

Abstract
AIM: To investigate the effect of glyceraldehyde-derived advanced glycation end-products (Glycer-AGEs) on hepatocellular carcinoma (HCC) cells.
METHODS: Two HCC cell lines (Hep3B and HepG2 cells) and human umbilical vein endothelial cells (HUVEC) were used. Cell viability was determined using the WST-8 assay. Western blotting, enzyme linked immunosorbent assay, and real-time reverse transcription-polymerase chain reactions were used to detect protein and mRNA. Angiogenesis was evaluated by assessing the proliferation, migration, and tube formation of HUVEC.
RESULTS: The receptor for AGEs (RAGE) protein was detected in Hep3B and HepG2 cells. HepG2 cells were not affected by the addition of Glycer-AGEs. Glycer-AGEs markedly increased vascular endothelial growth factor (VEGF) mRNA and protein expression, which is one of the most potent angiogenic factors. Compared with the control unglycated bovine serum albumin (BSA) treatment, VEGF mRNA expression levels induced by the Glycer-AGEs treatment were 1.00 ± 0.10 vs 1.92 ± 0.09 (P < 0.01). Similarly, protein expression levels induced by the Glycer-AGEs treatment were 1.63 ± 0.04 ng/mL vs 2.28 ± 0.17 ng/mL for the 24 h treatment and 3.36 ± 0.10 ng/mL vs 4.79 ± 0.31 ng/mL for the 48 h treatment, respectively (P < 0.01). Furthermore, compared with the effect of the control unglycated BSA-treated conditioned medium, the Glycer-AGEs-treated conditioned medium significantly increased the proliferation, migration, and tube formation of HUVEC, with values of 122.4% ± 9.0% vs 144.5% ± 11.3% for cell viability, 4.29 ± 1.53 vs 6.78 ± 1.84 for migration indices, and 71.0 ± 7.5 vs 112.4 ± 8.0 for the number of branching points, respectively (P < 0.01).
CONCLUSION: These results suggest that Glycer-AGEs-RAGE signaling enhances the angiogenic potential of HCC cells by upregulating VEGF expression.
Keywords: Advanced glycation end-products, Angiogenesis, Glyceraldehyde, Hepatocellular carcinoma, Nonalcoholic steatohepatitis
INTRODUCTION
Advanced glycation end-products (AGEs) are formed by the Maillard reaction, a non-enzymatic reaction between the ketones or aldehydes of sugars and the amino groups of proteins, which contributes to aging and the pathological complications of diabetes[1,2]. Recent studies have suggested that AGEs can be formed not only from sugars, but also from carbonyl compounds derived from the autoxidation of sugars and other metabolic pathways[3,4]. Among the different AGEs, there is evidence that glyceraldehyde-derived AGEs (Glycer-AGEs) are associated with the complications of diabetes, as well as Alzheimer’s disease, nonalcoholic steatohepatitis (NASH), and cancer[5-8].
NASH is recognized as a component of metabolic syndrome and is associated with insulin resistance and abnormalities in glucose and lipid metabolism[9-11]. NASH is one of a group of nonalcoholic fatty liver diseases (NAFLD) that range from simple steatosis to steatohepatitis[12]. However, although simple steatosis appears to be a benign and non-progressive condition, NASH is a potentially progressive disease that can lead to cirrhosis, liver failure, and hepatocellular carcinoma (HCC)[13,14]. In fact, several case series of NASH-associated HCC have been reported[15,16]. HCC, which accounts for more than 90% of all primary liver cancers, is one of the most common malignancies worldwide[17]. Its incidence is particularly high in the Asian population[18].
A recent study suggested that expression of the receptor for AGEs (RAGE) mRNA was lower in normal liver cells than in those of hepatitis and HCC patients[19]. Furthermore, we have demonstrated that Glycer-AGEs are present in significantly high concentrations in the sera of patients with NASH[8]. In addition, the interaction of Glycer-AGEs with the RAGE was found to increase C-reactive protein expression in Hep3B cells[20]. However, the effects of Glycer-AGEs on HCC cells remain poorly understood.
In the present study, we examined the effects of Glycer-AGEs on HCC cells and showed that Glycer-AGEs-RAGE signaling enhances the angiogenic potential of HCC cells by upregulating vascular endothelial growth factor (VEGF) expression.
MATERIALS AND METHODS
Preparation of glyceraldehyde-derived advanced glycation end-products
All chemicals were commercial samples of high purity and were used as supplied. Glycer-AGEs were prepared as described previously[21]. Briefly, 25 mg/mL of bovine serum albumin (BSA; A0281, Sigma-Aldrich) was incubated at 37 °C for 7 d under sterile conditions with 0.1 mol/L glyceraldehyde and 5 mmol/L diethylenetriaminepentaacetic acid (Dojindo Laboratories, Kumamoto, Japan) in 0.2 mol/L phosphate buffer (pH 7.4). As a control, unglycated BSA was incubated under the same conditions, but without glyceraldehyde. The unglycated and glycated albumin were purified using a PD-10 column (GE Healthcare UK Ltd., Buckinghamshire, England) and dialysis against PBS. All preparations were tested for endotoxin using the Endospecy ES-20S system (Seikagaku Co., Tokyo, Japan). Protein concentrations were determined using the Dc protein assay reagent (Bio-Rad Laboratories, Richmond, CA, United States), using BSA as a standard. In all experiments, control unglycated BSA and Glycer-AGEs were used at culture medium concentrations of 100 μg/mL.
