

Abstract
Hereditary hemorrhagic telangiectasia (HHT) is a rare autosomal-dominantly inherited disease that occurs in approximately one in 5000 to 8000 people. Clinical diagnosis of HHT is made when a person presents three of the following four criteria: family history, recurrent nosebleeds, mucocutaneous telangiectasis, and arteriovenous malformations (AVM) in the brain, lung, liver and gastrointestinal (GI) tract. Although epistaxis is the most common presenting symptom, AVMs affecting the lungs, brain and GI tract provoke a more serious outcome. Heterozygous mutations in endoglin, activin receptor-like kinase 1 (ACVRL1; ALK1), and SMAD4, the genes involved in the transforming growth factor-β family signaling cascade, cause HHT. We report here the case of a 63 year-old male patient who presented melena and GI bleeding episodes, proven to be caused by bleeding from multiple gastric angiodysplasia. Esophagogastroduodenoscopy revealed multiple angiodysplasia throughout the stomach. Endoscopic argon plasma coagulation was performed to control bleeding from a gastric angiodysplasia. The patient has been admitted several times with episodes of hemoptysis and hematochezia. One year ago, the patient was hospitalized due to right-sided weakness, which was caused by left basal ganglia hemorrhage as the part of HHT presentation. In family history, the patient’s mother and elder sister had died, due to intracranial hemorrhage, and his eldest son has been suffered from recurrent epistaxis for 20 years. A genetic study revealed a mutation in exon 3 of ALK1 (c.199C > T; p.Arg67Trp) in the proband and his eldest son presenting epistaxis.
Keywords: Hereditary hemorrhagic telangiectasia, Angiodysplasia, Intracranial hemorrhage, Epistaxis, Activin receptor-like kinase 1
INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT), also called Osler-Weber-Rendu syndrome, is a rare autosomal dominant genetic disorder occurring in about one in 5000 to 8000 people worldwide[1-3]. HHT is characterized by family history, recurrent nosebleeds, mucocutaneous telangiectasis, and arteriovenous malformations (AVMs) in the brain, lung, liver and gastrointestinal (GI) tract[4,5]. Clinical diagnosis of HHT is made when a person presents with three of these four symptoms[6]. However, due to the highly variable onset and penetrance of these clinical symptoms, HHT is often misdiagnosed. Epistaxis and skin telangiectasis are the most common presenting symptoms that appear in over 90% of HHT patients aged over 60 years[7]. Cerebral and pulmonary AVMs occur in around 10%-20% and 50% of HHT patients, respectively, and are associated with high mortality and morbidity due to stroke or brain abscess[8]. AVMs in the GI tract can lead to melena, brisk GI bleeding, and hematochezia. Most elderly HHT patients suffer from anemia due to epistaxis and GI bleeding.
Genetic studies have shown that heterozygous mutations in endoglin (ENG) or activin receptor-like kinase 1 (ALK1; ACVRL1) cause HHT[9-12]. HHT by ENG or ALK1 mutations is designated as HHT1 or HHT2, respectively, although they are clinically indistinguishable. Some juvenile polyposis (JP) patients with SMAD4 heterozygous mutations have shown to present HHT symptoms and are designated as JP-HHT[13,14]. Two additional genetic loci in chromosomes 5 (HHT3) and 7 (HHT4) have been identified[15,16]. HHT1 and HHT2 account for over 80% of HHT worldwide[17].
In this report, we describe a case of a patient presenting with multiple gastric angiodysplastic lesions, intracranial hemorrhage (ICH), and a family history of ICH and epistaxis. Through genetic testing, we found a known mutation in ALK1 (c.199 C > T; p.Arg67Trp) in the proband[18-23]. Additionally, the patient’s son had been suffering from epistaxis. To our knowledge, this is the first case in Korea of genetic confirmation of HHT2 with multiple gastric angiodysplasia.
