

Abstract
AIM: To investigate the effects of tectorigenin on human hepatocellular carcinoma (HCC) HepG2 cells.
METHODS: Tectorigenin, one of the main components of rhizome of Iris tectorum, was prepared by simple methods, such as extraction, filtration, concentration, precipitation and recrystallization. HepG2 cells were incubated with tectorigenin at different concentrations, and their viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Apoptosis was detected by morphological observation of nuclear change, agarose gel electrophoresis of DNA ladder, and flow cytometry with Hoechst 33342, Annexin V-EGFP and propidium iodide staining. Generation of reactive oxygen species was quantified using DCFH-DA. Intracellular Ca2+ was monitored by Fura 2-AM. Mitochondrial membrane potential was monitored using Rhodamine 123. Release of cytochrome c from mitochondria to cytosol was detected by Western blotting. Activities of caspase-3, -8 and -9 were investigated by Caspase Activity Assay Kit.
RESULTS: The viability of HepG2 cells treated by tectorigenin decreased in a concentration- and time-dependent manner. The concentration that reduced the number of viable HepG2 cells by 50% (IC50) after 12, 24 and 48 h of incubation was 35.72 mg/L, 21.19 mg/L and 11.06 mg/L, respectively. However, treatment with tectorigenin at 20 mg/L resulted in a very slight cytotoxicity to L02 cells after incubation for 12, 24 or 48 h. Tectorigenin at a concentration of 20 mg/L greatly inhibited the viability of HepG2 cells and induced the condensation of chromatin and fragmentation of nuclei. Tectorigenin induced apoptosis of HepG2 cells in a time- and dose-dependent manner. Compared with the viability rate, induction of apoptosis was the main mechanism of the anti-proliferation effect of tectorigenin in HepG2 cells. Furthermore, tectorigenin-induced apoptosis of HepG2 cells was associated with the generation of reactive oxygen species, increased intracellular [Ca2+]i, loss of mitochondrial membrane potential, translocation of cytochrome c, and activation of caspase-9 and -3.
CONCLUSION: Tectorigenin induces apoptosis of HepG2 cells mainly via mitochondrial-mediated pathway, and produces a slight cytotoxicity to L02 cells.
Keywords: Tectorigenin, Iris tectorum maxim, Apoptosis, Hepatocellular carcinoma, HepG2, Mitochondria, Liver cancer
INTRODUCTION
Hepatocellular carcinoma (HCC) is the third most common cause of cancer-related mortality worldwide, with 600 000 deaths per year[1]. It often develops in patients with chronic liver diseases associated with hepatitis B virus or hepatitis C virus infections[1]. Although surgical techniques have been improved and several non-surgical treatment modalities have been developed, there is still no effective therapy for significant improvement of extremely poor prognosis of HCC patients[2]. Therefore, efforts have been made to search for mechanism-based agents, such as sorafenib[3], chelidonine[4], 5-allyl-7-gen-difluoromethylenechrysin[5], troglitazone[6], chaga mushroom extract[7] and curcumin[8], for treatment of HCC. Sorafenib is the first substance that proved to significantly prolong the survival of HCC patients. And multikinase inhibitor has shown to have anti-proliferative and anti-angiogenic properties[3].
Apoptosis is a physiological process leading to cell deletion and regulates the balance between cell proliferation and death. The hallmark of cancer cells is the dysregulation of cell proliferation and apoptosis. The tumor growth depends on the cell proliferation rate and apoptosis. Therefore, induction of apoptosis of tumor cells has become a strategy in cancer treatment[5-7]. The integration of multiple survival and death signals determines whether a cell survives or undergoes apoptosis. In recent years, mitochondrial pathways and death receptor pathways have been identified as the two major mechanisms for induction of apoptosis[9,10].
Iris tectorum (I. tectorum), a traditional Chinese medicine, has been widely used for treating liver-related diseases, including hepatitis, liver fibrosis, liver cirrhosis and liver cancer. As one of the main ingredients of I. tectorum rhizome[11], tectorigenin has been reported to have in vivo and in vitro anti-angiogenic activities[12]. A previous study also showed that tectorigenin possessed anti-tumor activities in mice implanted with murine Lewis lung carcinoma or bearing sarcoma 180[12]. Our previous studies revealed that tectorigenin possessed anti-proliferative and pro-apoptotic effects on hepatic stellate cells (HSCs)[13]. However, until now there has been no report about the anti-tumor effect of tectorigenin on human HCC HepG2 cells and the associated mechanisms. Considering that tectorigenin is one of the main components in rhizome of I. tectorum which had been used for the treatment of liver cancer for centuries, we hypothesized that tectorigenin may have anti-proliferation and pro-apoptosis effects on human HCC HepG2 cells. The aim of the present study was to examine whether tectorigenin could suppress the proliferation of HepG2 cells and induce apoptosis of HepG2 cells.
MATERIALS AND METHODS
Reagents
All reagents and solvents were purchased from commercial suppliers and were used without further purification. Roswell Park Memorial Institute (RPMI)-1640 medium was purchased from HyClone (Hyclone, UT, United States). Fetal bovine serum (FBS) was purchased from Hangzhou Sijiqing Biological Engineering Materials Co., Ltd. (Hangzhou, China). Annexin V-EGFP was purchased from PharMingen (San Diego, CA, United States). Propidium iodide (PI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Hoechst H33258, DCFH-DA, ethidium bromide (EB), Proteinase K, RNase A and Rhodamine 123 were purchased from Sigma Aldrich Co. (St. Louis, MO, United States). Fura 2-AM was purchased from Dojindo Laboratories (Kumamoto, Japan). The kits used for caspase activity assays were obtained from Beyotime Institute of Biotechnology (Nantong, Jiangsu, China). All other chemicals were of analytic grade.
Plant materials
The rhizomes of I. tectorum were collected at Dafeng, Jiangsu Province, China. The voucher specimen (the registration number NJU-603) was identified by Prof. Gong ZN, Nanjing Normal University and deposited at the herbarium of Nanjing University, Nanjing, Jiangsu Province, China.
Preparation of tectorigenin
The rhizomes (400 g) of I. tectorum, after ground on an electrical grinder, were extracted twice with 2000 mL of 80% ethanol under reflux for 3 h. The filtrate of the obtained 80% ethanol extract was condensed in vacuum to afford a dark-brown residue called Crude-Extract (73.1 g). The Crude-Extract was dissolved in 4000 mL 3% Na2CO3 solution at 85 °C, and then the solution was filtrated. The pH of the filtrate was regulated to 3-4 by adding 18% HCl, and then the crude isoflavone part was precipitated from the acid liquor. The formed crude isoflavone (23.2 g) was collected by filtration and dissolved in 2000 mL 5% NaHCO3 solution at 85 °C. The 5% NaHCO3 solution was extracted with 2000 mL EtOAc twice. The EtOAc extract was condensed in vacuum to afford a light-brown residue (11.5 g). The residue was washed by 2000 mL hot distilled water and 2000 mL ligarine successively, and sequentially dried at 50 °C. The dried residue (7.3 g) was recrystallized in ethanol twice to afford pure compound (4.45 g).
