

Abstract
The contribution of the Wnt signaling pathway to HPV-induced carcinogenesis is poorly understood. In high-grade dysplastic lesions that are caused by high-risk human papilloma viruses (HR-HPVs), β-catenin is often located in the cell nucleus, which suggests that Wnt pathway may be involved in the development of HPV-related carcinomas. Most of the oncogenic potential of HR-HPVs resides on the E6 protein’s PDZ-binding domain. We hypothesized that the PDZ-binding domain of the HPV16-E6 oncoprotein induces the nuclear accumulation of β-catenin due to its capacity to degrade PDZ-containing cellular targets. To test this hypothesis, we evaluated the staining pattern of β-catenin in the skin epidermis of transgenic mice expressing the full-length E6 oncoprotein (K14E6 mice) and measured LacZ gene expression in K14E6 mice that were crossed with a strain expressing LacZ that was knocked into the Axin2 locus (Axin2+/LacZ mice). Here, we show that the E6 oncoprotein enhances the nuclear accumulation of β-catenin, the accumulation of cellularβ-catenin-responsive genes and the expression of LacZ. None of these effects were observed when a truncated E6 oncoprotein that lacks the PDZ-binding domain was expressed alone (K14E6ΔPDZ mice) or in combination with Axin2+/LacZ. Conversely, co-transfection with either E6 or E6ΔPDZ similarly enhanced canonical Wnt signaling in short-term in vitro assays that utilized a luciferase Wnt/β-catenin/TCF-dependent promoter. We propose that the activation of canonical Wnt signaling could be induced by the HPV16-E6 oncoprotein; however, the participation of the E6 PDZ-binding domain seems to be important in in vivo models only.
Keywords: HPV, Wnt/β-catenin, K14E6, K14E6-ΔPDZ, Axin2+/LacZ
Introduction
Human papillomaviruses (HPVs) are small DNA tumor viruses that cause the formation of warts and papillomas in several stratified epithelia such as those of the skin, genitalia and upper respiratory tract (1). Epidemiological and experimental data have shown that the “high-risk” HPVs (HR-HPVs), such as type 16 and 18, contribute to HPV-related cancer development (2). These HR-HPVs owe their oncogenic potential to the constitutive expression of the E6 and E7 oncogenes whose products enhance cell proliferation and perturb cell differentiation through several mechanisms (3). E7 promotes cell proliferation by associating with the tumor suppressor pRb and other cell cycle regulatory proteins (4), and E6 binds to and inactivates p53 tumor suppressor protein and several proteins that participate in cell-cell adhesion (5).
The progression of HPV-related cancers clearly involves dysplastic lesions that are caused by the E6 and E7 oncoproteins; nevertheless, some of the molecular events that are regulated by these oncoproteins are not fully understood (3). To date, cell culture experiments have demonstrated that the exogenous expression of HR-E6 proteins induces the immortalization of human mammary epithelial cells (6). Furthermore, transgenic mice that express the HPV16-E6 oncoprotein in the basal stratified epithelium under the control of the human keratin 14 promoter (K14E6 mice) develop papillomas and cancer of the skin epidermis (7). The in vivo tumor-capacity of E6 is dramatically reduced when a truncated version of E6 that lacks the PDZ-binding domain (K14E6ΔPDZ transgenic mice) is expressed in the basal stratified epithelium of transgenic mice (8). Although p53 is the most studied cellular target of E6, its participation in skin carcinogenesis seems to be minor in comparison with other E6 cellular targets, such as those that contain PDZ domains (7, 8). Recent experimental evidence suggests that the canonical Wnt signaling pathway may play a key role in E6’s oncogenic potential (9).
Wnt ligands control several differentiation processes during normal tissue homeostasis (10, 11), and they are implicated in pathological conditions such as in certain types of cancer (12, 13). In the absence of Wnt ligands, β-catenin forms a “degradation complex” with kinases and scaffold proteins, such as Glycogen Synthase Kinase-3β (GSK3β), Casein Kinase 1 and 2 (CK1 and CK2), Adenomatous Polyposis Coli (APC) and Axin2. This degradation complex phosphorylates β-catenin at serine and threonine residues inducing its ubiquitination by β-TcRP ubiquitin ligase and degradation. The activation of canonical Wnt signaling induces the phosphorylation of the intracellular Dishevelled (Dvl) protein, which eventually interacts with Axin2 and impedes the formation of the β-catenin degradation complex; this process leads to the accumulation and nuclear translocation of β-catenin. Once it is translocated into the nucleus, β-catenin binds members of the TCF/LEF family of transcription factors, of which T-cell factor 4 (TCF4) is the best characterized. β-catenin-TCF4 complexes control the expression of several target genes that regulate cell polarity, proliferation and differentiation (12) including c-jun (13), c-myc (14), cyclin D1 (15), multidrug resistance 1 (16), matrilysine (17), Axin2 (18) and survivin (13) and others (http://www.stanford.edu/~rnusse/pathways/targets.html).
The participation of canonical Wnt pathway in cervical cancer is evident because the nuclear accumulation of β-catenin correlates with tumor progression in human patients (19). Nuclear β-catenin is commonly found in human HPV16-positive invasive carcinoma biopsies, but it is uncommon in early dysplastic lesions (20–22). In addition, recent in vitro evidence supports the hypothesis that HR-E6 alone can enhance canonical Wnt signaling (9).
In this study, we present for the first time in vivo evidence supporting the hypothesis that the HR-E6 protein enhances canonical Wnt signaling. However, the importance of the E6 PDZ-binding domain remains controversial, while it is necessary for Wnt pathway activation in transgenic mice, it is not required in cultured COS7 cells or primary keratinocytes, indicating that other factors present in mice are required for Wnt signaling activation.