CASE REPORT
A 63-year-old male was admitted due to melena. Examination revealed stable vital signs with normal blood pressure and heart rate, but routine laboratory tests revealed mild anemia with a hemoglobin level of 9.6 g/dL. Esophagogastr oduodenoscopy (EGD) revealed multiple angiodysplastic lesions throughout the stomach (Figure 1A and B). Conservative treatment, including proton pump inhibitors, led to a stable clinical condition. The patient was subsequently hospitalized again due to hemoptysis, and then once more due to hematochezia; all were managed by conservative treatment regimens. We carefully investigated the patient’s medical and family history. The patient’s mother and older sister died from ICH. His eldest son had been suffering from recurrent epistaxis for 20 years (Figure 2A). Therefore, due to the presumptive diagnosis of HHT, we carried out genetic screening for the ENG and ALK1 genes. A heterozygous mutation in ALK1 was detected from the proband of both the patient and his eldest son, but not from a daughter who did not show any apparent HHT-related symptoms (Figure 2B). A single nucleotide substitution from “C” to “T” at the 199th coding nucleotide (c.199 C > T) in exon 3 changed the 67th amino acid, arginine, to tryptophan (p.Arg67Trp), resulting in loss of ALK1 function (Figure 2B)[18-23]. Abdomino-pelvic computed tomography (CT) scan of the proband, performed for evaluation of a gallbladder stone, revealed multiple intrahepatic AV shunts (Figure 3A). Later, the proband received peritoneoscopic cholecystectomy, and then the patient was hospitalized again due to the sudden onset of right-sided weakness. Brain CT revealed ICH at the left basal ganglion, and he was transferred to a local rehabilitation facility (Figure 3B). The patient was recently admitted for percutaneous endoscopic gastrostomy insertion.
Figure 1.
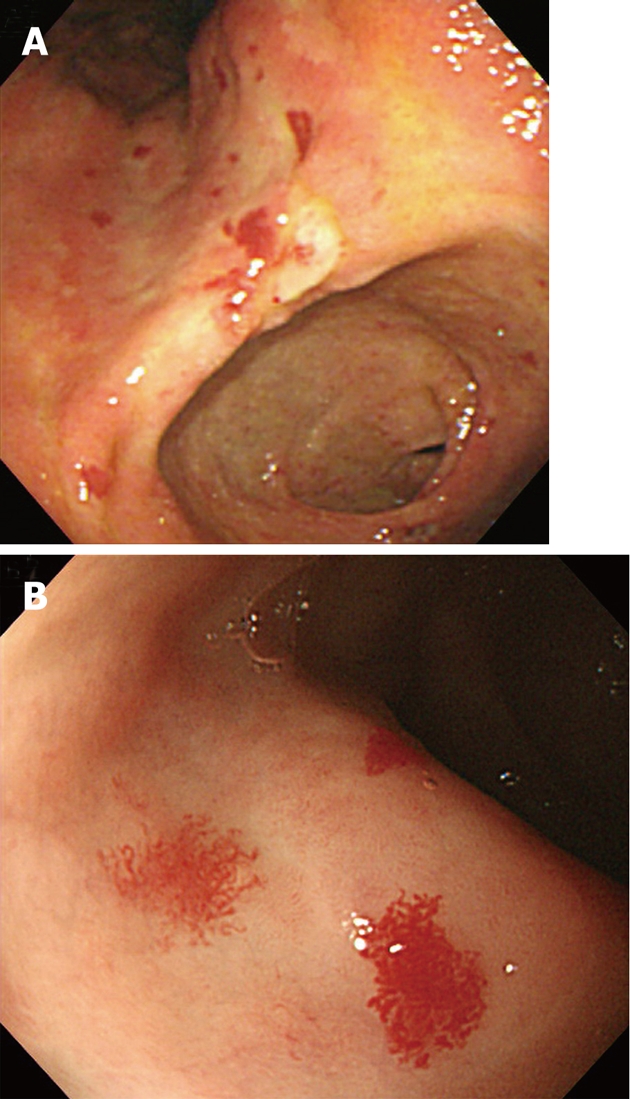
Multiple angiodysplastic lesions were noted in the entire stomach upon endoscopic examination. A: A view of the lesser curvature of the angle showing multiple scattered angiodysplastic lesions; B: A view of the anterior wall of the antrum, magnified view of angiodysplastic lesions showing vessel-pulled appearance.