Cell culture and treatment
Human liver carcinoma HepG2 cells were purchased from the Shanghai Institute of Cell Biology (Shanghai, China). These cells were grown in RPMI 1640 medium with 10% FBS in the presence of 100 U/mL penicillin and 100 μg/mL streptomycin and maintained at 37 °C in a humidified atmosphere containing 5% (v/v) CO2. L02 cells (a human hepatocyte cell line), purchased from Xiangya Central Experiment Laboratory, Central South University, China, were cultured in Dulbecco’s Modified Eagle Medium (Gibco, NY, United States) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% FBS, in a humidified atmosphere containing 5% (v/v) CO2 at 37 °C. Tectorigenin was dissolved in dimethyl sulfoxide (DMSO) at 20 mg/mL as a stock solution and diluted to the required concentration with fresh medium immediately before use. The final DMSO concentration in cultures was < 0.1% (v/v), which did not influence cell growth when compared with the vehicle-free controls. Cells grown in the media containing an equivalent amount of DMSO without tectorigenin served as control.
Cell viability assay
Cell viability was assessed by MTT method. Briefly, cells were seeded in 96-well plate at a density of 1 × 104 cells/well. After 24 h incubation, tectorigenin at different concentrations was added to the cells while only DMSO (solvent) was added as a negative control. After growing for 12, 24 and 48 h, cells were incubated with MTT (0.5 mg/mL) for 4 h at 37 °C. During this incubation period, water-insoluble formazan crystals were formed, which were dissolved by the addition of 100 μL/well DMSO. The optical densities (A) at 570 nm were measured using an enzyme-linked immunosorbent assay plate reader. Wells containing culture medium and MTT but no cells acted as blanks. The percentage of cell viability was calculated as follows: Adrug-blank/Acontrol-blank × 100%.
Morphological observation of nuclear change
Cell morphological assay was carried out according to Wu et al[13] with minor modification. In this assay, cells were seeded in 24-well plates and treated with tectorigenin or vehicle (control). After 48 h incubation, cells were washed carefully with phosphate buffered saline (PBS), fixed with 4% paraformaldehyde for 10 min at room temperature, then washed with pre-chilled PBS three times and exposed to 5 μg/mL Hoechst 33258 at room temperature in the dark for 20 min. Samples were observed under a fluorescent microscope (Nikon UFX-II, Japan). Cells showing cytoplasmic and nuclear shrinkage, chromatin condensation or fragmentation, were defined as apoptotic cells.
Determination of DNA fragmentation
The integrity of DNA was assessed by agarose gel electrophoresis. Cells were harvested after 48 h incubation with 20 μg/mL tectorigenin by centrifugation (300 × g, 5 min) and lysed in lysis buffer containing 10 mmol/L Tris-HCl (pH 7.4), 10 mmol/L ethylenediaminetetraacetic acid (EDTA), and 0.1% of Triton X-100. Then cells were incubated with RNase A at 37 °C for 60 min and proteinase K at 50 °C for 120 min, respectively. After centrifugation (2000 × g, 10 min), supernatants were transferred to new tubes and precipitated by the addition of 0.5 volume of 7.5 mol/L ammonium acetate and 2.5 volumes of ethanol overnight. After centrifugation at 600 × g for 5 min, the pellets were dissolved in Tris-HCl EDTA buffer (TE buffer) (10 mmol/L Tris-HCl, pH 8.0, 1 mmol/L EDTA) and loaded on 1.5% agarose gel for electrophoresis. The gel was stained with EB and photographed with ultraviolet illumination.
Assessment of apoptosis
Apoptosis could be determined by staining cells with Annexin V-EGFP and propidium iodide labeling. In this study, apoptosis was asssessed according to Hai et al[14]. Briefly, cells were harvested after having exposed to the indicated concentrations of tectorigenin for 24 or 48 h, washed twice with cold PBS and then resuspended in 1 mL binding buffer (10 mmol/L HEPES/NaOH (pH 7.4), 140 mmol/L NaCl, 2.5 mmol/L CaCl2) at a concentration of 1 × 106 cells/mL. The cells were incubated with 5 μL Annexin V-EGFP (300 mg/L) for 10 min, and then 10 μL of 20 mg/L PI for 30 min in the dark. Cell fluorescence was measured on FACScan flow cytometer (Becton Dickinson) using an argon ion laser (488 nm).
Measurement of reactive oxygen species generation
The cellular reactive oxygen species (ROS) was quantified using DCFH-DA according to Shi et al[15]. Cells were seeded in black 96-well plates and incubated with tectorigenin at indicated concentrations for 1, 3, 6 or 24 h. Then cells were washed with PBS twice and incubated with 100 μmol/L DCFH-DA in the loading medium in 5% CO2/95% air at 37 °C for 30 min. After DCFH-DA was removed, the cells were washed with PBS and DCFH-DA-loaded cells were read in a Safire (Tecan, Crailsheim, Germany) fluorescence plate reader (excitation, 485 ± 12 nm; emission, 530 ± 12 nm). The fold increase in fluorescence per well was calculated by the formula [Fti/Ft0], where Ft0 is the fluorescence without tectorigenin treatment and Fti is the fluorescence with the tectorigenin treatment at the indicated concentration.
Measurement of intracellular Ca2+
Intracellular Ca2+ ([Ca2+]i) was monitored using the fluorescent Ca2+-sensitive dye, Fura 2-acetoxymethy ester (Fura 2-AM)[15]. Cells were seeded in black 96-well plates and incubated with tectorigenin at the indicated concentration for 3, 6 or 24 h. The cells were washed with PBS twice and incubated with 1 μmol/L Fura 2-AM which was dissolved in HEPES buffer saline (20 mmol/L HEPES, 115 mmol/L NaCl, 5.4 mmol/L KCl, 1.8 mmol/L CaCl2, 0.8 mmol/L MgCl2, 13.8 mmol/L glucose, pH 7.4) for 30 min in the dark at 37 °C in a humidified incubator. They were then gently rinsed with HEPES buffer saline twice and incubated with HEPES buffer saline for 60 min at 37 °C in a humidified incubator. The fluorescence was measured at an emission wavelength of 510 nm and an excitation wavelength of 340 and 380 nm on a Safire (Tecan, Crailsheim, Germany) fluorescence plate reader. The ratio of fluorescence intensity of 340-380 nm (F340/F380) was used to estimate intracellular free calcium.
Measurement of mitochondrial membrane potential
As an index to determine mitochondrial dysfunction, mitochondrial membrane potential (MMP) was monitored using Rhodamine 123[16]. Cells were treated with tectorigenin at indicated concentration for 3, 6 or 24 h in a 6-well culture plate at 1 × 105 cells/mL. The medium was then removed and washed three times with serum-free RPMI 1640 medium followed by incubation in fresh serum-free medium containing 3 mg/L Rhodamine 123 at 37 °C in dark for 30 min. Finally, the cells were collected and washed twice with PBS and then analyzed by a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ, United States).