Materials and Methods
K14E6, K14E6ΔPDZ and Axin2+/LacZ mice and sample isolation
The construction of transgenic mice containing the full version of the HPV16-E6 oncogene (K14E6), the truncated version lacking the PDZ-binding domain (K14E6Δ146-151 or K14E6ΔPDZ), and the knock-in of Axin2+/LacZ has been described previously (7, 8, 23). The mice were housed and treated according to the American Association of Laboratory Animal Care (AALAC) regulations, and the research protocols were approved by the Research Unit for Laboratory Animal Care Committee (UPEAL-CINVESTAV-IPN, Mexico; NOM-062-ZOO-1999). Skin biopsies from the transgenic strains and FvB non-transgenic mice (NTG) were resected, fixed and paraffin-embedded for the histological procedures, or they were immediately frozen in liquid nitrogen for protein or RNA isolation (Trizol method). For the histological procedures (immunohistochemistry (IHC) and Immunofluorescence (IF)), five-micrometers-thick transversal sections were mounted on charged microscope slides (Fisher Scientific, U.S.A.) for immunohistochemistry (IHC), or immunofluorescence (IF) techniques.
Immunohistochemistry (IHC) and Immunofluorescence (IF) procedures
The skin sections were de-paraffinized and rehydrated as described previously (7). The protein detection for IHC was performed using the Mouse/Rabbit PolyDetector HRP/DAB Detection System (Bio SB, U.S.A.) according to manufacturer’s recommendations. The samples were incubated overnight with primary antibodies against PCNA, p53, Dlg1 or Scribble primary antibodies (Santa Cruz Biotechnology, U.S.A.) diluted at 1:100. Following the IHC procedures, the tissues were counterstained with hematoxylin and mounted in GVA-mount reagent (Zymed, U.S.A.). For the IF procedures, the skin sections were rinsed in 1X PBS and blocked for 2 hours at 4oC with 1X PBS that was supplemented with 0.3% Triton X-100 and 1% BSA; they were washed three times with 1X PBS and incubated for 1 h at 37°C with an anti-β-catenin antibody (Santa Cruz Biotechnology, U.S.A.). The sections were then incubated with a FITC-labeled secondary antibody (Zymed, U.S.A.) for 30 min at room temperature; they were rinsed above, counterstained with Propidium Iodide and mounted in Vectashield (Vector, Burlingame, CA). The preparations were examined by confocal microscopy using an SP2 (Leica Microsystems, Wetzlar, Germany). Captured images were imported into the ImageJ software program (version 1.37v, National Institutes of Health, Bethesda, MD) to produce maximum projections, and Adobe Photoshop (Adobe Systems, Mountain View, CA) was used to equalize the brightness and contrast in all of the images. The positive signals in the IHC and IF images were quantified from four different animals per experiment (Amplification 40X; 3 sections per mouse) using the Image-Pro Plus 7.0 software program (Media Cybernetics, U.S.A.) and “t student” statistical test (*p<0.05, **p<0.01 and ***p<0.001) were performed using the SPSS 13.0 software package (IBM, U.S.A.).
Wnt-responsive Axin2+/LacZ mice assays
Axin2+/LacZ mice were crossed to NTG, K14E6, or K14E6ΔPDZ mice. Skin tissues were resected from the resulting 7-month-old double transgenic mice and immediately embedded in paraffin for the histological procedures. The skin sections were fixed (0.5% glutaraldehyde, 1.25 mM EGTA and 2 mM MgCl2 in 1X PBS, pH=7.3), washed with 1X PBS that contained detergents (0.01% sodium deoxycholate and 0.02% Igepal CA-630) and 2 mM MgCl2, and then incubated in LacZ staining buffer (0.6 mg/ml X-gal, 4 mM potassium ferrocyanide and 4 mM potassium ferricyanide in wash buffer). The tissues were subsequently counter stained with Nuclear Fast Red and mounted with Permount.
Relative mRNA quantification by RT-qPCR and data analysis using the 2−ΔΔCT method
Real-time qPCR was performed using a LightCycler 2.0 apparatus (Roche, Germany) and a DNA Master SYBR Green I Kit (Roche, U.S.A.). The templates were amplified in 45 cycles of a 3-step PCR process, which included a 30-s of denaturation step at 95°C, a 30-s primer-dependent annealing phase (60°C), and a 30-s template-dependent elongation at 72°C. The amplification of each template was performed in duplicate in one PCR run. The differential expression of the investigated genes was calculated as a ratio normalized to GAPDH gene expression. The data were analyzed using the equation that was previously described by Livak (24) (amount of target = 2−ΔΔCT).
Luciferase assays
COS7 cells were plated in 12-well tissue culture plates and grown for 24 hr. The cells were then co-transfected with either E6, E6ΔPDZ (aa146-151 deleted), E7, Dvl2 (in the pCS2 vector that included a Flag tag and was kindly provided by Dr. Jeff Rubin), β-catenin S37A (pFlagCMV2, which was kindly provided by Dr. Stephen Byers) or the empty vector control alone or together with Super8XTOPFlash (which contains eight copies of TCF/LEF-binding sites upstream of the luciferase gene and was kindly provided by Dr. Randall Moon) and the Renilla-TK construct (transfection control, which was kindly provided by Dr. Stephen Byers) using FuGENE6 according to the manufacturer’s instructions (Roche Applied Science, Mannheim, Germany). All of the E6- and E7-gene-related constructs were cloned into pJS55 vector and tagged with an AU1 epitope at the C-terminus. The Dual-Luciferase Reporter assay was performed 24 h after the transfection according to the manufacturer’s protocol (Promega, U.S.A.).