Cell cultures
Hep3B and HepG2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS; Equitech-Bio, Kerrville, TX, United States) under standard cell culture conditions (humidified atmosphere, 5% CO2, 37 °C). Cells (1.5 × 104 cells/cm2) were then seeded in various plates or culture dishes (BD Biosciences, Franklin Lakes, NJ, United States) and incubated for 48 h before the start of all experiments, except the migration assay. The control unglycated BSA and Glycer-AGEs (100 μg/mL) treatments were carried out in serum free DMEM.
Human umbilical vein endothelial cells (HUVEC) were grown in endothelial cell growth medium (GM; Cell Applications, San Diego, CA, United States) under standard cell culture conditions.
Preparation of cell lysate
Cells were washed with ice-cold Ca2+ and Mg2+ free PBS [PBS (-)] and subjected to lysis buffer [25 mmol/L Tris-HCl (pH 7.6), 150 mmol/L sodium chloride, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 1 × protease inhibitor cocktail (complete, Mini; Roche)]. Subsequently, cell lysates were passed through a syringe several times for further homogenization and centrifuged at 10 000 × g for 10 min at 4 °C. Protein concentrations were measured using the Bradford assay (Bio-Rad Laboratories).
Western blotting
Cell lysates (30 μg of proteins/lane) were dissolved in SDS sample buffer [62.5 mmol/L Tris-HCl (pH 6.8), 2% SDS, 10% glycerol, and 0.01% bromophenol blue] containing 5% 2-mercaptoethanol, boiled for 3 min at 95 °C, separated by SDS-polyacrylamide gel electrophoresis, and then electro-transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, United States). Biotinylated markers (Cell Signaling, Beverly, MA, United States) were used as molecular weight markers. Membranes were blocked for 1 h using 5% skimmed milk in phosphate buffered saline (PBS) containing 0.05% polyoxyethylene sorbitan monolaurate (PBS-T). After being washed twice with PBS-T, membranes were incubated overnight with goat anti-RAGE antibody (N-16), mouse anti-β-actin antibody (Santa Cruz, Santa Cruz, CA, United States), or rabbit anti-cyclooxygenase-2 (anti-COX-2) antibody (Cayman Chemical, Ann Arbor, MI, United States). Subsequently, membranes were washed twice with PBS-T and incubated with anti-goat IgG antibody (Santa Cruz), anti-mouse Ig’s antibody (Biosource, Camarillo, CA, United States), or anti-rabbit IgG and anti-biotin antibodies (Cell Signaling) for 1 h. After being washed a further three times with PBS-T, immunoreactive proteins were detected with ECL Plus Western Blotting Detection Reagents (GE Healthcare) using a luminescent image analyzer (LAS-1000UVmini; Fujifilm, Tokyo, Japan). The density of the bands was analyzed using a Multi Gauge version 3.0 (Fujifilm).
Cell viability
Cell viability was determined using the WST-8 assay, which measures metabolic activity. After removing the medium from a 96-well microplate that had been used to culture cells as above, 100 μL/well of 10% FBS/DMEM and 10 μL/well of WST-8 solution (Dojindo Laboratories) were added, and cells were incubated for 2 h. Absorbance was then measured at 450 nm and 650 nm using a microplate reader (Labsystems Multiskan Ascent, Model No. 354; Thermo Fisher Scientific, Kanagawa, Japan). The net difference (A450-A650) was used as a measure of cell viability.
Real-time reverse transcription-polymerase chain reaction analysis
Total RNA was isolated from cells with ISOGEN (Nippon Gene, Tokyo, Japan), and 50 ng of RNA were reverse transcribed into cDNA with the PrimeScript™ reverse transcription (RT) reagent kit (Takara, Shiga, Japan) using a GeneAmp® 9700 polymerase chain reaction (PCR) System (Perkin-Elmer Applied Biosystems, Foster City, CA, United States). Real-time polymerase chain reaction was performed with SYBR Premix Ex Taq™ (Takara) using a Smart Cycler® II System (Takara). The reaction mixture (25 μL) contained 1 × SYBR Premix Ex Taq™, 0.2 μmol/L PCR forward primers, 0.2 μmol/L PCR reverse primers, and 10 ng of cDNA as a template. The primers used were as follows: COX-2: 5’-GAGTACCGCAAACGCTTTATGC-3’ and 5’-GCCGAGGCTTTTCTACCAGAA-3’, VEGF: 5’-TGCAGATTATGCGGATCAAACC-3’ and 5’-TGCATTCACATTTGTTGTGCTGTAG-3’, and β-actin: 5’-TCCACCTCCAGCAGATGTGG-3’ and 5’-GCATTTGCGGTGGACGAT-3’. All processes were performed according to the manufacturer’s instructions. Expression levels of the target genes were calculated using a relative quantification method. β-Actin was used as an endogenous control gene to normalize target gene expression values. Product specificity was determined by a melting curve analysis.
Enzyme linked immunosorbent assay
Cells were incubated with control unglycated BSA or Glycer-AGEs for 24 h or 48 h. The culture medium was collected and centrifuged at 200 × g for 10 min to remove any particles, and the resultant supernatant was analyzed using the VEGF enzyme-linked immunosorbent assay kit (Ray Biotech, Norcross, GA, United States). All processes were performed according to the manufacturer’s instructions.
