiology. 1998;275:H1191–H1199. doi: 10.1152/ajpheart.1998.275.4.H1191. [DOI] [PubMed] [Google Scholar]
- 157.Arai AE, Kasserra CE, Territo PR, Gandjbakhche AH, Balaban RS. Myocardial oxygenation in vivo: optical spectroscopy of cytoplasmic myoglobin and mitochondrial cytochromes. Am J Physiol. 1999;277:H683–H697. doi: 10.1152/ajpheart.1999.277.2.H683. [DOI] [PubMed] [Google Scholar]
- 158.Jobsis PD, Ashikaga H, Wen H, Rothstein EC, Horvath KA, McVeigh ER, Balaban RS. The visceral pericardium: macromolecular structure and contribution to passive mechanical properties of the left ventricle. Am J Physiol Heart Circ Physiol. 2007;293:H3379–H3387. doi: 10.1152/ajpheart.00967.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wan B, Doumen C, Duszynski J, Salama G, Vary TC, LaNoue KF. Effect of cardiac work on electrical potential gradient across mitochondrial membrane in perfused rat hearts. Amer J Physiol. 1993;265:453–460. doi: 10.1152/ajpheart.1993.265.2.H453. [DOI] [PubMed] [Google Scholar]
- 160.Kauppinen RA, Hassinen IE. Monitoring of mitochondrial membrane potential in isolated perfused rat heart. American Journal of Physiology - Heart and Circulatory Physiology. 1984;247:H508–H516. doi: 10.1152/ajpheart.1984.247.4.H508. [DOI] [PubMed] [Google Scholar]
- 161.Kauppinen RA, Hassinen IE. Monitoring of mitochondrial membrane potential in isolated perfused rat heart. Am J Physiol. 1984;247:H508–H516. doi: 10.1152/ajpheart.1984.247.4.H508. [DOI] [PubMed] [Google Scholar]
- 162.Piwnica-Worms DP, Kronauge JF, LeFurgey A, Backus M, Hockett D, Ingram P, Lieberman M, Holman BL, Jones AG, Davison A. Mitochondrial localization and characterization of 99Tc-SESTAMIBI in heart cells by electron probe X-ray microanalysis and 99Tc-NMR spectroscopy. Magn Reson Imaging. 1994;12:641–652. doi: 10.1016/0730-725x(94)92459-7. [DOI] [PubMed] [Google Scholar]
- 163.Verzijlbergen JF, van OD, Cramer MJ, Ascoop CA, Zwinderman AH, Niemeyer MG, van der Wall EE, Pauwels EK. Quantitative analysis of planar technetium-99m Sestamibi myocardial perfusion images. Clinical application of a modified method for the subtracton of tissue crosstalk. Eur Heart J. 1994;15:1217–1226. doi: 10.1093/oxfordjournals.eurheartj.a060656. [DOI] [PubMed] [Google Scholar]
- 164.Phillips D, Aponte AM, Covian RG, Balaban RS. Intrinsic Protein Kinase Activity of Mitochondrial Oxidative Phosphorylation Complexes. Biochemistry. 2011;50:2515–2529. doi: 10.1021/bi101434x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem. 1955;217:409–427. [PubMed] [Google Scholar]
- 166.Chance B, Williams GR. The respiratory chain and oxidative phosphorylation. Adv Enzymol Relat Subj Biochem. 1956;17:65–134. doi: 10.1002/9780470122624.ch2. [DOI] [PubMed] [Google Scholar]
- 167.Bender E, Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 2000;466:130–134. doi: 10.1016/s0014-5793(99)01773-1. [DOI] [PubMed] [Google Scholar]
- 168.Brown GC. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995;369:136–139. doi: 10.1016/0014-5793(95)00763-y. [DOI] [PubMed] [Google Scholar]
- 169.Steenaart NA, Shore GC. Mitochondrial cytochrome c oxidase subunit IV is phosphorylated by an endogenous kinase. FEBS Lett. 1997;415:294–298. doi: 10.1016/s0014-5793(97)01145-9. [DOI] [PubMed] [Google Scholar]
- 170.Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, Huttemann M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem. 2005;280:6094–6100. doi: 10.1074/jbc.M411335200. [DOI] [PubMed] [Google Scholar]
- 171.Acin-Perez R, Gatti DL, Bai Y, Manfredi G. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: coupled mechanisms of energy metabolism regulation. Cell Metab. 2011;13:712–719. doi: 10.1016/j.cmet.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kirichenko A, pfitzner U, Ludwig B, Soares CM, Vygodina TV, Konstantinov A. Cytochrome c Oxidase as a Calcium Binding Protein. Studies on the Role of a Conserved Aspartate in Helices XI-XII Cytoplasmic Loop in Cation Binding. Biochemistry. 2005;44:12391–12401. doi: 10.1021/bi050376v. [DOI] [PubMed] [Google Scholar]
- 173.Aliev MK, Saks VA. Compartmentalized energy transfer in cardiomyocytes: use of mathematical modeling for analysis of in vivo regulation of respiration. Biophys J. 1997;73:428–445. doi: 10.1016/S0006-3495(97)78082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Saks V, Beraud N, Wallimann T. Metabolic compartmentation - a system level property of muscle cells: real problems of diffusion in living cells. Int J Mol Sci. 2008;9:751–767. doi: 10.3390/ijms9050751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Saks V, Dzeja P, Schlattner U, Vendelin M, Terzic A, Wallimann T. Cardiac system bioenergetics: metabolic basis of the Frank-Starling law. J Physiol. 2006;571:253–273. doi: 10.1113/jphysiol.2005.101444. [DOI] [PMC free article] [PubMed] [Google Scholar]