Immunoprecipitation
COS7 cells were cotransfected with either: Dvl2, E6, E6ΔPDZ or the empty vector alone or together by using FuGENE6. Twenty-four hours after transfection the cells were lysed with phospholysis buffer (50 mM Hepes, pH 7.9, 100 mM NaCl, 4 mM NaPP, 10 mM EDTA, 10 mM NaF, 1% TritonX100, fresh 2 mM fresh vanadate, 1 mM PMSF, 2 μg/mL aprotinin and 2 μg/mL leupeptin). One milligram of the protein lysate protein was subjected to overnight immunoprecipitation with an anti-Dvl2 antibody (Santa Cruz, U.S.A.) and this was followed with a 1-h incubation with protein G agarose beads (Invitrogen, U.S.A.). The immunoprecipitates were subjected to SDS/PAGE and transferred to an Immobilon-P membrane (Millipore). The membrane was blocked in 5% nonfat dry milk in 1X TBS (20 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.5% Tween-20) for 2 h, blotted with an anti-Dvl2 (1:1000) or anti-AU1 (1:1000; Convance, U.S.A.) primary antibody overnight, and then incubated with an HRP-linked anti-mouse or anti-rabbit secondary antibody (GE Healthcare) for 1 h. The membranes were developed using the Millipore Immobilon Western Chemiluminescent HRP Substrate according to the manufacturer’s instructions. Chemiluminescense was detected using a Fujifilm LAS-3000 imaging system.
Results
The E6’s PDZ-binding domain is required to maintain skin hyperplasia in adult transgenic mice
K14E6 and K14E6ΔPDZ mice were previously reported to express the complete and truncated E6 oncoprotein in the basal layer of stratified epithelia, respectively (7, 8). However, of the two transgenic strains, only the K14E6 mice developed skin hyperplasia and skin cancer at advanced ages (7, 8). Based on this observation, we first compared the cell proliferation pattern in the skin epidermis for the E6 transgenic strains and the NTG mice at juvenile and adult ages (Figure 1A).
Figure 1. The E6 PDZ-binding domain is essential for maintaining suprabasal cell proliferation in the adult mouse epidermis.

(A) Immunodetection of PCNA, a cell proliferation marker, in the dorsal skin biopsies of 8-days-old and 4-months-old mice. At 8 days of age, mice expressing either a full-length or truncated E6 oncoprotein (E6 or E6ΔPDZ) display suprabasal cell proliferation and hyperplasia compared to non-transgenic (NTG) mice. However, only the mice containing the complete version of E6 retains suprabasal cell proliferation at 4 months of age. Therefore, the E6 PDZ-binding domain may exert differential oncogenic effects on adult skin. (B) Immunodetection of the p53 tumor suppressor protein in 8-days-old mice 8 h after they were irradiated with 500 mJ/cm2 of UVB. Both forms of the E6 oncoprotein can degrade the p53 protein, which suggests that the differences in their oncogenic effects are independent of p53 protein-degradation capacity. Magnification: 40X. Ep = Epidermis. Dashed line = Basal membrane. Scale bar = 20μm. Arrows = Positive nuclear signal.
The Immunostaining of PCNA, a cell proliferation marker shows, that cell proliferation at a young age was quite similar in the K14E6 and K14E6ΔPDZ mice. However, unlike the K14E6ΔPDZ or NTG mice, only the K14E6 adult mice retained suprabasal cell proliferation (Figure 1A). It was previously reported that both transgenic E6 strains display equally diminished p53 protein expression in adult animals (8). We also found that both of the transgenic strains can degrade the p53 protein in younger animals after ultraviolet light irradiation (Figure 1B). Therefore, the cell proliferation differences between the two transgenic strains are not related to differences in p53 degradation.
To test the functionality of the E6’s PDZ-binding domain in adult mice, we examined the levels of Dlg1 and Scribble (Scrib), which are two well-described PDZ-cellular-targets of HR-E6. Importantly, these targets participate processes such as cell-cell junction formation and canonical Wnt signaling activation (25). We observed that the K14E6 mice displayed lower levels of both cellular proteins compared to the K14E6ΔPDZ counterpart and NTG mice (Figure 2). Therefore, it is possible that the oncogenic effects that were observed in the adult skin epidermis of the K14E6 mice could be partially related, in part, to cellular processes regulated by those PDZ-containing targets including the Wnt pathway.
Figure 2. Functional validation of the role of the E6 PDZ-binding domain in adult mice strains.

Two PDZ-containing E6 targets (Dlg1 and Scribble) were evaluated in dorsal skin biopsies from 4-months-old mice by immunohistochemistry (A) or immunoblotting (B). As we show in graphs, only the K14E6 strain significantly suppressed the Dlg1 or Scribble (Scrib) proteins compared to K14E6ΔPDZ and the non-transgenic (NTG) mice. Magnification: 40X. Ep = Epidermis. Dashed line = Basal membrane. Scale bar = 20 μm.
The E6’s PDZ-binding domain induces nuclear β-catenin protein accumulation and enhances Wnt signaling pathway
To evaluate whether canonical Wnt signaling is activated by the E6 PDZ-binding domain, we detected β-catenin in skin slices from mice at different ages. In eight-day-old mice, β-catenin was not apparent in the cell nucleus; however, from the age of four months onward, an increased amount of β-catenin level was detected in the nuclei of the K14E6 mice and the most pronounced accumulation was observed in 1-year-old mice (Figure 3A). We next asked whether the nuclear β-catenin detected in the skin epidermis of adult mice was transcriptionally active. Axin2 is a negative feedback regulator of the Wnt pathway and is expressed in response to Wnt signaling (26). The insertion of the LacZ gene into the Axin2 locus (Axin2+/LacZ mice) mimics the expression pattern of Axin2, but it does not lead to a detectable axin-deficient phenotype in the heterozygous state (27). Therefore, we crossed Axin2+/LacZ mice with either K14E6, K14E6ΔPDZ or control NTG mice to measure Wnt signaling activity in adult skin samples. LacZ was expressed more strongly in the skin epidermis of the Axin2+/LacZ/K14E6 double transgenic mice compared to the Axin2+/LacZ/K14E6ΔPDZ or Axin2+/LacZ/NTG mice (Figure 3B). We also measured the mRNA levels of three well-described β-catenin targets that participate in cell cycle progression or cell survival c-myc, Birc5 and ccnd1 genes in adult skin. As shown in Figure 4A, the mRNA induction of these targets genes was enhanced in adult skin tissue of the K14E6 mice compared to the K14E6ΔPDZ mice. c-Myc expression was also validated by RT-PCR and Western blotting (Figure 4B). Therefore, we concluded that Wnt signaling may be enhanced in vivo by the HPV16-E6 oncogene, and that the PDZ-binding domain plays an important role in this induction.