Migration assay
The migratory capacity of Hep3B cells was evaluated using the Oris™ Cell Migration Assay (Platypus Technologies, Madison, WI, United States). Cells (1.5×105 cells/mL) were incubated with 10% FBS/DMEM for 24 h. After removing the stopper covering the center of the well, fluorescently-labeled cells were incubated with control unglycated BSA or Glycer-AGEs for 24 h. The number of cells that had migrated to the center was assessed at excitation and emission wavelengths of 485 nm and 530 nm, respectively, using a fluorescence microplate reader (Labsystems Fluoroskan Ascent CF, Type 374; Thermo Fisher Scientific).
Preparation of conditioned medium
Hep3B cells were incubated with control unglycated BSA or Glycer-AGEs for 48 h. The culture medium was collected and filtered to remove any particles. The CM was then frozen at -80 °C until it was used in the experiments.
Human umbilical vein endothelial cells proliferation assay
HUVEC (0.75× 104 cells/cm2) were incubated with GM for 24 h, before being cultured in DMEM or CM in 10% FBS for 72 h. HUVEC proliferation was determined using the WST-8 assay.
Human umbilical vein endothelial cells migration assay
The migratory capacity of HUVEC was evaluated using the BD BioCoat™ Angiogenesis System-Endothelial Cell Migration assay (BD Biosciences). In this assay, the upper and lower culture compartments were separated by fluorescence blocking polyethylene terephthalate filters (3 μm pore size) coated with human fibronectin. Cells (3.3 × 105 cells/mL) in serum-free DMEM were added to each of the upper chambers for 20 h. Then, 0.5% FBS/CM was added to the lower chamber and used as a chemoattractant. The number of fluorescently-labeled cells that had migrated to the opposite side of the chamber was then assessed at excitation and emission wavelengths of 485 nm and 530 nm, respectively, using a fluorescence microplate reader (Thermo Fisher Scientific).
Tube formation assay
The tube formation assay was performed with the BD BioCoat™ Angiogenesis System-Endothelial Cell Tube Formation assay (BD Biosciences). Before the start of the assay, the Matrigel matrix was polymerized for 30 min at 37 °C under a 5% CO2 environment. Cells (4 × 105 cells/mL) were incubated with 0.5% FBS/CM for 12 h and then photographed under a microscope, and the number of branch points was counted in five randomly chosen fields.
Statistical analysis
All experiments were performed in duplicate and repeated at least two or three times, with each experiment yielding essentially identical results. Data are expressed as the mean ± SD. The significance of the differences between group means was determined by a one-way analysis of variance. P values of less than 0.05 were considered significant.
RESULTS
To investigate whether RAGE proteins were present in Hep3B and HepG2 cells, we carried out Western blotting using an anti-RAGE antibody. RAGE proteins of different molecular weights were detected in Hep3B and HepG2 cells (Figure 1). In full-length RAGE cDNA-transfected Hep3B cells, the major band (57 kDa) (indicated by an arrow in Figure 1) represented the full-length RAGE protein. Likewise, the full-length RAGE protein was also detected in Hep3B and HepG2 cells, and there was no difference in its expression level between the two cell types.
Figure 1.
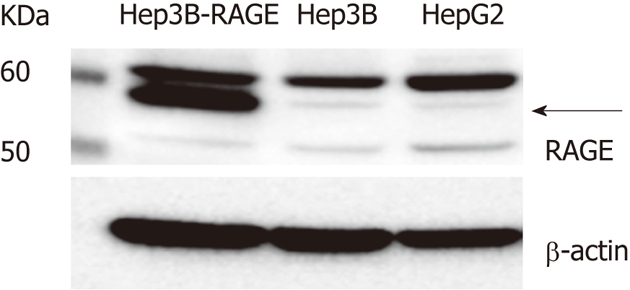
The receptor for advanced glycation end-products expression in hepatocellular carcinoma cells. The receptor for advanced glycation end-products (RAGE) expression as measured by Western blotting. Cell lysates (30 μg of proteins/lane) were loaded onto a 10% polyacrylamide gel. Size markers (kDa) are shown on the left. Equal protein loading was verified using an anti-β-actin antibody. The arrow indicates full-length RAGE.
We examined the effect of Glycer-AGEs on the viability of Hep3B and HepG2 cells. Cell viabilities resulting from treatment with the control unglycated BSA or the Glycer-AGEs for 24 h were 100% ± 3.5% vs 97.8% ± 3.3% in Hep3B cells (Figure 2A), and 100% ± 4.3% vs 102.3% ± 6.7% in HepG2 cells (Figure 2B). Thus, Glycer-AGEs did not have any effect on the viability of Hep3B and HepG2 cells.
Figure 2.
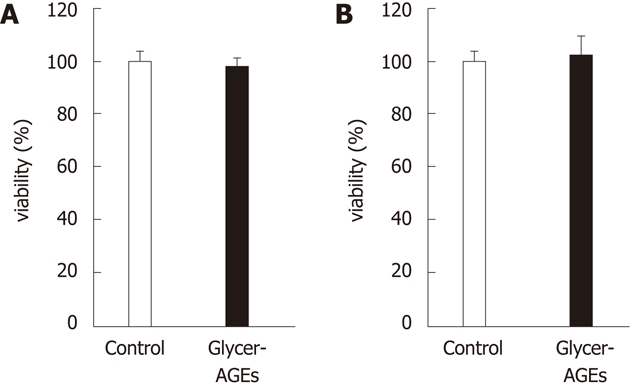
Effect of glyceraldehyde-derived advanced glycation end-products on the viability of hepatocellular carcinoma cells. Cell viability was determined using the WST-8 assay. Hep3B (A) and HepG2 (B) cells were incubated with control unglycated bovine serum albumin (BSA) or glyceraldehyde-derived advanced glycation end-products (Glycer-AGEs) (100 μg/mL) for 24 h. The open and filled bars represent results for cells treated with control unglycated BSA and Glycer-AGEs, respectively. Data are shown as the mean ± SD (n = 6).