Figure 2.
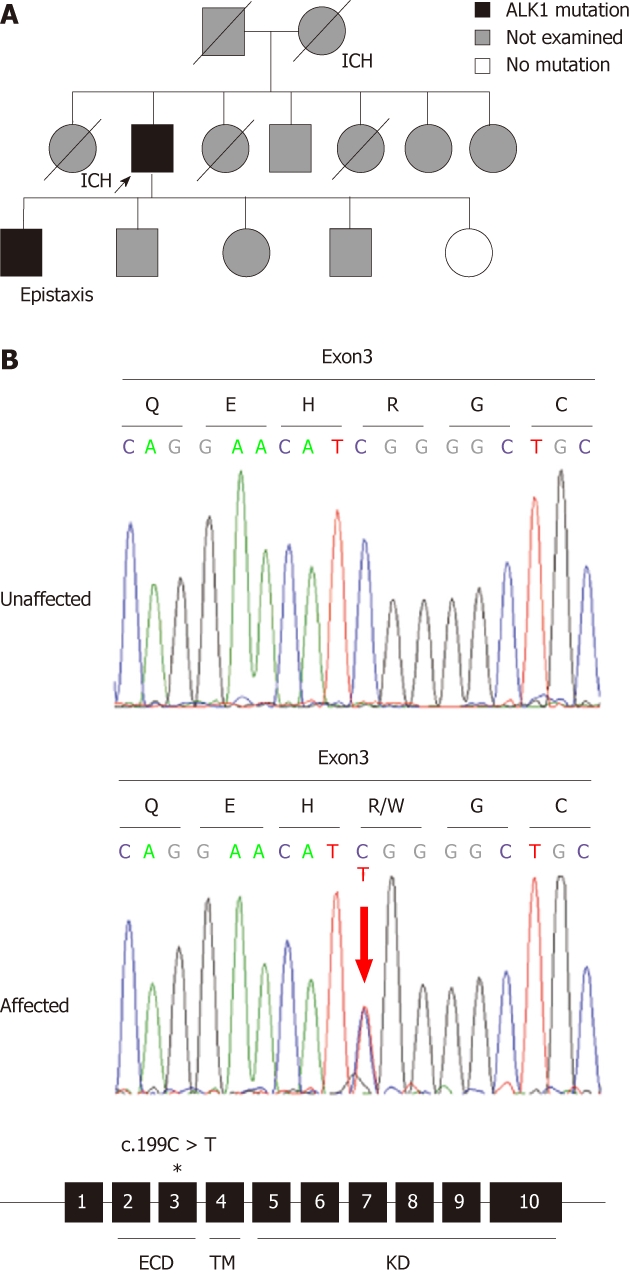
Pedigree and genetic analysis of an hereditary hemorrhagic telangiectasia family. A: Pedigree of a family with genetic mutations and/or symptoms of hereditary hemorrhagic telangiectasia (HHT) and intracranial hemorrhage (ICH). The proband is indicated by an arrow. A divided symbol represents the individual with ICH. Deceased individuals are indicated by a slash; B: Genetic studies of unaffected and affected family members. The affected member had a heterozygous activin receptor-like kinase 1 (ALK1) mutation (c.199C > T; p.Arg67Trp). The amino acid translation is shown above each codon. The mutation was found in exon 3, indicated by an asterisk; 3. Protein domains of ALK1 are indicated under the exons: extracellular domain (ECD), transmembrane domain (TM), and kinase domain (KD).
Figure 3.