Figure 3. β-catenin nuclear cell accumulation and Wnt signaling induction.

Skin tissues from the indicated mice were paraffin embedded. (A) Immunofluorescence detection of the β-catenin protein was performed in 5-μm-thick dorsal skin samples from mice of three 3 different ages as indicated. Note that only the K14E6 mice gradually accumulated nuclear β-catenin, and the highest accumulation occurred at 1 year of age. (B) Wnt/β-catenin signaling induction was evaluated by the Axin2-dependent expression of LacZ gene in the skin epithelia of 7-months-old double transgenic mice. Note that only the Axin2+/LacZ/K14E6 mice show maximal LacZ expression. In (A): Green signal = β-catenin, Red signal = Propidium Iodide (PI) counterstaining, Yellow signal = Merged signal. In (B): Blue signal represents LacZ activity, and counterstaining was performed with Fast Red. Magnification: 40X. Ep = Epidermis. Dashed Line = Basal membrane. Scale bar = 30μm. Arrows = Positive LacZ nuclear signal.
Figure 4. The PDZ-binding domain of E6 is necessary to activate β-catenin-responsive genes in adult dorsal skin.

(A) Relative mRNA expression of three Wnt signaling-responsive target genes (ccnd1, c-myc and birc5) that were evaluated by RT-qPCR. All of the samples were obtained as described in the methods section. (B) RT-PCR and Western blot of the c-Myc gene in non-injured skin from the indicated mice, and from wild type-injured skin (positive control). Note that only the entire E6 protein induces c-Myc protein in a manner that is similar to the wound stimulus.
The E6 oncoprotein associates with Dvl2 and induces canonical Wnt signaling in vitro independently of the PDZ-binding domain in vitro
To analyze Wnt signaling induction in an in vitro system, we transiently transfected COS7 cells with a well-established functional assay vector that contains several copies of the TCF-binding site and regulates a luciferase reporter gene (the TOPFLASH construct). The transfection of the HPV16-E6 expression vector alone results in the minimal expression of the TOPFLASH vector; however, the combination of HPV16-E6 and a Wnt signaling inducer, such as β-catenin or Dvl2 (the Dvl isoform that is preferentially expressed in the skin) clearly enhances TOPFLASH activity (Supplemental Figure 1). Interestingly, only transfected HPV16-E6, but not HPV16-E7 cooperates with β-catenin or Dvl2 to trigger TOPFLASH induction (Supplemental Figure 1).
To determine whether the E6 PDZ-binding domain is involved in Wnt pathway induction in vitro, we co-transfected COS7 cells with constructs expressing the complete HPV-E6 protein, or a truncated version lacking the last 6 amino acids which comprise the PDZ-binding domain. In contrast to our in vivo observations, and consistent with the report of Lichtig et al. data (9), both versions of the E6 oncoprotein cooperated with β-catenin or Dvl2 and induced Wnt signaling (Figure 5A). We also performed these in vitro experiments with primary human foreskin keratinocytes and obtained similar results to those that were observed in the COS7 cells (Supplemental Figure 2).
Figure 5. HPV16-E6 enhances canonical Wnt/β-catenin pathway activation and interacts with the Dvl2 protein independently of the E6 PDZ-binding domain.