To investigate whether Glycer-AGEs affected the malignancy of HCC cells, we examined COX-2 mRNA and protein expression. COX-2 mRNA expression levels induced by the control unglycated BSA or the Glycer-AGEs treatment were 1.00 ± 0.27 vs 2.16 ± 0.34 (P < 0.01) (Figure 3A), and COX-2 protein expression levels were increased by Glycer-AGEs at 24 h in Hep3B cells (Figure 3B), whereas no such change was detected in HepG2 cells (Figure 3B).
Figure 3.

Effect of glyceraldehyde-derived advanced glycation end-products on the malignancy of hepatocellular carcinoma cells. Hep3B and HepG2 cells were incubated with control unglycated bovine serum albumin (BSA) or glyceraldehyde-derived advanced glycation end-products (Glycer-AGEs) for 24 h. A: In Hep3B cells, cyclooxygenase-2 (COX-2) mRNA expression levels were analyzed using real-time reverse transcription-polymerase chain reactions, and results were normalized to the β-actin mRNA level (n = 3), bP < 0.01 vs control unglycated BSA; B: COX-2 expression as measured by Western blotting. Cell lysates (30 μg of proteins/lane) were loaded onto a 10% polyacrylamide gel. Size markers (kDa) are shown on the left. Equal protein loading was verified using an anti-β-actin antibody. As a positive control, A549 cells were incubated with phorbol 12-myristate 13-acetate (PMA: 100 nmol/L) for 6 h; C: The migratory capacity of Hep3B cells was evaluated using the Oris cell migration assay (n = 8). Cells were incubated with control unglycated BSA or Glycer-AGEs for 24 h, and the number of migrating cells was then assessed using a fluorescence microplate reader. RFU: Relative fluorescence units. The open and filled bars represent results for cells treated with control unglycated BSA and Glycer-AGEs, respectively. Data are shown as the mean ± SD.
We also evaluated the influence of Glycer-AGEs on cell migration, which is an index of malignancy in Hep3B cells. However, migration indices for the control unglycated BSA or the Glycer-AGEs treatment were 18.2 ± 0.6 vs 18.7 ± 0.5 (Figure 3C), and Glycer-AGEs did not affect the migratory capacity of Hep3B cells; i.e., Glycer-AGEs did not increase the malignancy of Hep3B cells.
To investigate whether Glycer-AGEs affected the angiogenesis of HCC cells, we examined the expression levels of VEGF mRNA and protein. VEGF mRNA expression levels induced by the control unglycated BSA or the Glycer-AGEs treatment were 1.00 ± 0.10 vs 1.92 ± 0.09 in Hep3B cells (P < 0.01) (Figure 4A), and 1.00 ± 0.11 vs 0.87 ± 0.10 in HepG2 cells (Figure 4B). The expression levels of the VEGF protein induced by the control unglycated BSA or the Glycer-AGEs treatment for 24 and 48 h were 1.63 ± 0.04 ng/mL vs 2.28 ± 0.17 ng/mL (24 h, P < 0.01), and 3.36 ± 0.10 ng/mL vs 4.79 ± 0.31 ng/mL (48 h, P < 0.01), respectively, in Hep3B cells (Figure 4C). In HepG2 cells, the results were 1.15 ± 0.19 ng/mL vs 1.04 ± 0.03 ng/mL (24 h), and 2.70 ± 0.10 ng/mL vs 2.53 ± 0.32 ng/mL (48 h), respectively, (Figure 4D). Thus, the VEGF mRNA expression of Hep3B cells was increased by Glycer-AGEs at 24 h, and VEGF protein expression levels in these cells were also increased by Glycer-AGEs at 24 and 48 h. However, no such changes were observed in HepG2 cells.
Figure 4.

Effect of glyceraldehyde-derived advanced glycation end-products on the angiogenesis of hepatocellular carcinoma cells. A and B: Hep3B and HepG2 cells were incubated with control unglycated bovine serum albumin (BSA) or glyceraldehyde-derived advanced glycation end-products (Glycer-AGEs) for 24 h. Vascular endothelial growth factor (VEGF) mRNA expression was analyzed using real-time reverse transcription-polymerase chain reactions, and results were normalized to the β-actin mRNA level; C and D: Hep3B and HepG2 cells were incubated with control unglycated BSA or Glycer-AGEs for 24 or 48 h. The conditioned medium was collected, and VEGF expression levels of the cells were determined by enzyme-linked immunosorbent assay. The open and filled bars represent results for cells treated with control unglycated BSA and Glycer-AGEs, respectively. Data are shown as the mean ± SD (n = 3), bP < 0.01 vs control unglycated BSA.