Western blotting
HepG2 cells were seeded into 60-mm dishes (1 × 106 cells/dish). On the next day, after treated for 48 h with tectorigenin at 0, 5, 10 and 20 mg/L, respectively, HepG2 cells were harvested, resuspended in an ice-cold lysis buffer containing 50 mmol/L Tris-HCl, pH 8.0, 50 mmol/L KCl, 5 mmol/L dithiothreitol (DTT), 1 mmol/L EDTA, 0.1% sodium dodecyl sulfate (SDS), 0.5% Triton X-100, and protease inhibitor cocktail tablets (Roche, IN), incubated for 10 min on ice, disrupted in a microultrasonic cell disrupter for 10 s and centrifuged at 750 × g for 15 min at 4 °C. The supernatant (cytosolic fraction) was removed and maintained at -80 °C. The pellet containing mitochondria was resolved in a lysis buffer. Protein level was measured using a standard colorimetric assay kit (BCA kit). Proteins were separated by polyacrylamide/SDS gel electrophoresis and transferred onto polyvinylidene fluoride membranes (Roche, IN). The membranes were probed with antibody (cytochrome c diluted at 1:1000, Cell Signaling Technology, MA, United States) overnight at 4 °C, and incubated with a Horseradish peroxidase (HRP)-coupled secondary antibody (HRP; 1:5000, Cell Signaling Technology, MA, United States). Detection was performed using a LumiGLO chemiluminescent subtract system (KPL, Guildford, United Kingdom). β-actin (1:200, Boster, Wuhan, China) as a loading control. Results were quantified with a scanning densitometer (Bio-Rad, United States).
Caspase-3, -8 and -9 activity assay
Caspases activities were measured using Caspase Activity Assay Kit (Beyotime, C1115, C1151 and C1157) according to the manufacturer’s instructions. Briefly, cultured HepG2 cells (5 × 106) were washed with cold PBS twice, resuspended in lysis buffer and left on ice for 20 min. The lysate was centrifuged at 16 000 × g at 4 °C for 3 min. Supernatants were collected and protein concentrations were measured with a BCA kit. Caspase-3, -8 and -9 activities were measured by reaction buffer (containing DTT) and caspase substrate peptides Ac-DEVD-pNA, Ac-IETD-pNA and Ac-LEHD-pNA, respectively. The release of p-nitroanilide (pNA) was qualified by determining the absorbance with Tecan SUNRISE at 405 nm. The fold increase in absorbance was calculated by the formula [ODi/OD0], where OD0 is the absorbance without tectorigenin treatment and ODi is the absorbance with tectorigenin treatment at the indicated concentration.
Statistical analysis
All data were expressed as mean ± SD. Origin Pro 7.0 statistical package was used to determine statistical significance. Difference between two groups was analyzed by two-tailed Student’s t test, and difference among three or more groups was analyzed by one-way analysis of variance multiple comparisons. P < 0.05 was considered statistically significant.
RESULTS
Preparation of tectorigenin
As illustrated in Figure 1A, the high performance liquid chromatography (HPLC) profile for Crude-Extract revealed that tectorigenin is one of the main components of the rhizomes of I. tectorum. The compound was identified as tectorigenin (CAS registry number: 548-77-6) by chemical and spectral approaches. The structure is shown in Figure 1A. The purity of tectorigenin was above 98% (HPLC analysis, Figure 1B).
Figure 1.

The high-performance liquid chromatography profiles of the aqueous ethanol (80%) extract of Iris tectorum rhizomes (A) and tectorigenin (B). High-performance liquid chromatography analysis was accomplished at 25 °C on an instrument consisting of L-7110 (Hitachi) pump, L-7420 (Hitachi) ultraviolet detector (set at 254 nm) and Alltech Apollo C18 (4.6 mm × 250 mm) column using MeOH:EtOAc:H2O (60:1:40) mixture as a mobile phase at a flow rate of 1.0 mL/min. Sample injection: 10 μL of extract (2.5 g/L) or tectorigenin (100 mg/L) in MeOH.
Effect of tectorigenin on viability of HepG2 and L02 cells
The effect of different concentrations of tectorigenin on the viability of HepG2 cells for 12, 24 and 48 h was assessed by the MTT assay. The number of viable HepG2 cells treated by tectorigenin decreased in a concentration- and time-dependent manner (Figure 2A). When HepG2 cells were treated with tectorigenin at 5, 10 and 20 mg/L for 24 h, the viability rate was 91%, 79% and 62%, respectively (Figure 2A). Whereas when HepG2 cells were treated with tectorigenin at 5, 10 and 20 mg/L for 48 h, the viability rate was reduced to 83%, 57% and 33%, respectively (Figure 2A). The concentration that reduced the number of viable HepG2 cells by 50% (IC50) after 12h, 24 h and 48h of incubation was 35.72, 21.19 and 11.06 mg/L, respectively. However, treatment with tectorigenin at 20 mg/L resulted in a very slight cytotoxicity to L02 cells after incubation for 12, 24 or 48 h (Figure 2B).
Figure 2.
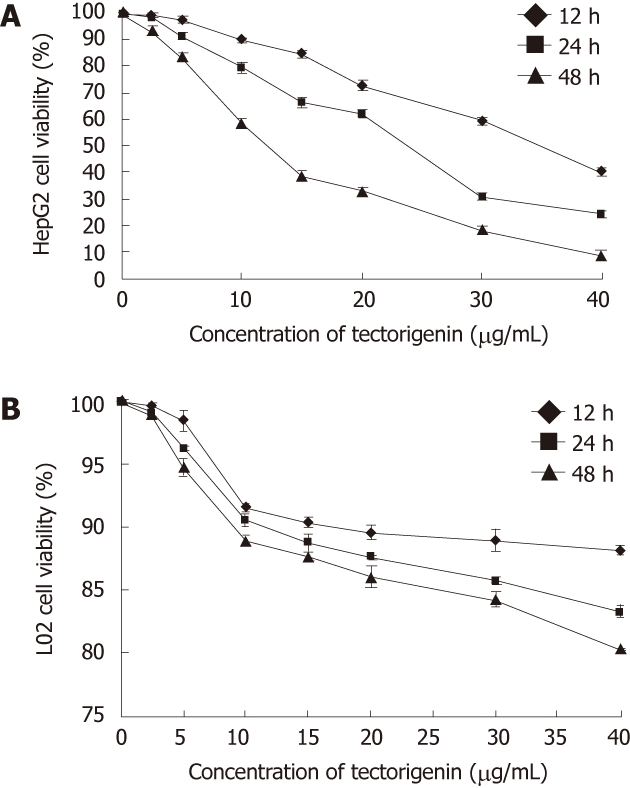
Effects of tectorigenin on the viability of HepG2 (A) and L02 cells (B). Cells were treated for 12, 24 and 48 h with tectorigenin at 2.5, 5, 10, 15, 20, 30 and 40 mg/L, respectively, followed by assessing the cell viability relative to that of untreated cells (control). Bars represent mean ± SD.