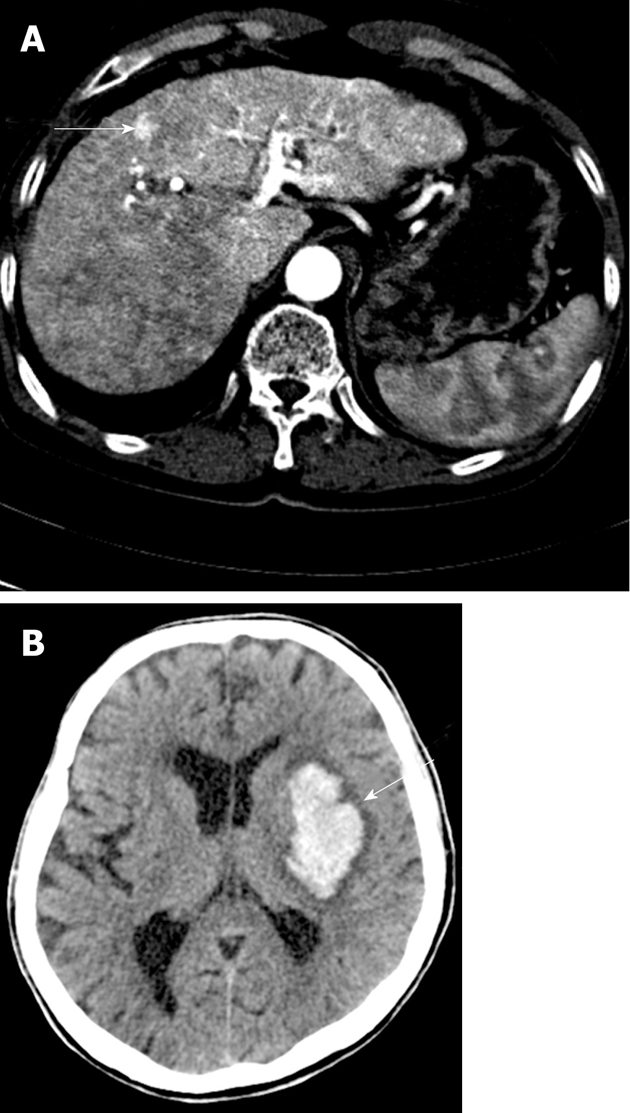
Hepatic arterio-venous shunt and cerebral hemorrhage. A: Abdominal computed tomography (CT) showing an intra-hepatic arterio-venous shunt; B: Cerebral hemorrhage was noted in the left basal ganglia on brain CT.
DISCUSSION
In the present case, we report a patient with multiple angioplastic lesions in the stomach, chronic hemorrhages in the GI tract, and stroke by ICH. Presence of ICH and epistaxis in his immediate family members (mother, sister and son) led us to diagnose HHT. Genetic screening of two HHT genes, ALK1 and ENG, revealed a substitution mutation at the 199th coding nucleotide of the ALK1 gene, specifically in the proband and the son presenting as epistaxis, but not in the daughter who had no apparent symptoms[18-23]. These data confirmed the clinical diagnosis and showed that the patient carries the HHT2 subtype. HHT1 and HHT2 are clinically similar in presentation, but genotype-phenotype correlation studies have shown that occurrence of pulmonary AVMs is significantly higher in HHT1, whereas liver AVM tend to be more common in HHT2[20,21,24-28]. Cerebral involvement and spinal AVMs are reported in 10%-20% and 1%-2% of HHT1 and HHT2 cases, respectively[8,27]. Whether genetic or environmental factors contributed to this observation in this family or the wider Korean HHT population remains to be determined. The mutation found in this family (c.199 C > T; p.Arg67Trp) has previously been reported by five other groups[18-23]. The 67th amino acid is located in the ligand binding domain of ALK1. Previous biochemical analysis of the Arg67Gln substitution resulted in a deficiency in signal transduction, suggesting a null mutation[29]. Life-threatening cerebral and pulmonary AVMs that form during development and the neonatal period are often asymptomatic for a prolonged period. Manifestations of epistaxis, skin telangiectasis, and GI AVMs are generally absent at birth, but develop over puberty and progressively worsen with age[7,30,31]. Genetic screening of asymptomatic family members would allow detection of deadly forms of vascular lesions in advance, before complications arise.