We then examined the effect of CM-Glycer-AGEs on the viability of HUVEC. Cell viabilities resulting from treatment with the control unglycated BSA or the Glycer-AGEs for 72 h were 100% ± 6.4% vs 96.2% ± 5.4%, and the control unglycated BSA-treated CM (CM-BSA) or the CM-Glycer-AGEs for 72 h were 122.4% ± 9.0% vs 144.5% ± 11.3% (P < 0.01) (Figure 5). There was no difference in viability between the Glycer-AGEs-treated and control unglycated BSA-treated cells, whereas cell viability in cells treated with CM-Glycer-AGEs was significantly higher than that of those treated with CM-BSA.
Figure 5.
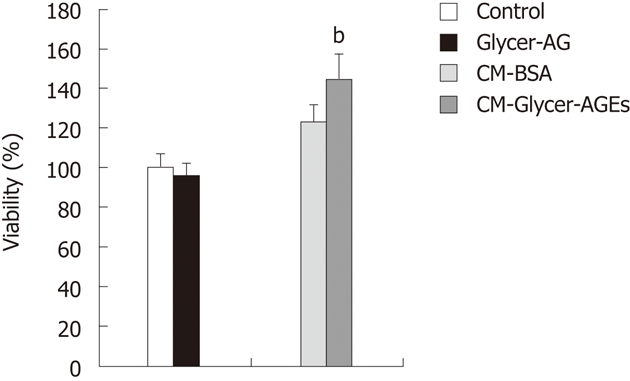
Effect of glyceraldehyde-derived advanced glycation end-products-treated CM on human umbilical vein endothelial cells proliferation. Cell viability was determined with the WST-8 assay. Human umbilical vein endothelial cells were incubated with control unglycated bovine serum albumin (BSA), glyceraldehyde-derived advanced glycation end-products (Glycer-AGEs) (100 μg/mL), CM-BSA, or CM-Glycer-AGEs for 72 h. The open and filled bars represent results for cells treated with control unglycated BSA and Glycer-AGEs, respectively, and the light grey and the black grey bars represent results for cells treated with CM-BSA and CM-Glycer-AGEs, respectively. Data are shown as the mean ± SD (n = 6), bP < 0.01 vs CM-BSA.
Finally, we examined the effect of CM-Glycer-AGEs on the migration and tube formation of HUVEC. Migration and tube formation of endothelial cells play key roles in tumor angiogenesis. Migration indices for the CM-BSA or the CM-Glycer-AGEs treatment were 4.29 ± 1.53 vs 6.78 ± 1.84 (P < 0.05) (Figure 6A), and cell migration with the CM-Glycer-AGEs treatment was significantly higher than that of the CM-BSA treatment. In tube formation, the CM-Glycer-AGEs-treated HUVEC showed an increased number of tube-like structures and larger tube networks (Figure 6B). Furthermore, the number of branching points for the CM-BSA or the CM-Glycer-AGEs treatment were 71.0% ± 7.5% vs 112.4% ± 8.0% (P < 0.01) (Figure 6C), and the number of branching points with the CM-Glycer-AGEs treatment was significantly higher than that of the CM-BSA treatment (Figure 6C).
Figure 6.
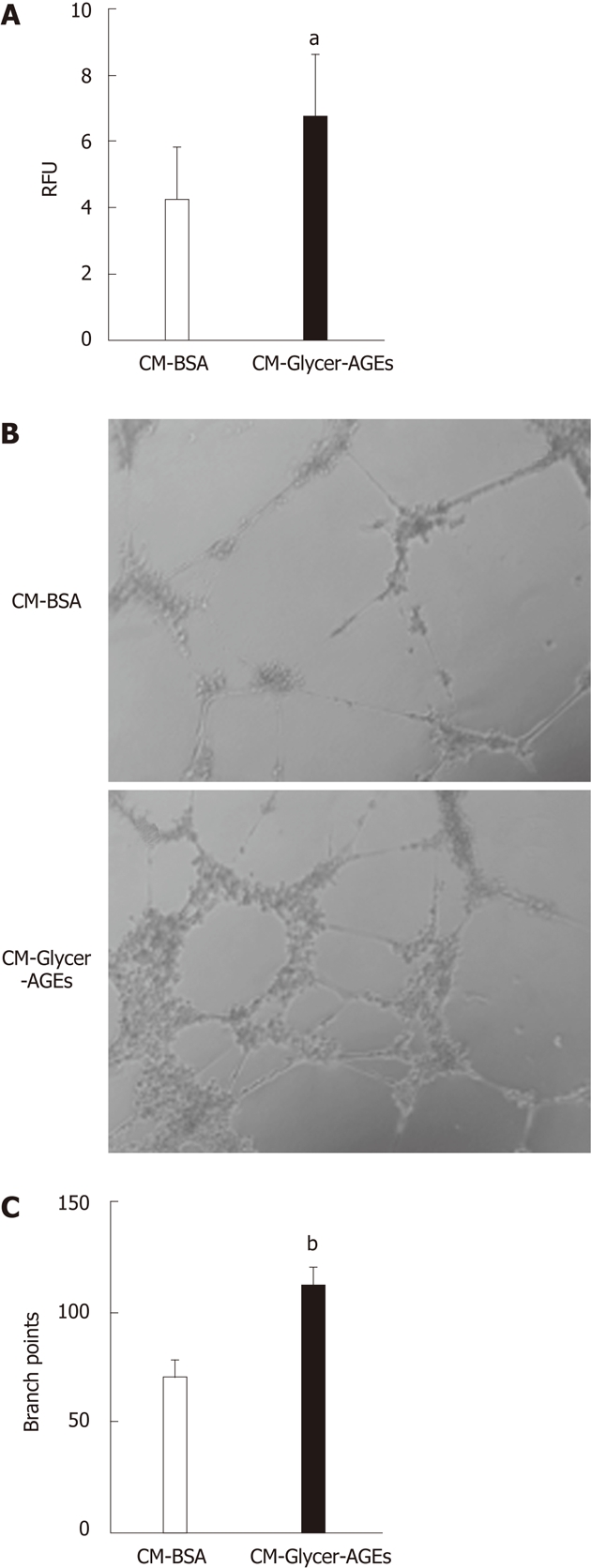
Effect of glyceraldehyde-derived advanced glycation end-products-treated CM on human umbilical vein endothelial cells angiogenesis. A: The migratory capacity of human umbilical vein endothelial cells (HUVEC) was evaluated using the endothelial cell migration assay. Cells were incubated with CM-bovine serum albumin (BSA) or glyceraldehyde-derived advanced glycation end-products-treated CM (CM-Glycer-AGEs) for 22 h, and the number of migrating cells was assessed using a fluorescence microplate reader. RFU: relative fluorescence units (n = 8); B and C: The tube formation of HUVEC was evaluated using the endothelial cell tube formation assay (n = 5). Cells were incubated with CM-BSA or CM-Glycer-AGEs for 12 h and then photographed under a microscope (B); the number of branch points was counted (C). Magnification = 100 ×. The open and filled bars represent results for cells treated with CM-BSA and CM-Glycer-AGEs, respectively. Data are shown as the mean ± SD, aP < 0.05, bP < 0.01 vs CM-BSA.
These results showed that Glycer-AGEs enhance the angiogenic potential of HCC cells by upregulating VEGF expression.
DISCUSSION
The incidence of HCC in developed countries has been increasing over the last 20 years[22]. Although hepatitis C virus is responsible for half of the recent increase in the prevalence of HCC, the etiologies of 15%-50% of new HCC cases remain unclear[23]. NASH, a component of metabolic syndrome, is thought to be responsible for some of these cases[15,16]. AGEs are one possible mechanistic link between metabolic syndrome and NASH, and Glycer-AGEs have been reported to be involved in NASH[8,24]; however, their role in HCC has barely been investigated.