Morphological changes of HepG2 cells after exposure to tectorigenin
To verify tectorigenin-induced apoptosis of HepG2 cells, we observed the changes in cell morphology after tectorigenin exposure by Hoechst 33 258 staining. As shown in Figure 3, regular and round-shaped nuclei were observed in the control HepG2 cells (Figure 3A). After treated with 5, 10 and 20 mg/L (Figure 3B-D) tectorigenin for 48 h, the blue emission light became much brighter in apoptotic cells than in control cells. Condensed chromatin could also be found in many tectorigenin-treated cells and the structure of apoptotic bodies was formed in some cells, which is one of the classic characteristics of apoptotic cells. Moreover, after treated with 20 mg/L tectorigenin, the nuclei of HepG2 cells were further condensed with the number of apoptotic bodies sharply increased.
Figure 3.
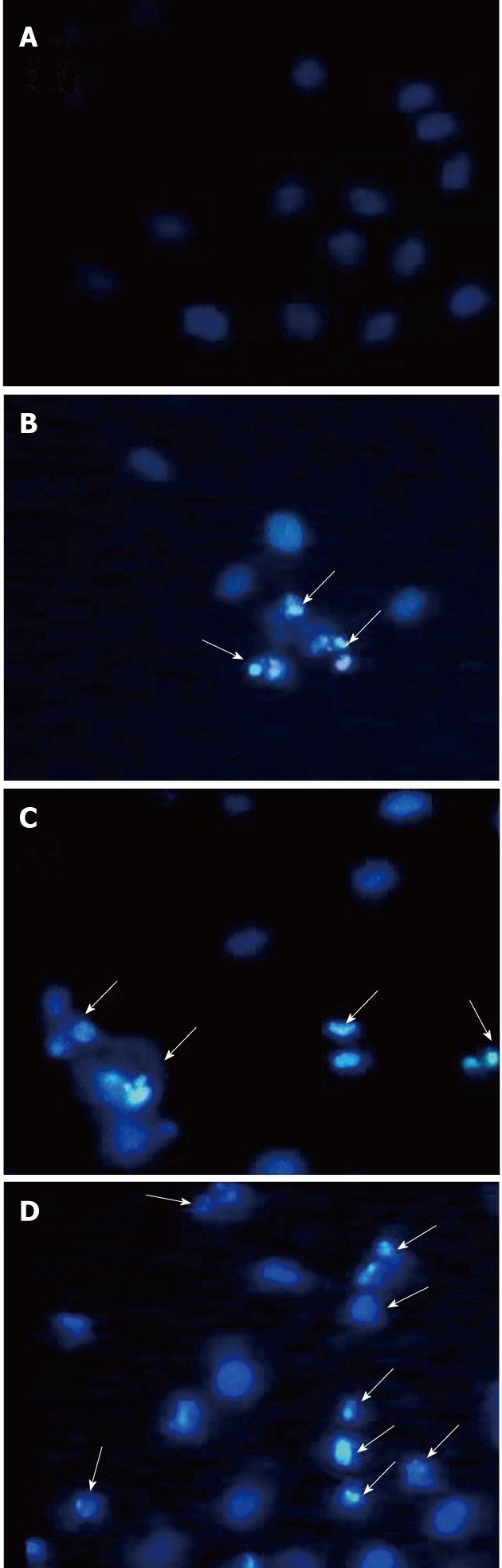
Morphological changes of HepG2 cells after exposure to tectorigenin. HepG2 cells were treated with vehicle (A) and tectorigenin at 5 (B), 10 (C), 20 (D) mg/L for 48 h, the cells were then fixed and stained with Hoechst 33258 and observed under a fluorescent microscope (Leica DM IRB) (Magnification × 400). Tectorigenin significantly induced condensed chromatin and fragmented nuclei. The arrows in B-D indicate cells undergoing apoptosis.
DNA ladder of HepG2 cells treated with tectorigenin
Genomic DNA was prepared from HepG2 cells that had been incubated in the presence or absence of 20 mg/L tectorigenin for 48 h. The integrity of the DNA was assessed by agarose gel electrophoresis. DNA isolated from HepG2 cells cultured with 20 mg/L tectorigenin for 48 h showed a ‘‘ladder’’ pattern of apoptosis (Figure 4). A comparison with molecular weight markers indicated that the fragments were multiples of approximately 180-200 base pairs.
Figure 4.

DNA fragmentation of HepG2 cells induced by tectorigenin. Cells were treated with tectorigenin at 20 mg/L for 48 h, then total DNA was isolated as described in Methods and analyzed by eletrophoresis for detection of DNA fragmentation. 1: Marker; 2: Control; 3: Tectorigenin.
Apoptotic rate of HepG2 cells treated with tectorigenin
Treatment of HepG2 cells with tectorigenin resulted in significant increase of apoptotic cells in a time- and dose-dependent manner compared with that of non-treated cells quantified by Annexin V analysis (Figure 5B), as shown in Figure 5A. Apoptotic rate of HepG2 cells treated with 5, 10 and 20 mg/L tectorigenin for 24 h was 8%, 14%, and 30%, respectively. However, when HepG2 cells were treated with 5, 10 and 20 mg/L tectorigenin for 48 h, its apoptotic rate was 16%, 42% and 58%, respectively. Compared with the viability rate in Figure 2, these results suggested that induction of apoptosis was the main mechanism of the anti-proliferative effect of tectorigenin in HepG2 cells.
Figure 5.

Flow cytometric analysis of apoptosis in HepG2 cells treated with tectorigenin. HepG2 cells were incubated for 24 and 48 h with tectorigenin at 0, 5, 10, 20 mg/L, respectively. And then the cells were stained with EGFP-conjugated Annexin V and propidium iodide (PI). The EGFP and PI fluorescence was measured using flow cytometer with FL1 and FL3 filters, respectively. A: Representative dot plots of Annexin V/PI staining. a: Control, 24 h; b: 5 mg/L, 24 h; c: 10 mg/L, 24 h; d: 20 mg/L, 24 h; e: Control, 48h; f: 5 mg/L, 48 h; g: 10 mg/L, 48 h; h: 20 mg/L, 48 h. The lower left quadrant contains the vital (double negative) population. The lower right quadrant contains the early apoptotic (Annexin V+/PI-) population and upper right quadrant contains the late apoptotic/necrotic (Annexin V+/PI+) population; B: Data pooled from three independent experiments show the percentage of apoptotic cells. Difference was considered statistically significant when aP < 0.05 and bP < 0.01 vs control.