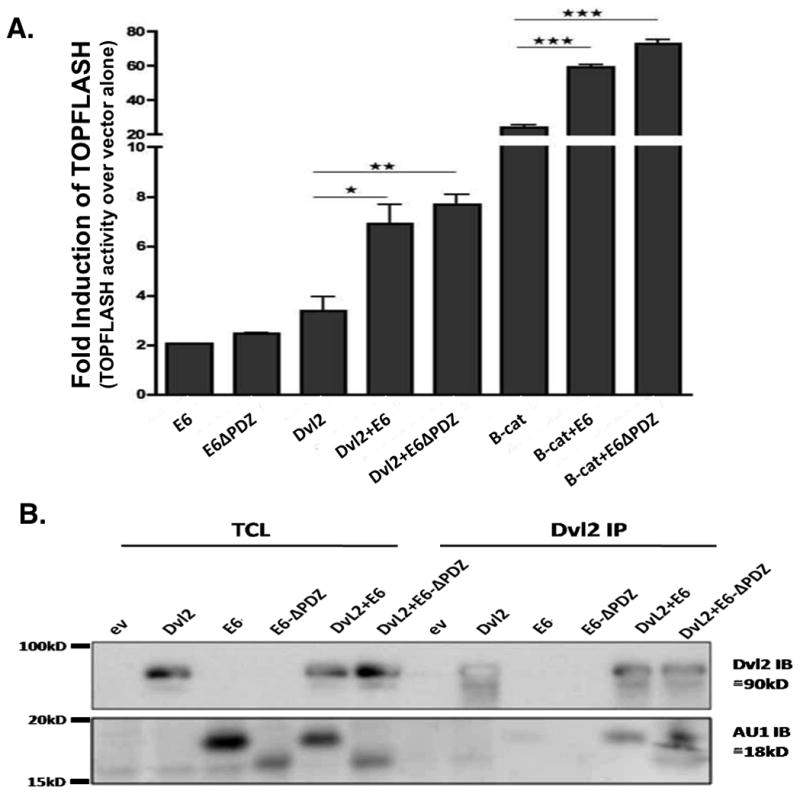
(A) Wnt/β-catenin pathway activation was assessed using the TOPFLASH construct. COS7 cells were co-transfected with, E6, HPV16-E6Δ141-151 (E6ΔPDZ), Dvl2 and β-catenin (B-cat) expression vectors. Both full-lenght E6 and E6 lacking the PDZ-binding domain (E6ΔPDZ) minimally activated the canonical Wnt pathway. Both constructs show very significant enhancement when co-transfected with Dvl2 or B-cat, which suggests that this effect was independent of the PDZ-binding domain in our short-term in vitro assays. (B) COS7 cells were transfected with expression vectors encoding Dvl2, E6 and E6ΔPDZ alone or in combination. The cells were lysed 24 h after the transfection, and the total cell lysate (TCL) was subjected to immunoprecipitation (IP) with an anti-Dvl2 antibody. The immunoprecipitates were then subjected to SDS-PAGE and blotted with anti-Dvl2 and anti-AU1 antibodies. Dvl2 was detected at the expected 90-kD size (upper panel). E6 and E6ΔPDZ, which contain AU1 tags, were detected with the anti-AU1 antibody at the expected size (18-kD). All of the constructs were expressed at comparable levels. Both, E6 and E6ΔPDZ interact with Dvl2.
Dvl proteins play a central role in the regulation of both canonical and non-canonical Wnt signaling. There are three Dvl isoforms in mammals (28), all them containing PDZ domains (29). Because E6 contains a PDZ-binding domain, it was possible to consider a possible PDZ-connection with Dvl isoforms. To test this possibility, we immunoprecipitated E6 and blotted against the three Dvl isoforms (and vice versa) E6 co-immunoprecipitated E6 with the Dvl2 protein but not with the Dvl1 or 3 isoforms (Supplemental Figure 3) which suggests that the E6 oncoprotein preferentially binds the Dvl isoform that is expressed in the skin (30). We next tested the ability of E6 to interact with the Dvl2 protein via its PDZ-domain using the E6 or E6ΔPDZ expression vectors. Interestingly, both of the E6 oncoproteins co-immunoprecipitated with Dvl2 (Figure 5B), which suggests the presence of a PDZ-independent interaction. Therefore, E6 proteins that contain or lack the PDZ domain could enhance Wnt signaling and interact with the Dvl2 protein in short-term in vitro assays.
Discussion
The K14E6 mouse strains has been established as a model of papilloma-induced skin cancer (7). In this model, the expression of the HPV16-E6 oncogene was directed to the basal layer of stratified epithelia which includes the skin epidermis, lens and cervical tissue, among others (7, 31). Although E6 is virtually expressed in all stratified epithelia, it only develops spontaneous dysplasias or tumors in the skin epidermis (not in other tissues such as the cervix) (32, 33), which suggests that tissue-specific factors are critical for mediating the oncogenic potential of E6 (7, 8, 33). In an attempt to explain discrepancies that occur due to tissue type, we recently reported the global gene-expression differences in the skin epidermis and cervical tissue in adult K14E6 mice (33). We found that the expression of genes that participate in cell adhesion and Wnt signaling were preferentially altered in the skin epidermis compared to the cervical tissue (33). Based on these observations, we used the skin epidermis of K14E6 mice as a functional model to study the relationship between the E6 oncoprotein and Wnt signaling.
The in vivo data that were obtained with the K14E6ΔPDZ mouse model suggests that E6’s oncogenic potential in skin epidermis relies on its PDZ-binding domain (8, 34). In this study, we show that although K14E6 and K14E6ΔPDZ mice have a quite similar hyperproliferative skin phenotype at eight days of age (Figure 1A) only K14E6 mice retain the hyperproliferative skin phenotype, at four months of age, which suggests that E6’s early hyperproliferative effects on the skin are independent of the PDZ-binding domain. However, the PDZ-binding domain is indeed required to maintain suprabasal cell proliferation in adult mice (Figure 1).
Most of the previous studies regarding HPV-positive cervical lesions support the view that Wnt signaling represents a late-stage step in cervical carcinogenesis and that other factors are needed to activate this pathway (19–22, 35). Unlike its role in cervical tissues, E6 is sufficient to induce skin papillomas and cancer in K14E6 mice (7); however, similarly as to cervical tissue, Wnt signaling occurs only after PDZ-independent hyperproliferation. As demonstrated in Figure 3A, the adult K14E6 animals accumulated β-catenin in their skin-cell nuclei but the precise mechanism by which E6 enhances Wnt pathway is beyond the scope of this study. A possible mechanism by which E6 induces Wnt pathway activation may involve its effects on cellular targets that containin PDZ domains such as Dlg1 and Scribble. It is well-known that Dlg1 and Scribble proteins (both containing PDZ domains) participate in the zonula adherens formation (36), and they also interact whit APC protein in a region necessary for tumor suppressor activity (37, 38). Furthermore, APC-hDlg complexes were shown to negatively regulate cell cycle progression in NIH3T3 mouse fibroblast (39). Therefore, E6-induced degradation of the Dlg1 and Scribble (5) may lead to decreased APC activity and the nuclear accumulation of β-catenin.