A recent study suggested that the expression of RAGE mRNA was lower in normal liver tissue than in the liver cells of hepatitis and HCC patients. In addition, in HCC, RAGE mRNA expression was high in well and moderately differentiated tumors, but declined as the tumors dedifferentiated to poorly differentiated HCC[19]. The Hep3B and HepG2 cells used in this experiment were well-differentiated HCC cell lines. RAGE protein was detected in both Hep3B and HepG2 cells and there was no difference in its expression between the two cell lines. However, the effects of Glycer-AGEs differ between Hep3B and HepG2 cells. A previous report found that Hep3B cells activate AGEs signaling through RAGE[20]. On the other hand, it was reported that HepG2 cells did not express the RAGE protein on their cell surfaces and were not affected by Glycer-AGEs[25]. HepG2 cells were also not affected by Glycer-AGEs in our study.
In this study, Glycer-AGEs did not increase cell growth or migration of Hep3B cells. However, Glycer-AGEs slightly increased COX-2 protein expression levels in these cells. This protein plays important roles in HCC malignancy by producing prostaglandin E2 (PGE2), including cell growth, migration, and invasion[26,27], and PGE2 promotes the migration of HCC cell lines in > 1.5 μg/mL[28-30]. Indeed, PGE2 protein expression levels induced by COX-2 increased by Glycer-AGEs, but the quantity was < 6 pg/mL (data not shown). The results imply that although Glycer-AGEs induced increases in the expression levels of COX-2 and PGE2 protein, they were not sufficient to increase cell growth or migration.
On the other hand, Glycer-AGEs markedly increased the VEGF protein expression levels of Hep3B cells. VEGF is one of the most potent angiogenic factors[31], and angiogenesis plays a significant role in HCC progression[32-34]. Proliferation, migration, and tube formation of endothelial cells are important events in angiogenesis[35]. In addition, CM-Glycer-AGEs significantly increased the proliferation, migration, and tube formation of HUVEC. The results suggested that Glycer-AGEs indirectly increased angiogenesis in HCC. The formation of new blood vessels is initiated by hypoxic or ischemic conditions[36]. Interestingly, it was reported that HCC cell lines that are resistant to hypoxia displayed higher levels of RAGE expression, and RAGE expression in these cell lines increased under hypoxic conditions[19]. These results suggest that Glycer-AGEs-RAGE signaling is increased during the early stages of tumorigenesis, which occurs in hypoxic conditions.
In summary, we have demonstrated that Glycer-AGEs-RAGE signaling enhances the angiogenic potential of HCC cells by upregulating VEGF expression. These results suggest that Glycer-AGEs-RAGE signaling plays a critical role in the progression of HCC, and hence, is a potential target for therapeutic intervention.
COMMENTS
Background
Advanced glycation end-products (AGEs) are formed by the Maillard reaction, a non-enzymatic reaction between the ketones or aldehydes of sugars and the amino groups of proteins that contributes to aging and the pathological complications of diabetes. Among the different AGEs, there is evidence that Glycer-AGEs are associated with the complications of diabetes, as well as nonalcoholic steatohepatitis (NASH) and cancer. NASH is a potentially progressive disease that can lead to cirrhosis, liver failure and hepatocellular carcinoma (HCC). In fact, several case series of NASH-associated HCC have been reported. However, the effects of Glycer-AGEs on HCC cells remain poorly understood.