Intracellular accumulation of ROS induced by tectorigenin
To determine whether tectorigenin could induce intracellular ROS generation, levels of ROS production in HepG2 cells were determined using the fluorescence probe DCFH-DA. As shown in Figure 6, HepG2 cells exposed to tectorigenin at 10 and 20 mg/L for 1 or 6 h displayed a significant increase in the intracellular level of ROS as compared with that in the control cells. When HepG2 cells were treated with tectorigenin at 5, 10 and 20 mg/L for 3 h, they displayed a maximal increase in the intracellular level of ROS. However, HepG2 cells incubated with tectorigenin at 5, 10 and 20 mg/L for 24 h, showed a mild but not significant increase in the intracellular level of ROS compared with that in the control cells.
Figure 6.
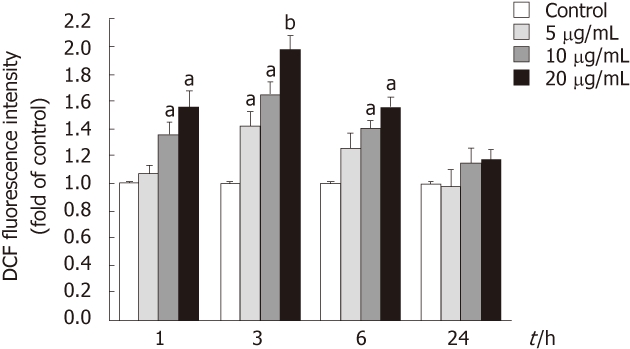
Stimulation by tectorigenin of reactive oxygen species generation in HepG2 cells. Reactive oxygen species was measured using an oxidation-sensitive fluorescent probe, DCFH-DA. Cells were treated with tectorigenin for 1, 3, 6 and 24 h, and then incubated for 30 min with DCFH-DA at 37 °C. After washing with phosphate buffered saline, the fluorescence intensity of the cells was analyzed with a fluorescence plate reader. Data are expressed as the mean of three individual experiments ± SD. Difference was considered statistically significant when aP < 0.05 and bP < 0.01 vs control. DCF: Dichlorodihydrofluorescein.
Tectorigenin-induced increase of [Ca2+]i in HepG2 cells
To determine whether tectorigenin influences the level of intracellular Ca2+, the level of intracellular Ca2+ was measured with Fura 2-AM staining. As shown in Figure 7, treatment of HepG2 cells with tectorigenin at the concentrations of 5, 10 and 20 mg/L for 3 h increased the fluorescence ratio (F340/F380) by 1.11 ± 0.09, 1.32 ± 0.11 and 1.86 ± 0.13 fold as compared with the control. When HepG2 cells were treated with tectorigenin for 6 and 24 h, they displayed a higher increase of the fluorescence ratio (F340/F380) in a dose- and time-dependent manner (Figure 7). Moreover, when the incubation time was prolonged from 3 h to 24 h with the incubation concentration of 20 mg/L, the F340/F380 value increased from 1.86 ± 0.13 to 2.52 ± 0.17.
Figure 7.
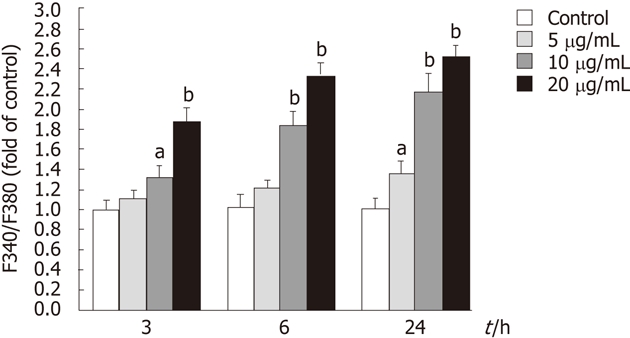
Effects of tectorigenin on the increase of intracellular Ca2+ in HepG2 cells. Cells were treated with tectorigenin at 5, 10 or 20 mg/L for different time points, and then loaded with Fura 2-acetoxymethy ester. Intracellular Ca2+ was measured by fluorescence analysis. The data were presented as mean ± SD (n = 6). Difference was considered statistically significant when aP < 0.05 and bP < 0.01 vs control.
Tectorigenin-induced loss of MMP in HepG2 cells
To assess the effect of tectorigenin on the changes of MMP in HepG2 cells, flow cytometric analysis was carried out to detect the fluorescence intensity of Rhodamine 123. As shown in Figure 8B, treatment of HepG2 cells with tectorigenin at the concentrations of 5 and 10 mg/L for 3 h and 6 h caused a moderate depolarization of MMP. However, HepG2 cells treated with tectorigenin at the concentrations of 5, 10 and 20 mg/L for 24 h (Figure 8A and B), as well as for 3 and 6h, displayed a remarkable depolarization of MMP corresponding to a much lower fluorescence intensity compared with the control, suggesting the collapse of the inner mitochondrial membrane and mitochondrial dysfunction. Compared with the control cells (without treatment of tectorigenin), HepG2 cells treated with 5, 10 and 20 mg/L tectorigenin for 24 h decreased the MMP from 95.39% ± 5.47% to 81.39% ± 4.28%, 72.55% ± 3.97% and 69.45% ± 3.28%, respectively.
Figure 8.

Effects of tectorigenin on integrity of mitochondrial membrane in HepG2 cells. Cells were treated with tectorigenin for different time points, then incubated with Rhodamine 123 and analyzed by flow cytometry. A: HepG2 cells were treated with 0 (a), 5 (b), 10 (c) and 20 (d) mg/L tectorigenin for 24 h. Mitochondrial membrane potential was measured by Rh123, a fluorescent probe, on FACSCalibur at FL2 channel with an excitation wavelength of 488 nm. Each test was performed 3 times and images presented were typical of 3 independent experiments; B: The data were presented as mean ± SD (n = 3). Difference was considered statistically significant when aP < 0.05 and bP < 0.01 vs control.
Effect of tectorigenin on cytochrome c release in HepG2 cells
The cytosolic and mitochondrial levels of cytochrome c were measured to confirm apoptosis via the mitochondrial pathway in tectorigenin-treated HepG2 cells. The results indicated that mitochondrial cytochrome c was released into the cytosol in a dose-dependent manner (Figure 9A). Compared with control cells (without treatment of tectorigenin), HepG2 cells treated with tectorigenin at 5, 10 and 20 mg/L for 48 h showed an increase in levels (cytochrome c/β-actin ratio) of cytosolic cytochrome c from 0.08 ± 0.007 to 0.21 ± 0.03, 0.47 ± 0.04 and 0.89 ± 0.05, respectively (Figure 9B).
Figure 9.

Western blotting analysis. After 48-h exposure to tectorigenin at 0, 5, 10 and 20 mg/L, respectively, levels of cytochrome c in HepG2 cells (A), along with those of cytochrome c (B) in cytosol, were evaluated. Protein (50 μg) from each sample was resolved on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and β-actin was used as a loading control.
Effect of tectorigenin on caspase-3, -8 and -9 activities in HepG2 cells
The apoptotic process included the activation of cysteine proteases, which represent both initiators and executors of cell death. Tectorigenin treatment caused a significant time- and dose-dependent increase in caspase-3 and -8 proteolytic activity in HepG2 cells (Figure 10A and C). However, HepG2 cells treated with tectorigenin displayed a very slight increase in caspase-8 activity (Figure 10B).