It is difficult to know whether the slight reduction in Dlg1 that was observed in the K14E6 mice (Figure 2) is sufficient to activate Wnt signaling or whether the partial loss of Dlg1 synergizes with the more sharply reduced Scribble protein levels (Figure 2). Other evidence suggests that the reduction of Dlg1 alone may not lead to β-catenin nuclear accumulation (39). Therefore, Dlg1 reduction may not be the primary cause of Wnt signaling enhancement by E6. However, considering current data, we cannot completely discount a potential role for Dlg1 in Wnt signaling induction (at least in in vivo models). Importantly, in vitro models may not reflect the mechanisms that occur in vivo. Consistent with these findings, Cavatorta et al. also reported the presence of only a slight reduction in the hDlg protein in benign HPV-infected cervical lesions (in which nuclear β-catenin is uncommon), but they reported a more dramatic reduction in cervical cancers (in which nuclear β-catenin is frequent) (40). Therefore, low levels of the hDlg protein and nuclear β-catenin may be considered as late-stage markers in HPV-related carcinogenesis (22, 40).
Short-term in vitro experiments in COS7 cells and primary human foreskin keratinocytes (Figure 5 and Supplemental Figures 1–2) demonstrated that exogenous expression of the HPV16-E6 oncoprotein alone only weakly induced Wnt signaling, which was indicated by TOPFLASH induction, but it demonstrated a significant effect in combination with exogenous β-catenin or Dvl2 expression. Lichtig et al. reported similar results in HEK293T cells although their short-term experiments were performed in a Wnt3a-enriched medium. Therefore, the E6 oncoprotein appears to enhance, but not to initiate Wnt signaling (9). Consistent with this view, we did not observe any nuclear accumulation of β-catenin in the young K14E6 animals (Figure 3A).
Concerning the role of the E6 PDZ-binding domain, the discrepancies between our in vitro and in vivo observations may be due to the use of transient (as opposed to stable) transfections and by the fact that E6 may alter cellular pathways in younger animals in a PDZ-independent manner, which is not necessarily represented in short-term cell cultures. In this study, we also demonstrate an interaction between E6 and the Dvl2 protein isoform, which is a key player in both canonical and non-canonical Wnt signaling (Supplemental Figure 3 and Figure 5B). The in E6 interaction with Dvl2 seems to be independent of its PDZ-binding domain. This interaction may provide a possible mechanism by which E6 initiates Wnt signaling in a PDZ-independent manner, although the ultimate accumulation and translocation of β-catenin may require the E6 PDZ-binding domain in vivo.
This work demonstrates for the first time that HPV16-E6 can enhance canonical Wnt signaling in vivo and in vitro. However, our paradoxical findings regarding the E6’s PDZ-binding domain suggests that the interaction between E6 and the Wnt pathway is complex, and it may be dependent on the status of other signaling pathways. The precise mechanism for Wnt signaling activation remains unknown, and more studies are needed to clarify the mechanism by which E6 enhances Wnt signaling.
Supplementary Material
(A) COS7 cells were transiently transfected with the TCF/B-catenin dependent luciferase reporter construct (TOPFLASH) and Renilla together with one or two of the following expression vectors encoding the following: HPV16-E6 (E6), HPV16-E7 (E7), Dvl2 or β-catenin (B-cat). Luciferase activity from culture cells was analyzed 48 h after transfection and is shown as normalized to total protein. HPV16-E6 but not E7 significantly induced TOPFLASH activity with or without B-cat cotransfection (A) or Dvl2 cotransfection (B) co-transfection, which suggests that E6 may positively modulate canonical Wnt signaling. The Renilla signal and empty vector were used to normalize luciferase activity.
Wnt/β-catenin pathway activation was assessed using the TOPFLASH construct. Human foreskin keratinocytes were co-transfected with, E6, HPV16-E6Δ141-151 (E6ΔPDZ), Dvl2 and B-cat expression vectors. Both, E6 and the mutated form lacking the PDZ-binding domain (E6ΔPDZ), minimally activated the canonical Wnt pathway. However, both of the constructs induced very significant enhancement when they were co-transfected with Dvl2 or β-catenin (B-cat), which suggests that this effect was independent of the PDZ-binding domain.
COS7 cells were transiently transfected with HPV-E6 and one of the Dvl cDNAs. HPV-E6 was tagged with AU1, and the Dvl genes included Flag tags. Cell lysates were prepared 48 h after transfection, and immunoprecipitation was performed with either an anti-flag antibody or anti-AU1 antibody. The immunoprecipitates were separated on a 10% acrylamide gel and transferred to membranes, and western blots were performed using anti-Flag and anti-AU1 antibodies. The Dvl proteins are indicated by arrows and the E6 proteins are indicated by the arrowheads. All three of the Dvl proteins (upper left panel) and HPV-E6 (lower right panel) were expressed at the expected size. The upper-right and lower-left panels show that Dvl2 and E6 may exist in the same protein complex. The first lane on each panel represents the resulting transfection with the empty vector as the negative control.
Acknowledgments
The authors thank Tzitzijanik Madrigal-Domínguez, Rodolfo Ocádiz-Delgado, Enrique Garcia-Villa and Elizabeth Álvares-Ríos for their technical support. We also thank Dr. Gustavo Acosta-Altamirano and Dr. Martín Antonio-Manrique for their financial support. Finally, we thank Gabriela Mora-Macias for her secretarial assistance.
Grant Support:
This work was supported by CONACyT (Grant numbers: 83597 and 127335). During this work, JBD was the recipient of a fellowship from CONACyT and COMECyT.
Contributor Information
José Bonilla-Delgado, Email: jbonilla@cinvestav.mx.
Gülay Bulut, Email: gb243@georgetown.edu.
Xuefeng Liu, Email: xl24@georgetown.edu.
Enoc M. Cortés-Malagón, Email: cormalagon@yahoo.com.mx.
Richard Schlegel, Email: schleger@georgetown.edu.