Research frontiers
Angiogenesis plays a significant role in HCC progression, and vascular endothelial growth factor (VEGF) is one of the most potent angiogenic factors. In this study, the authors examined the effects of Glycer-AGEs on the angiogenesis of HCC cells.
Innovations and breakthroughs
This study reported the Glycer-AGEs enhanced the angiogenic potential of HCC cells by upregulating VEGF expression.
Applications
The experimental data can be used in further studies as a potential target for therapeutic intervention.
Terminology
AGEs: AGEs are formed by the Maillard reaction, a non-enzymatic reaction between the ketones or aldehydes of sugars and the amino groups of proteins that contributes to aging and the pathological complications of diabetes; RAGE: RAGE is a multi-ligand member of the immunoglobulin superfamily of cell surface molecules, and interacts with distinct molecules implicated in homeostasis, development, and inflammation properties; NASH: NASH is a disease having the histopathological findings typical of alcoholic liver disease in patients without a history of significant alcohol abuse; VEGF: VEGF, which is also known as vascular permeability factor, is a specific mitogen to endothelial cells.
Peer review
The work presented is very interesting, and I am sure further work in the future will extend to clinical studies as a potential target for therapeutic intervention.
Footnotes
Supported by Grants from the Japan Society for the Promotion of Science, Grant-in-Aid for Scientific Research (B), No. 22300264
Peer reviewer: Xue-Wu Zhang, Professor, Basic Medical College, Yanbian University, No. 997 Gongyuan Lu, Yanji 133002, Jilin Province, China
S- Editor Gou SX L- Editor Stewart GJ E- Editor Xiong L
References
- 1.al-Abed Y, Kapurniotu A, Bucala R. Advanced glycation end products: detection and reversal. Methods Enzymol. 1999;309:152–172. doi: 10.1016/s0076-6879(99)09013-8. [DOI] [PubMed] [Google Scholar]
- 2.Vlassara H, Palace MR. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251:87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 3.Glomb MA, Monnier VM. Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the Maillard reaction. J Biol Chem. 1995;270:10017–10026. doi: 10.1074/jbc.270.17.10017. [DOI] [PubMed] [Google Scholar]
- 4.Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J. 1999;344 Pt 1:109–116. [PMC free article] [PubMed] [Google Scholar]
- 5.Takeuchi M, Bucala R, Suzuki T, Ohkubo T, Yamazaki M, Koike T, Kameda Y, Makita Z. Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J Neuropathol Exp Neurol. 2000;59:1094–1105. doi: 10.1093/jnen/59.12.1094. [DOI] [PubMed] [Google Scholar]
- 6.Abe R, Shimizu T, Sugawara H, Watanabe H, Nakamura H, Choei H, Sasaki N, Yamagishi S, Takeuchi M, Shimizu H. Regulation of human melanoma growth and metastasis by AGE-AGE receptor interactions. J Invest Dermatol. 2004;122:461–467. doi: 10.1046/j.0022-202X.2004.22218.x. [DOI] [PubMed] [Google Scholar]
- 7.Sato T, Iwaki M, Shimogaito N, Wu X, Yamagishi S, Takeuchi M. TAGE (toxic AGEs) theory in diabetic complications. Curr Mol Med. 2006;6:351–358. doi: 10.2174/156652406776894536. [DOI] [PubMed] [Google Scholar]
- 8.Hyogo H, Yamagishi S, Iwamoto K, Arihiro K, Takeuchi M, Sato T, Ochi H, Nonaka M, Nabeshima Y, Inoue M, et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22:1112–1119. doi: 10.1111/j.1440-1746.2007.04943.x. [DOI] [PubMed] [Google Scholar]
- 9.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 10.Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, Karim R, Lin R, Samarasinghe D, Liddle C, et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35:373–379. doi: 10.1053/jhep.2002.30692. [DOI] [PubMed] [Google Scholar]
- 11.Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. doi: 10.1053/jhep.2003.50161. [DOI] [PubMed] [Google Scholar]
- 12.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 13.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 14.Dam-Larsen S, Franzmann M, Andersen IB, Christoffersen P, Jensen LB, Sørensen TI, Becker U, Bendtsen F. Long term prognosis of fatty liver: risk of chronic liver disease and death. Gut. 2004;53:750–755. doi: 10.1136/gut.2003.019984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 16.Takuma Y, Nouso K. Nonalcoholic steatohepatitis-associated hepatocellular carcinoma: our case series and literature review. World J Gastroenterol. 2010;16:1436–1441. doi: 10.3748/wjg.v16.i12.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu AS, Keeffe EB. Management of hepatocellular carcinoma. Rev Gastroenterol Disord. 2003;3:8–24. [PubMed] [Google Scholar]