Figure 10.

Caspase activities in tectorigenin-treated HepG2 cells. Cells were treated with tectorigenin at the indicated concentrations for 24 and 48 h. Cytosolic extracts were prepared and assayed for caspase activities as described in “Materials and Methods”. A: Alteration of caspase-3 activity in tectorigenin-induced HepG2 cells for indicated time; B: Effect of tectorigein on caspase-8 activity in HepG2 cells; C: Changes of caspase-9 activity in tectorigenin-treated HepG2 cells at 5, 10 and 20 mg/L for 24 and 48 h. Data are expressed as the mean of three individual experiments ± SD. Difference was considered statistically significant when aP < 0.05 and bP < 0.01 vs control.
DISCUSSION
I. tectorum is a kind of widely planted flower, which has been used in traditional Chinese medicine as a folk proven prescription for curing liver-related illnesses. By chemical analysis, we found that tectorigenin is one of the main components in the rhizome of I. tectorum (Figure 1) and another main component was tectoridin[11,17]. However, it has been reported that tectorigenin could be transformed from tectoridin by intestinal microflora[17]. Therefore, we hypothesized that the main active ingredient in the rhizome of I. tectorum might be tectorigenin. In fact, tectorigenin has been reported to have in vivo hepatoprotective[18], estrogenic[19] and anti-inflammation activities[20], and pro-apoptotic effect on hepatic stellate cells[13]. Previous studies also have demonstrated a role for tectorigenin in regulating prostate cancer cell number by inhibiting proliferation through cell cycle regulation[21]. Tectorigenin also has pro-apoptotic effects and decreases tissue invasion by up-regulation of tissue inhibitor of metalloproteinase-3 in prostate cancer[22]. Therefore, its hepatoprotective activity and pro-apoptotic effect on hepatic stellate cells made us believe that tectorigenin is the substance base of the utilization of I. tectorum rhizome in the treatment of liver-related diseases. Considering that tectorigenin is one of the main components in the rhizome of I. tectorum (Figure 1) and I. tectorum has been used as a folk proven prescription for the treatment of liver cancer, we supposed that tectorigenin could be used for the treatment of HCC. The present study revealed that tectorigenin possessed considerable pro-apoptotic effects on human HCC HepG2 cells and reinforced our hypothesis that tectorigenin is the substance base of the utilization of I. tectorum rhizome in the treatment of liver cancer. Base on the results in the present study, we will further investigate the in vivo anticancer effects of tectorigenin. After 48 h of incubation, IC50 of tectorigenin against HepG2 cells was 11.06 mg/L and treatment with tectorigenin at 10 mg/L resulted in about 10% cell death of the normal L02 cells (Figure 2). The dosage of tectorigenin 10-15 mg/kg body weight is large enough to induce in vivo apoptosis of cancer cells.
In this study, compared with the viability rate (Figure 2), the high apoptotic cell percentage (Figure 5) suggested that induction of apoptosis was the main mechanism of the anti-proliferative effect of tectorigenin in HepG2 cells. The upstream apoptotic mechanism of tectorigenin in HepG2 cells was investigated based on the intracellular accumulation of ROS. A previous study has found that mitochondria are the major organelle where ROS was generated and excessive ROS could lead to lipid peroxidation, oxidation of proteins and DNA damage[23]. Theoretically, as a consequence of excessive ROS generation in cells, mitochondrial dysfunction should occur[24]. The intracellular ROS peaked when HepG2 cells were treated with different concentrations of tectorigenin for 3 h, then declined and when treated for 24 h it declined to the approximately normal level (Figure 6). As expected, significant dissipation of MMP (Figure 8) was observed in the current study, indicative of mitochondrial dysfunction. Excessive ROS could raise the Ca2+ concentration in the cytoplasm[25,26]. Marked elevation in Ca2+ then causes the new ROS formation[27-29]. On the other hand, the increased Ca2+ may also impair mitochondrial function[29-31], leading to a significant decrease in MMP. In this study, tectorigenin could significantly increase the [Ca2+]i in HepG2 cells.
In normal situation, cytochrome c resides in the mitochondrial intermembrane and serves as a transducer of electrons in the respiratory chain. However, the increment of ROS and [Ca2+]i , and subsequent mitochondrial dysfunction result in cytochrome c release[32]. In the present study, release of cytochrome c from mitochondria to cytosol was detected (Figure 9). After release from mitochondria, cytochrome c could bind with Apaf- 1 and participate in the activation of caspase-9 (the initiator). The activated caspase-9 then activates caspase-3 (the effector). Initiator caspases and effector caspases act together to augment the death signal and finally lead to apoptosis[31-36]. In this study, we detected enhanced caspase-3 and -9 activities in tectorigenin-treated HepG2 cells (Figure 10A and C), which validated mitochondria-mediated apoptosis pathway. Similarly, in the study of Zhou et al[6], troglitazone inhibited growth and induced apoptosis of HepG2 cells in a dose-dependent manner, and induced activation of caspase-3 expression. Caspase-8 is an initiating caspase, which modulates the death receptor-dependent pathway. We found very mild increase in caspase-8 activity in tectorigenin-treated HepG2 cells (Figure 10B), which suggested that a death receptor-mediated pathway might be excluded in the apoptosis of HepG2 cells induced by tectorigenin. In the study of Tsagarakis et al[37], 10-8 mol/L octreotide could significantly inhibit the proliferation of HepG2 cells and significantly increase caspase-3, caspase-8 and caspase-2 activities. In this study, tectorigenin increased the ROS production and Ca2+ concentration in cytoplasm of HepG2 cells, the intracellular accumulation of ROS and Ca2+ further induced the loss of MMP. The disruption of MMP caused release of cytochrome c from mitochondria to cytosol. Cytosolic cytochrome c activated the pro-caspase-9 and subsequently, caspase-9 activated the downstream effector caspases-3, eventually triggered apoptosis of HepG2 cells (Figure 11). In the study of Tan et al[5], 5-allyl-7-gen-difluoromethylenechrysin was found to exert its apoptotic effect by activation of Peroxisome proliferator-activated receptor gamma, down-regulation of nuclear factor kappa B and Bcl-2 protein expression, up-regulation of Bax protein expression, and reduction of the ratio of Bcl-2 to Bax.
Figure 11.