GI bleeding associated with HHT occurs in about 15%-30% of patients, and GI telangiectasia, including angiodysplasia, is a relatively common manifestation of HHT[7,32,33]. However, reports of this syndrome in the form of angiodysplasia on EGD in the GI tract, including the stomach, are rare.
Together with epistaxis, GI bleeding is a serious issue for elderly HHT patients. Recent studies indicate that environmental factors such as injury and inflammation, in addition to genetic predisposition (ALK1 or ENG mutations), are critical for development of AVMs in adults[34,35]. Multiple anti-inflammatory, anti-angiogenic, and anti-oxidants drugs such as thalidomide, bevacizumab and N-acetyl-cysteine have been tested as potential therapies for ameliorating epistaxis and GI bleeding[7,36]. We report here that the proband had multiple angiodysplastic lesions in the stomach and also hematochezia, indicating that vascular lesions had spread to the lower GI tract. In addition, close monitoring for GI bleeding is needed. Capsule endoscopy and conventional endoscopy may provide important information on whether these drugs can induce regression of existing vascular lesions.
In summary, we report the case of a patient in whom we found multiple gastric angiodysplastic lesions. With both clinical evaluation and genetic screening, we confirmed that the patient was suffering from HHT2. Family history and other HHT symptoms should be carefully evaluated when a patient shows multiple angiodysplastic lesions. Genetic screening has tremendous benefit not only for confirming the diagnosis but also in preventing asymptomatic family members developing life-threatening complications.
ACKNOWLEDGMENTS
The authors would like to thank all patients and families who participated in this study.
Footnotes
Supported by A grant of the South Korea Healthcare technology R and D Project, Ministry for Health, Welfare and Family Affairs, South Korea, No. A080588-23; and in part by a grant from the World Class University (WCU by Korean Ministry of Education, Science and Technology) (to Oh SP)
Peer reviewers: Antonello Trecca, Digestive Endoscopy, Usi Group, Via Machiavelli, 22, 00184 Rome, Italy; Anastasios Koulaouzidis, Centre for Liver and Digestive Disorders, Endoscopy Unit, Centre of Liver and Digestive Disorders, Royal Infirmary of Edinburgh, Little France Crescent, Edinburgh EH16 4SA, Scotland, United Kingdom
S- Editor Gou SX L- Editor O’Neill M E- Editor Xiong L
References
- 1.Bideau A, Brunet G, Heyer E, Plauchu H, Robert JM. An abnormal concentration of cases of Rendu-Osler disease in the Valserine valley of the French Jura: a genealogical and demographic study. Ann Hum Biol. 1992;19:233–247. doi: 10.1080/03014469200002112. [DOI] [PubMed] [Google Scholar]
- 2.Kjeldsen AD, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med. 1999;245:31–39. doi: 10.1046/j.1365-2796.1999.00398.x. [DOI] [PubMed] [Google Scholar]
- 3.Dakeishi M, Shioya T, Wada Y, Shindo T, Otaka K, Manabe M, Nozaki J, Inoue S, Koizumi A. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat. 2002;19:140–148. doi: 10.1002/humu.10026. [DOI] [PubMed] [Google Scholar]
- 4.Abdalla SA, Letarte M. Hereditary haemorrhagic telangiectasia: current views on genetics and mechanisms of disease. J Med Genet. 2006;43:97–110. doi: 10.1136/jmg.2005.030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lenato GM, Guanti G. Hereditary Haemorrhagic Telangiectasia (HHT): genetic and molecular aspects. Curr Pharm Des. 2006;12:1173–1193. doi: 10.2174/138161206776361291. [DOI] [PubMed] [Google Scholar]