- 18.Simonetti RG, Liberati A, Angiolini C, Pagliaro L. Treatment of hepatocellular carcinoma: a systematic review of randomized controlled trials. Ann Oncol. 1997;8:117–136. doi: 10.1023/a:1008285123736. [DOI] [PubMed] [Google Scholar]
- 19.Hiwatashi K, Ueno S, Abeyama K, Kubo F, Sakoda M, Maruyama I, Hamanoue M, Natsugoe S, Aikou T. A novel function of the receptor for advanced glycation end-products (RAGE) in association with tumorigenesis and tumor differentiation of HCC. Ann Surg Oncol. 2008;15:923–933. doi: 10.1245/s10434-007-9698-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida T, Yamagishi S, Nakamura K, Matsui T, Imaizumi T, Takeuchi M, Ueno T, Sata M. Pigment epithelium-derived factor (PEDF) inhibits advanced glycation end product (AGE)-induced C-reactive protein expression in hepatoma cells by suppressing Rac-1 activation. FEBS Lett. 2006;580:2788–2796. doi: 10.1016/j.febslet.2006.04.050. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi M, Makita Z, Bucala R, Suzuki T, Koike T, Kameda Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol Med. 2000;6:114–125. [PMC free article] [PubMed] [Google Scholar]
- 22.Bosch FX, Ribes J, Díaz M, Cléries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 23.Gomaa AI, Khan SA, Toledano MB, Waked I, Taylor-Robinson SD. Hepatocellular carcinoma: epidemiology, risk factors and pathogenesis. World J Gastroenterol. 2008;14:4300–4308. doi: 10.3748/wjg.14.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kimura Y, Hyogo H, Yamagishi S, Takeuchi M, Ishitobi T, Nabeshima Y, Arihiro K, Chayama K. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J Gastroenterol. 2010;45:750–757. doi: 10.1007/s00535-010-0203-y. [DOI] [PubMed] [Google Scholar]
- 25.Sakuraoka Y, Sawada T, Okada T, Shiraki T, Miura Y, Hiraishi K, Ohsawa T, Adachi M, Takino J, Takeuchi M, et al. MK615 decreases RAGE expression and inhibits TAGE-induced proliferation in hepatocellular carcinoma cells. World J Gastroenterol. 2010;16:5334–5341. doi: 10.3748/wjg.v16.i42.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cervello M, Montalto G. Cyclooxygenases in hepatocellular carcinoma. World J Gastroenterol. 2006;12:5113–5121. doi: 10.3748/wjg.v12.i32.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu T. Cyclooxygenase-2 in hepatocellular carcinoma. Cancer Treat Rev. 2006;32:28–44. doi: 10.1016/j.ctrv.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Mayoral R, Fernández-Martínez A, Boscá L, Martín-Sanz P. Prostaglandin E2 promotes migration and adhesion in hepatocellular carcinoma cells. Carcinogenesis. 2005;26:753–761. doi: 10.1093/carcin/bgi022. [DOI] [PubMed] [Google Scholar]
- 29.Han C, Michalopoulos GK, Wu T. Prostaglandin E2 receptor EP1 transactivates EGFR/MET receptor tyrosine kinases and enhances invasiveness in human hepatocellular carcinoma cells. J Cell Physiol. 2006;207:261–270. doi: 10.1002/jcp.20560. [DOI] [PubMed] [Google Scholar]
- 30.Bai XM, Zhang W, Liu NB, Jiang H, Lou KX, Peng T, Ma J, Zhang L, Zhang H, Leng J. Focal adhesion kinase: important to prostaglandin E2-mediated adhesion, migration and invasion in hepatocellular carcinoma cells. Oncol Rep. 2009;21:129–136. [PubMed] [Google Scholar]
- 31.Toi M, Matsumoto T, Bando H. Vascular endothelial growth factor: its prognostic, predictive, and therapeutic implications. Lancet Oncol. 2001;2:667–673. doi: 10.1016/S1470-2045(01)00556-3. [DOI] [PubMed] [Google Scholar]
- 32.Poon RT, Ng IO, Lau C, Yu WC, Yang ZF, Fan ST, Wong J. Tumor microvessel density as a predictor of recurrence after resection of hepatocellular carcinoma: a prospective study. J Clin Oncol. 2002;20:1775–1785. doi: 10.1200/JCO.2002.07.089. [DOI] [PubMed] [Google Scholar]
- 33.Lee TK, Poon RT, Yuen AP, Man K, Yang ZF, Guan XY, Fan ST. Rac activation is associated with hepatocellular carcinoma metastasis by up-regulation of vascular endothelial growth factor expression. Clin Cancer Res. 2006;12:5082–5089. doi: 10.1158/1078-0432.CCR-05-2794. [DOI] [PubMed] [Google Scholar]
- 34.Jie S, Li H, Tian Y, Guo D, Zhu J, Gao S, Jiang L. Berberine inhibits angiogenic potential of Hep G2 cell line through VEGF down-regulation in vitro. J Gastroenterol Hepatol. 2011;26:179–185. doi: 10.1111/j.1440-1746.2010.06389.x. [DOI] [PubMed] [Google Scholar]
- 35.Lu N, Gao Y, Ling Y, Chen Y, Yang Y, Gu HY, Qi Q, Liu W, Wang XT, You QD, et al. Wogonin suppresses tumor growth in vivo and VEGF-induced angiogenesis through inhibiting tyrosine phosphorylation of VEGFR2. Life Sci. 2008;82:956–963. doi: 10.1016/j.lfs.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 36.Coulon S, Heindryckx F, Geerts A, Van Steenkiste C, Colle I, Van Vlierberghe H. Angiogenesis in chronic liver disease and its complications. Liver Int. 2011;31:146–162. doi: 10.1111/j.1478-3231.2010.02369.x. [DOI] [PubMed] [Google Scholar]