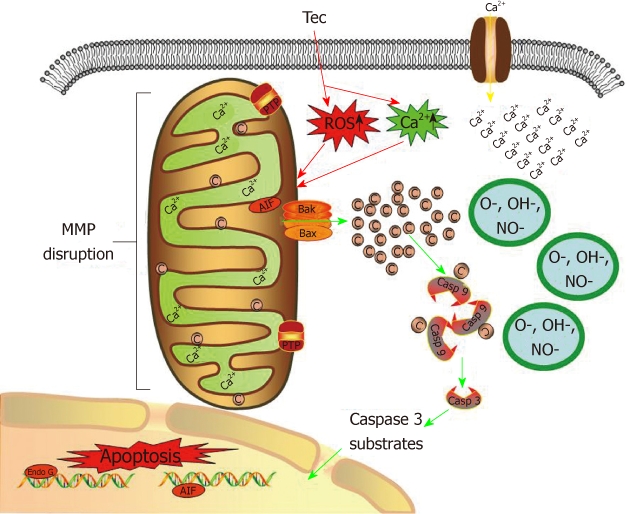
Possible mechanisms by which tectorigenin induces apoptosis in human hepatocellular carcinoma HepG2 cells. Tectorigenin produces reactive oxygen species and increases Ca2+ concentration in cytoplasm of HepG2 cells, leading to the depletion of mitochondrial membrane potential and release of cytochrome c from mitochondria. Cytochrome c facilitates apoptosis of HepG2 cells by activating caspase cascade. Tec: Tectorigenin; ROS: Reactive oxygen species; C: Cytochrome c; Casp: Caspase; AIF: Apoptosis inducing factor; MMP: Mitochondrial membrane potential.
Our previous research showed that tectorigenin suppressed the proliferation of HSC-T6 cells and induced apoptosis of HSC-T6 cells in a time- and dose-dependent manner[13]. Tectorigenin at a concentration of 100 mg/L greatly inhibited the viability of HSC-T6 cells and induced the condensation of chromatin and fragmentation of nuclei[13]. In this study, tectorigenin could inhibit the viability of HepG2 cells and induce apoptosis at a lower concentration of 20 mg/L. Furthermore, tectorigenin-induced apoptosis of HSC-T6 cells was associated with the generation of ROS, increased intracellular [Ca2+]i, loss of mitochondrial membrane potential, translocation of cytochrome c, and activation of caspase-9 and -3[13]. In this study, tectorigenin could induce apoptosis of HepG2 cells by similar mechanisms. In contrast, in the study of Zhou et al[6], troglitazone not only drove apoptosis-inhibiting factor survivin to translocate incompletely from the nucleus to the cytoplasm, but also inhibited expression of survivin, while it did not affect expression of apoptosis-promoting factor Bax.
In conclusion, tectorigenin significantly inhibits the proliferation and induces apoptosis in human HCC HepG2 cells mainly via mitochondrial-mediated pathway. These observations indicated that tectorigenin is a promising chemotherapeutic and chemopreventive agent for the treatment of HCC.
COMMENTS
Background
Human hepatocellular carcinoma (HCC) is the fifth most common cancer in the world. Unfortunately, the disease is often diagnosed at a late stage. The hallmark of cancer cells is the dysregulation of cell proliferation and apoptosis. The growing of a tumor is determined by the rate of cell proliferation and the apoptosis. Therefore, the induction of apoptosis in tumor cells has become a strategy in cancer treatment and the mitochondrial-mediated pathways have been identified as one of the major mechanisms for induction of apoptosis. Iris tectorum (I. tectorum), a traditional Chinese medicine, has been widely used for treating liver-related diseases, including hepatitis, liver fibrosis, liver cirrhosis and liver cancer.
Research frontiers
Recent studies have revealed that tectorigenin possessed anti-proliferative and pro-apoptotic effects on hepatic stellate cells and have potent in vivo anti-liver fibrosis activity. However, until now there is no report about the anti-tumor effect of tectorigenin on human HCC HepG2 cells and the associated mechanisms. In this study, tectorigenin was found to suppress the proliferation of HepG2 cells and induce apoptotis of HepG2 cells.
Innovations and breakthroughs
This study revealed that tectorigenin possessed considerable pro-apoptotic effects on human HCC HepG2 cells, which confirmed that tectorigenin is the substance base of the utilization of I. tectorum rhizome in the treatment of liver cancer. To be known, this is the first study to report the apoptosis induction effects of tectorigenin on human HCC HepG2 cells.
Applications
Tectorigenin significantly inhibits the proliferation and induces apoptosis in human HCC HepG2 cells mainly via mitochondrial-mediated pathway, and produces a slight cytotoxicity to L02 cells. These observations indicated that tectorigenin is a promising chemotherapeutic and chemopreventive agent for the treatment of HCC.
Peer review
The paper investigates the effect of tectorigenin on proliferation and apoptosis in human hepatocellular carcinoma HepG2 cells. The study is of interest.
Footnotes
Supported by The National Natural Science Foundation of China, No. NSFC30801417; Natural Science Foundation of Jiangsu Province, No. BK2009010 and BK2008267; Doctoral Fund of Ministry of Education of China, No. RFDP200802841004; and Science Fund of Ministry of Health of China, No. LW201008
Peer reviewers: Rui Tato Marinho, MD, PhD, Department of Gastroenterology and Hepatology, Hospital Santa Maria, Medical School of Lisbon, Av. Prof. Egas Moniz, 1649-035 Lisboa, Portugal; Heitor Rosa, Professor, Department of Gastroenterology and Hepatology, Federal University School of Medicine, Rua 126 n.21, Goiania, GO 74093-080, Brazil; Dario Conte, Professor, GI Unit - IRCCS Osp. Maggiore, Chair of Gastroenterology, Via F. Sforza, 35, 20122 Milano, Italy
S- Editor Tian L L- Editor Ma JY E- Editor Li JY
References
- 1.But DY, Lai CL, Yuen MF. Natural history of hepatitis-related hepatocellular carcinoma. World J Gastroenterol. 2008;14:1652–1656. doi: 10.3748/wjg.14.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dienstag JL, Isselbacher KJ. Tumors of the Liver and Biliary Tract. In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, et al., editors. Harrison’s Principles of Internal Medicine. 15th ed. New York: McGraw-Hill; 2001. pp. 588–591. [Google Scholar]
- 3.Faivre S, Bouattour M, Raymond E. Novel molecular therapies in hepatocellular carcinoma. Liver Int. 2011;31 Suppl 1:151–160. doi: 10.1111/j.1478-3231.2010.02395.x. [DOI] [PubMed] [Google Scholar]