- 6.Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome) Am J Med Genet. 2000;91:66–67. doi: 10.1002/(sici)1096-8628(20000306)91:1<66::aid-ajmg12>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 7.Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010;24:203–219. doi: 10.1016/j.blre.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17:860–871. doi: 10.1038/ejhg.2009.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 10.McDonald MT, Papenberg KA, Ghosh S, Glatfelter AA, Biesecker BB, Helmbold EA, Markel DS, Zolotor A, McKinnon WC, Vanderstoep JL. A disease locus for hereditary haemorrhagic telangiectasia maps to chromosome 9q33-34. Nat Genet. 1994;6:197–204. doi: 10.1038/ng0294-197. [DOI] [PubMed] [Google Scholar]
- 11.Vincent P, Plauchu H, Hazan J, Fauré S, Weissenbach J, Godet J. A third locus for hereditary haemorrhagic telangiectasia maps to chromosome 12q. Hum Mol Genet. 1995;4:945–949. doi: 10.1093/hmg/4.5.945. [DOI] [PubMed] [Google Scholar]
- 12.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 13.Gallione CJ, Richards JA, Letteboer TG, Rushlow D, Prigoda NL, Leedom TP, Ganguly A, Castells A, Ploos van Amstel JK, Westermann CJ, et al. SMAD4 mutations found in unselected HHT patients. J Med Genet. 2006;43:793–797. doi: 10.1136/jmg.2006.041517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 15.Cole SG, Begbie ME, Wallace GM, Shovlin CL. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42:577–582. doi: 10.1136/jmg.2004.028712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bayrak-Toydemir P, McDonald J, Akarsu N, Toydemir RM, Calderon F, Tuncali T, Tang W, Miller F, Mao R. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006;140:2155–2162. doi: 10.1002/ajmg.a.31450. [DOI] [PubMed] [Google Scholar]
- 17.Lesca G, Burnichon N, Raux G, Tosi M, Pinson S, Marion MJ, Babin E, Gilbert-Dussardier B, Rivière S, Goizet C, et al. Distribution of ENG and ACVRL1 (ALK1) mutations in French HHT patients. Hum Mutat. 2006;27:598. doi: 10.1002/humu.9421. [DOI] [PubMed] [Google Scholar]
- 18.Olivieri C, Mira E, Delù G, Pagella F, Zambelli A, Malvezzi L, Buscarini E, Danesino C. Identification of 13 new mutations in the ACVRL1 gene in a group of 52 unselected Italian patients affected by hereditary haemorrhagic telangiectasia. J Med Genet. 2002;39:E39. doi: 10.1136/jmg.39.7.e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdalla SA, Cymerman U, Rushlow D, Chen N, Stoeber GP, Lemire EG, Letarte M. Novel mutations and polymorphisms in genes causing hereditary hemorrhagic telangiectasia. Hum Mutat. 2005;25:320–321. doi: 10.1002/humu.9312. [DOI] [PubMed] [Google Scholar]
- 20.Kuehl HK, Caselitz M, Hasenkamp S, Wagner S, El-Harith el-HA, Manns MP, Stuhrmann M. Hepatic manifestation is associated with ALK1 in hereditary hemorrhagic telangiectasia: identification of five novel ALK1 and one novel ENG mutations. Hum Mutat. 2005;25:320. doi: 10.1002/humu.9311. [DOI] [PubMed] [Google Scholar]
- 21.Bossler AD, Richards J, George C, Godmilow L, Ganguly A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): correlation of genotype with phenotype. Hum Mutat. 2006;27:667–675. doi: 10.1002/humu.20342. [DOI] [PubMed] [Google Scholar]
- 22.Argyriou L, Twelkemeyer S, Panchulidze I, Wehner LE, Teske U, Engel W, Nayernia K. Novel mutations in the ENG and ACVRL1 genes causing hereditary hemorrhagic teleangiectasia. Int J Mol Med. 2006;17:655–659. [PubMed] [Google Scholar]