Catalina Flores-Maldonado, Email: ceflores@fisio.cinvestav.mx.
Rubén G. Contreras, Email: rcontrer@fisio.cinvestav.mx.
Sang-Hyuk Chung, Email: schung2@central.uh.edu.
Paul F. Lambert, Email: plambert@wisc.edu.
Aykut Üren, Email: au26@georgetown.edu.
Patricio Gariglio, Email: vidal@cinvestav.mx.
References
- 1.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 2.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–9. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 3.Narisawa-Saito M, Kiyono T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci. 2007;98:1505–11. doi: 10.1111/j.1349-7006.2007.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLaughlin-Drubin ME, Munger K. The human papillomavirus E7 oncoprotein. Virology. 2009;384:335–44. doi: 10.1016/j.virol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tungteakkhun SS, Duerksen-Hughes PJ. Cellular binding partners of the human papillomavirus E6 protein. Arch Virol. 2008;153:397–408. doi: 10.1007/s00705-007-0022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wazer DE, Liu XL, Chu Q, Gao Q, Band V. Immortalization of distinct human mammary epithelial cell types by human papilloma virus 16 E6 or E7. Proc Natl Acad Sci U S A. 1995;92:3687–91. doi: 10.1073/pnas.92.9.3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song S, Pitot HC, Lambert PF. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J Virol. 1999;73:5887–93. doi: 10.1128/jvi.73.7.5887-5893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen ML, Nguyen MM, Lee D, Griep AE, Lambert PF. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6’s induction of epithelial hyperplasia in vivo. J Virol. 2003;77:6957–64. doi: 10.1128/JVI.77.12.6957-6964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lichtig H, Gilboa DA, Jackman A, Gonen P, Levav-Cohen Y, Haupt Y, et al. HPV16 E6 augments Wnt signaling in an E6AP-dependent manner. Virology. 2010;396:47–58. doi: 10.1016/j.virol.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 10.Goodwin AM, D’Amore PA. Wnt signaling in the vasculature. Angiogenesis. 2002;5:1–9. doi: 10.1023/a:1021563510866. [DOI] [PubMed] [Google Scholar]
- 11.Millar SE, Willert K, Salinas PC, Roelink H, Nusse R, Sussman DJ, et al. WNT signaling in the control of hair growth and structure. Dev Biol. 1999;207:133–49. doi: 10.1006/dbio.1998.9140. [DOI] [PubMed] [Google Scholar]
- 12.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 13.Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, et al. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci U S A. 1999;96:1603–8. doi: 10.1073/pnas.96.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 15.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 16.Yamada T, Takaoka AS, Naishiro Y, Hayashi R, Maruyama K, Maesawa C, et al. Transactivation of the multidrug resistance 1 gene by T-cell factor 4/beta-catenin complex in early colorectal carcinogenesis. Cancer Res. 2000;60:4761–6. [PubMed] [Google Scholar]
- 17.Crawford HC, Fingleton BM, Rudolph-Owen LA, Goss KJ, Rubinfeld B, Polakis P, et al. The metalloproteinase matrilysin is a target of beta-catenin transactivation in intestinal tumors. Oncogene. 1999;18:2883–91. doi: 10.1038/sj.onc.1202627. [DOI] [PubMed] [Google Scholar]
- 18.Yan D, Wiesmann M, Rohan M, Chan V, Jefferson AB, Guo L, et al. Elevated expression of axin2 and hnkd mRNA provides evidence that Wnt/beta -catenin signaling is activated in human colon tumors. Proc Natl Acad Sci U S A. 2001;98:14973–8. doi: 10.1073/pnas.261574498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shinohara A, Yokoyama Y, Wan X, Takahashi Y, Mori Y, Takami T, et al. Cytoplasmic/nuclear expression without mutation of exon 3 of the beta-catenin gene is frequent in the development of the neoplasm of the uterine cervix. Gynecol Oncol. 2001;82:450–5. doi: 10.1006/gyno.2001.6298. [DOI] [PubMed] [Google Scholar]
- 20.Imura J, Ichikawa K, Takeda J, Fujimori T. Beta-catenin expression as a prognostic indicator in cervical adenocarcinoma. Int J Mol Med. 2001;8:353–8. [PubMed] [Google Scholar]
- 21.Pereira-Suarez AL, Meraz MA, Lizano M, Estrada-Chavez C, Hernandez F, Olivera P, et al. Frequent alterations of the beta-catenin protein in cancer of the uterine cervix. Tumour Biol. 2002;23:45–53. doi: 10.1159/000048688. [DOI] [PubMed] [Google Scholar]
- 22.Uren A, Fallen S, Yuan H, Usubutun A, Kucukali T, Schlegel R, et al. Activation of the canonical Wnt pathway during genital keratinocyte transformation: a model for cervical cancer progression. Cancer Res. 2005;65:6199–206. doi: 10.1158/0008-5472.CAN-05-0455. [DOI] [PubMed] [Google Scholar]
- 23.Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22:1184–93. doi: 10.1128/MCB.22.4.1184-1193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Nagafuchi A. Molecular architecture of adherens junctions. Curr Opin Cell Biol. 2001;13:600–3. doi: 10.1016/s0955-0674(00)00257-x. [DOI] [PubMed] [Google Scholar]
- 26.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–83. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soshnikova N, Zechner D, Huelsken J, Mishina Y, Behringer RR, Taketo MM, et al. Genetic interaction between Wnt/beta-catenin and BMP receptor signaling during formation of the AER and the dorsal-ventral axis in the limb. Genes Dev. 2003;17:1963–8. doi: 10.1101/gad.263003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakanaka C, Sun TQ, Williams LT. New steps in the Wnt/beta-catenin signal transduction pathway. Recent Prog Horm Res. 2000;55:225–36. [PubMed] [Google Scholar]