- 4.Noureini SK, Wink M. Transcriptional down regulation of hTERT and senescence induction in HepG2 cells by chelidonine. World J Gastroenterol. 2009;15:3603–3610. doi: 10.3748/wjg.15.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan XW, Xia H, Xu JH, Cao JG. Induction of apoptosis in human liver carcinoma HepG2 cell line by 5-allyl-7-gen-difluoromethylenechrysin. World J Gastroenterol. 2009;15:2234–2239. doi: 10.3748/wjg.15.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou YM, Wen YH, Kang XY, Qian HH, Yang JM, Yin ZF. Troglitazone, a peroxisome proliferator-activated receptor gamma ligand, induces growth inhibition and apoptosis of HepG2 human liver cancer cells. World J Gastroenterol. 2008;14:2168–2173. doi: 10.3748/wjg.14.2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Youn MJ, Kim JK, Park SY, Kim Y, Kim SJ, Lee JS, Chai KY, Kim HJ, Cui MX, So HS, et al. Chaga mushroom (Inonotus obliquus) induces G0/G1 arrest and apoptosis in human hepatoma HepG2 cells. World J Gastroenterol. 2008;14:511–517. doi: 10.3748/wjg.14.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoysungnoen P, Wirachwong P, Changtam C, Suksamrarn A, Patumraj S. Anti-cancer and anti-angiogenic effects of curcumin and tetrahydrocurcumin on implanted hepatocellular carcinoma in nude mice. World J Gastroenterol. 2008;14:2003–2009. doi: 10.3748/wjg.14.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 10.Sheikh MS, Huang Y. Death receptors as targets of cancer therapeutics. Curr Cancer Drug Targets. 2004;4:97–104. doi: 10.2174/1568009043481597. [DOI] [PubMed] [Google Scholar]
- 11.Wu YX, Xu LX. Analysis of isoflavones in Belamcanda chinensis (L.)DC. and Iris tectorum Maxim by square wave voltammetry. Yaoxue Xuebao. 1992;27:64–68. [PubMed] [Google Scholar]
- 12.Jung SH, Lee YS, Lee S, Lim SS, Kim YS, Ohuchi K, Shin KH. Anti-angiogenic and anti-tumor activities of isoflavonoids from the rhizomes of Belamcanda chinensis. Planta Med. 2003;69:617–622. doi: 10.1055/s-2003-41125. [DOI] [PubMed] [Google Scholar]
- 13.Wu JH, Wang YR, Huang WY, Tan RX. Anti-proliferative and pro-apoptotic effects of tectorigenin on hepatic stellate cells. World J Gastroenterol. 2010;16:3911–3918. doi: 10.3748/wjg.v16.i31.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hai J, Lin Q, Lu Y, Zhang H, Yi J. Induction of apoptosis in rat C6 glioma cells by panaxydol. Cell Biol Int. 2007;31:711–715. doi: 10.1016/j.cellbi.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Shi da H, Wu JH, Ge HM, Tan RX. Protective effect of hopeahainol A, a novel acetylcholinesterase inhibitor, on hydrogen peroxide-induced injury in PC12 cells. Environ Toxicol Pharmacol. 2009;28:30–36. doi: 10.1016/j.etap.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Gong Y, Han XD. Nonylphenol-induced oxidative stress and cytotoxicity in testicular Sertoli cells. Reprod Toxicol. 2006;22:623–630. doi: 10.1016/j.reprotox.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 17.Park EK, Shin YW, Lee HU, Lee CS, Kim DH. Passive cutaneous anaphylaxis-inhibitory action of tectorigenin, a metabolite of tectoridin by intestinal microflora. Biol Pharm Bull. 2004;27:1099–1102. doi: 10.1248/bpb.27.1099. [DOI] [PubMed] [Google Scholar]
- 18.Lee HW, Choo MK, Bae EA, Kim DH. Beta-glucuronidase inhibitor tectorigenin isolated from the flower of Pueraria thunbergiana protects carbon tetrachloride-induced liver injury. Liver Int. 2003;23:221–226. doi: 10.1034/j.1600-0676.2003.00830.x. [DOI] [PubMed] [Google Scholar]
- 19.Morito K, Aomori T, Hirose T, Kinjo J, Hasegawa J, Ogawa S, Inoue S, Muramatsu M, Masamune Y. Interaction of phytoestrogens with estrogen receptors alpha and beta (II) Biol Pharm Bull. 2002;25:48–52. doi: 10.1248/bpb.25.48. [DOI] [PubMed] [Google Scholar]
- 20.Shin KH, Kim YP, Lim SS, Lee S, Ryu N, Yamada M, Ohuchi K. Inhibition of prostaglandin E2 production by the isoflavones tectorigenin and tectoridin isolated from the rhizomes of Belamcanda chinensis. Planta Med. 1999;65:776–777. doi: 10.1055/s-2006-960868. [DOI] [PubMed] [Google Scholar]
- 21.Morrissey C, Bektic J, Spengler B, Galvin D, Christoffel V, Klocker H, Fitzpatrick JM, Watson RW. Phytoestrogens derived from Belamcanda chinensis have an antiproliferative effect on prostate cancer cells in vitro. J Urol. 2004;172:2426–2433. doi: 10.1097/01.ju.0000143537.86596.66. [DOI] [PubMed] [Google Scholar]
- 22.Thelen P, Seseke F, Ringert RH, Wuttke W, Seidlová-Wuttke D. Pharmacological potential of phytoestrogens in the treatment of prostate cancer. Urologe A. 2006;45:195–196. doi: 10.1007/s00120-005-0932-3. [DOI] [PubMed] [Google Scholar]
- 23.Götz ME, Künig G, Riederer P, Youdim MB. Oxidative stress: free radical production in neural degeneration. Pharmacol Ther. 1994;63:37–122. doi: 10.1016/0163-7258(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 24.Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem. 2003;278:33714–33723. doi: 10.1074/jbc.M302559200. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Joseph JA. Mechanisms of hydrogen peroxide-induced calcium dysregulation in PC12 cells. Free Radic Biol Med. 2000;28:1222–1231. doi: 10.1016/s0891-5849(00)00241-0. [DOI] [PubMed] [Google Scholar]
- 26.Li G, Xiao Y, Zhang L. Cocaine induces apoptosis in fetal rat myocardial cells through the p38 mitogen-activated protein kinase and mitochondrial/cytochrome c pathways. J Pharmacol Exp Ther. 2005;312:112–119. doi: 10.1124/jpet.104.073494. [DOI] [PubMed] [Google Scholar]
- 27.Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakraborti T, Das S, Mondal M, Roychoudhury S, Chakraborti S. Oxidant, mitochondria and calcium: an overview. Cell Signal. 1999;11:77–85. doi: 10.1016/s0898-6568(98)00025-4. [DOI] [PubMed] [Google Scholar]
- 29.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 30.Trump BF, Berezesky IK. Calcium-mediated cell injury and cell death. FASEB J. 1995;9:219–228. doi: 10.1096/fasebj.9.2.7781924. [DOI] [PubMed] [Google Scholar]
- 31.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 32.Chandra D, Liu JW, Tang DG. Early mitochondrial activation and cytochrome c up-regulation during apoptosis. J Biol Chem. 2002;277:50842–50854. doi: 10.1074/jbc.M207622200. [DOI] [PubMed] [Google Scholar]
- 33.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 34.Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J Biol Chem. 1999;274:2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- 36.Zhang P, Xu Y, Li L, Jiang Q, Wang M, Jin L. In vitro protective effects of pyrroloquinoline quinone on methylmercury-induced neurotoxicity. Environ Toxicol Pharmacol. 2009;27:103–110. doi: 10.1016/j.etap.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Tsagarakis NJ, Drygiannakis I, Batistakis AG, Kolios G, Kouroumalis EA. Octreotide induces caspase activation and apoptosis in human hepatoma HepG2 cells. World J Gastroenterol. 2011;17:313–321. doi: 10.3748/wjg.v17.i3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]