- 23.Argyriou L, Pfitzmann R, Wehner LE, Twelkemeyer S, Neuhaus P, Nayernia K, Engel W. ALK-1 mutations in liver transplanted patients with hereditary hemorrhagic telangiectasia. Liver Transpl. 2005;11:1132–1135. doi: 10.1002/lt.20544. [DOI] [PubMed] [Google Scholar]
- 24.McAllister KA, Lennon F, Bowles-Biesecker B, McKinnon WC, Helmbold EA, Markel DS, Jackson CE, Guttmacher AE, Pericak-Vance MA, Marchuk DA. Genetic heterogeneity in hereditary haemorrhagic telangiectasia: possible correlation with clinical phenotype. J Med Genet. 1994;31:927–932. doi: 10.1136/jmg.31.12.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berg JN, Guttmacher AE, Marchuk DA, Porteous ME. Clinical heterogeneity in hereditary haemorrhagic telangiectasia: are pulmonary arteriovenous malformations more common in families linked to endoglin. J Med Genet. 1996;33:256–257. doi: 10.1136/jmg.33.3.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Tsao G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT) J Hepatol. 2007;46:499–507. doi: 10.1016/j.jhep.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 27.Letteboer TG, Mager JJ, Snijder RJ, Koeleman BP, Lindhout D, Ploos van Amstel JK, Westermann CJ. Genotype-phenotype relationship in hereditary haemorrhagic telangiectasia. J Med Genet. 2006;43:371–377. doi: 10.1136/jmg.2005.035451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kjeldsen AD, Møller TR, Brusgaard K, Vase P, Andersen PE. Clinical symptoms according to genotype amongst patients with hereditary haemorrhagic telangiectasia. J Intern Med. 2005;258:349–355. doi: 10.1111/j.1365-2796.2005.01555.x. [DOI] [PubMed] [Google Scholar]
- 29.Lux A, Attisano L, Marchuk DA. Assignment of transforming growth factor beta1 and beta3 and a third new ligand to the type I receptor ALK-1. J Biol Chem. 1999;274:9984–9992. doi: 10.1074/jbc.274.15.9984. [DOI] [PubMed] [Google Scholar]
- 30.Plauchu H, de Chadarévian JP, Bideau A, Robert JM. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am J Med Genet. 1989;32:291–297. doi: 10.1002/ajmg.1320320302. [DOI] [PubMed] [Google Scholar]
- 31.Porteous ME, Burn J, Proctor SJ. Hereditary haemorrhagic telangiectasia: a clinical analysis. J Med Genet. 1992;29:527–530. doi: 10.1136/jmg.29.8.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Begbie ME, Wallace GM, Shovlin CL. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syndrome): a view from the 21st century. Postgrad Med J. 2003;79:18–24. doi: 10.1136/pmj.79.927.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sadick H, Sadick M, Götte K, Naim R, Riedel F, Bran G, Hörmann K. Hereditary hemorrhagic telangiectasia: an update on clinical manifestations and diagnostic measures. Wien Klin Wochenschr. 2006;118:72–80. doi: 10.1007/s00508-006-0561-x. [DOI] [PubMed] [Google Scholar]
- 34.Park SO, Wankhede M, Lee YJ, Choi EJ, Fliess N, Choe SW, Oh SH, Walter G, Raizada MK, Sorg BS, et al. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest. 2009;119:3487–3496. doi: 10.1172/JCI39482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jerkic M, Peter M, Ardelean D, Fine M, Konerding MA, Letarte M. Dextran sulfate sodium leads to chronic colitis and pathological angiogenesis in Endoglin heterozygous mice. Inflamm Bowel Dis. 2010;16:1859–1870. doi: 10.1002/ibd.21288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rohrmeier C, Sachs HG, Kuehnel TS. A retrospective analysis of low dose, intranasal injected bevacizumab (Avastin) in hereditary haemorrhagic telangiectasia. Eur Arch Otorhinolaryngol. 2012;269:531–536. doi: 10.1007/s00405-011-1721-9. [DOI] [PubMed] [Google Scholar]