- 29.Wong HC, Bourdelas A, Krauss A, Lee HJ, Shao Y, Wu D, et al. Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol Cell. 2003;12:1251–60. doi: 10.1016/s1097-2765(03)00427-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Charpentier E, Lavker RM, Acquista E, Cowin P. Plakoglobin suppresses epithelial proliferation and hair growth in vivo. J Cell Biol. 2000;149:503–20. doi: 10.1083/jcb.149.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song S, Liem A, Miller JA, Lambert PF. Human papillomavirus types 16 E6 and E7 contribute differently to carcinogenesis. Virology. 2000;267:141–50. doi: 10.1006/viro.1999.0106. [DOI] [PubMed] [Google Scholar]
- 32.Elson DA, Riley RR, Lacey A, Thordarson G, Talamantes FJ, Arbeit JM. Sensitivity of the cervical transformation zone to estrogen-induced squamous carcinogenesis. Cancer Res. 2000;60:1267–75. [PubMed] [Google Scholar]
- 33.Mendoza-Villanueva D, Diaz-Chavez J, Uribe-Figueroa L, Rangel-Escareao C, Hidalgo-Miranda A, March-Mifsut S, et al. Gene expression profile of cervical and skin tissues from human papillomavirus type 16 E6 transgenic mice. BMC Cancer. 2008;8:347. doi: 10.1186/1471-2407-8-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simonson SJ, Difilippantonio MJ, Lambert PF. Two distinct activities contribute to human papillomavirus 16 E6’s oncogenic potential. Cancer Res. 2005;65:8266–73. doi: 10.1158/0008-5472.CAN-05-1651. [DOI] [PubMed] [Google Scholar]
- 35.Rampias T, Boutati E, Pectasides E, Sasaki C, Kountourakis P, Weinberger P, et al. Activation of Wnt signaling pathway by human papillomavirus E6 and E7 oncogenes in HPV16-positive oropharyngeal squamous carcinoma cells. Mol Cancer Res. 2010;8:433–43. doi: 10.1158/1541-7786.MCR-09-0345. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen MM, Rivera C, Griep AE. Localization of PDZ domain containing proteins Discs Large-1 and Scribble in the mouse eye. Mol Vis. 2005;11:1183–99. [PubMed] [Google Scholar]
- 37.Matsumine A, Ogai A, Senda T, Okumura N, Satoh K, Baeg GH, et al. Binding of APC to the human homolog of the Drosophila discs large tumor suppressor protein. Science. 1996;272:1020–3. doi: 10.1126/science.272.5264.1020. [DOI] [PubMed] [Google Scholar]
- 38.Takizawa S, Nagasaka K, Nakagawa S, Yano T, Nakagawa K, Yasugi T, et al. Human scribble, a novel tumor suppressor identified as a target of high-risk HPV E6 for ubiquitin-mediated degradation, interacts with adenomatous polyposis coli. Genes Cells. 2006;11:453–64. doi: 10.1111/j.1365-2443.2006.00954.x. [DOI] [PubMed] [Google Scholar]
- 39.Ishidate T, Matsumine A, Toyoshima K, Akiyama T. The APC-hDLG complex negatively regulates cell cycle progression from the G0/G1 to S phase. Oncogene. 2000;19:365–72. doi: 10.1038/sj.onc.1203309. [DOI] [PubMed] [Google Scholar]
- 40.Cavatorta AL, Fumero G, Chouhy D, Aguirre R, Nocito AL, Giri AA, et al. Differential expression of the human homologue of drosophila discs large oncosuppressor in histologic samples from human papillomavirus-associated lesions as a marker for progression to malignancy. Int J Cancer. 2004;111:373–80. doi: 10.1002/ijc.20275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) COS7 cells were transiently transfected with the TCF/B-catenin dependent luciferase reporter construct (TOPFLASH) and Renilla together with one or two of the following expression vectors encoding the following: HPV16-E6 (E6), HPV16-E7 (E7), Dvl2 or β-catenin (B-cat). Luciferase activity from culture cells was analyzed 48 h after transfection and is shown as normalized to total protein. HPV16-E6 but not E7 significantly induced TOPFLASH activity with or without B-cat cotransfection (A) or Dvl2 cotransfection (B) co-transfection, which suggests that E6 may positively modulate canonical Wnt signaling. The Renilla signal and empty vector were used to normalize luciferase activity.
Wnt/β-catenin pathway activation was assessed using the TOPFLASH construct. Human foreskin keratinocytes were co-transfected with, E6, HPV16-E6Δ141-151 (E6ΔPDZ), Dvl2 and B-cat expression vectors. Both, E6 and the mutated form lacking the PDZ-binding domain (E6ΔPDZ), minimally activated the canonical Wnt pathway. However, both of the constructs induced very significant enhancement when they were co-transfected with Dvl2 or β-catenin (B-cat), which suggests that this effect was independent of the PDZ-binding domain.
COS7 cells were transiently transfected with HPV-E6 and one of the Dvl cDNAs. HPV-E6 was tagged with AU1, and the Dvl genes included Flag tags. Cell lysates were prepared 48 h after transfection, and immunoprecipitation was performed with either an anti-flag antibody or anti-AU1 antibody. The immunoprecipitates were separated on a 10% acrylamide gel and transferred to membranes, and western blots were performed using anti-Flag and anti-AU1 antibodies. The Dvl proteins are indicated by arrows and the E6 proteins are indicated by the arrowheads. All three of the Dvl proteins (upper left panel) and HPV-E6 (lower right panel) were expressed at the expected size. The upper-right and lower-left panels show that Dvl2 and E6 may exist in the same protein complex. The first lane on each panel represents the resulting transfection with the empty vector as the negative control.