

Abstract
Introduction
Muscle pain is a common side effect of statin medications, however, the cause is poorly understood.
Methods
We characterized phosphocreatine (PCr) exercise recovery kinetics in 10 patients with hypercholesterolemia before and after a 4 week regimen of statin therapy using 31P magnetic resonance spectroscopy (31P-MRS). 31P spectra were obtained before, during, and following exercise on a calf flexion pedal ergometer. Creatine kinase (CK) serum levels were drawn before and after statin therapy.
Results
The mean metabolic recovery time constant in subjects increased from 28.1s (SE=6.5s) to 55.4s (SE=7.4s) following statin therapy. The unweighted mean of the pre-post recovery time difference was -27.3s (SE=12.4s); (p-value = 0.02). Pre- and post-therapy CK levels were not significantly different (p-value = 0.50).
Discussion
Metabolic recovery time in the calf is prolonged in patients following statin use. This suggests that statins impair mitochondrial oxidative function, and 31P –MRS is a potential study model for statin-associated myopathy.
Keywords: Statin medications, MR spectroscopy, myopathy, phosphocreatine exercise recovery kinetics, mitochondrial disease
Introduction
Statins, 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA) inhibitors, are among the most commonly prescribed medications in the world. They are used for the prevention and treatment of atherosclerotic and cardiovascular disease.1 These drugs are generally safe, however, myotoxicity, including fatal rhabdomyolysis can occur.2, 3 Although severe muscle-related side effects occur in <0.1% of statin users4, less severe symptoms, such as myalgia and muscle cramps, occur in 1–7% of users.5–8 Since statins must be taken long-term, muscle pains can affect their tolerability, and adherence is a major concern.9 Previous studies suggest that <40% of patients take ≥80% of prescribed statin doses 1 year after starting therapy and about half discontinue the drugs within 6 months of starting them.10 The mechanisms for statin-associated myopathy remain unclear, and numerous hypotheses exist. Among the most convincing are that the effect of statins on muscle may be due to inhibition of the HMG-CoA reductase pathway which synthesizes not only cholesterol but also isoprenyl end products. These are required for numerous proteins, cofactors and signaling molecules that are important for skeletal muscle cell function. Defects in mitochondrial energy production and/or function have received much attention, because the HMG-CoA reductase pathway is important for the synthesis of the isoprenyl tail of Coenzyme Q10, an integral component of oxidative phosphorylation in mitochondria ATP production.6, 7, 11, 12 Preliminary studies show that statin therapy may reduce mitochondrial number and/or volume.13, 14 Additionally, evidence linking the atrogin-1 gene to statin-associated muscle injury shows that the gene and its muscle damaging effects may be blocked by PGC-1alpha, which is thought to be due to its ability to augment mitochondrial number and function.15, 16
Non-invasive objective tools to study statin-associated muscle effects and response to treatment are lacking. Creatine kinase (CK) is a biomarker for muscle damage, but it can be normal in patients with symptomatic, biopsy proven statin-associated myopathy.17, 18 Imaging with MRI and MIBI scintigraphy fail to show visible changes in patients with statin-associated myopathy.18, 19 There are case reports of radiotracer uptake in skeletal muscle on bone scintigraphy and 18F-FDG PET, but only in patients with severe myotoxicity with rhabdomyolysis.20, 21 Muscle biopsy with morphological/biochemical analysis is currently the reference standard. Unfortunately, biopsies are invasive, painful, and provide only a metabolic “snapshot” of the muscle. Muscle biopsy cannot evaluate the dynamic nature of the various muscle metabolites.
31P magnetic resonance spectroscopy (31P-MRS) is unique in its ability to study, continuously and non-invasively, the biochemical pathways for the supply and utilization of energy in muscle tissue.22 The post-exercise metabolic recovery rate of skeletal muscle phosphocreatine (PCr) as measured by 31P-MRS has been used as an index of mitochondrial oxidative capacity in vivo.23–26 During repeated muscle contraction, the concentration of adenosine triphosphate (ATP) is maintained at a constant level through glycolysis, oxidative phosphorylation, and the CK reaction, which results in a decrease in the concentration of PCr. 31P-MRS in muscle can provide insights into the pathophysiology of certain diseases as well as normal physiology25 and has been used as an outcome measure for clinical trials.27 Although not in routine clinical use, 31P-MRS has been used to study muscle metabolite changes in athletes and a wide range of diseases.22, 24, 28–30 Blunted PCr recovery time has been described in patients with peripheral vascular disease, diabetes, and myopathies,24, 26 and increased PCr recovery can be seen in professional cyclists30.
If oxidative phosphorylation and ATP synthesis in mitochondria are impaired by statins, then one may also see prolongation of the metabolic recovery time constant. The purpose of this pilot study was to characterize phosphocreatine (PCr) exercise recovery kinetics in patients with hypercholesterolemia before and after initiation of statin therapy using 31P-MRS.
Materials and Methods
Study Subjects
Ten subjects (5 females and 5 males with a mean age of 52 years, range 35–69) with hypercholesterolemia were recruited from preventive cardiology and primary care clinics at Beth Israel Deaconess Medical Center in Boston Massachusetts. Individuals were eligible if they were statin naïve or had been off statin medication for 2 weeks or longer and had been prescribed a statin by their physician at a dose approximately equivalent to or higher than simvastatin 20 mg daily. A minimum two week period off statin was required in order to allow clearance of the medication from the systemic circulation. All subjects were without muscle symptoms at the time of their baseline measurement. Potential subjects with established peripheral vascular disease, diabetes, or use of other lipid lowering agents or coenzyme Q10 in the last 30 days were excluded. Patients were excluded if they were using medications associated with mitochondrial dysfunction or an increased risk of myopathy, including propranolol, niacin, and fibrates.2, 31 Moreover, all subjects were instructed to not alter exercise habits or diets during the 4 weeks on statin medication. This study was approved by our institutional review board, and informed consent was obtained from all subjects.
31 P MRS equipment/protocol
31P-MRS was performed on the posterior calf using a surface coil during exercise on a custom-built MRI-compatible pedal ergometer. The 10 subjects were studied before and 4 weeks after statin therapy. The exercise ergometer was designed to allow the study subjects to perform plantar flexion exercise by pressing against a foot pedal while lying in the supine position. The subject’s lower extremity was secured to the MRI table with straps across the mid thigh and mid lower leg in order to isolate usage of the posterior calf muscles. The force required to depress the pedal was supplied by a pressurized tank of nitrogen gas with an adjustable pressure regulator, which was connected to a piston cylinder attached to the foot pedal (Fig. 1). The maximum pressure (PSI) was determined during 1 trial by incrementally increasing the pressure on the pedal to a point where the subject could only depress the pedal through the full range of motion once. 40% of this maximum pressure was then used for both the pre- and post-therapy studies. The exercise protocol consisted of pressing the pedal down for one second and relaxing back for one second as cued by a metronome with the pressure reduced to 40% of the maximum value. The subjects performed 30 plantar flexions per minute for a maximum of 7 minutes, or until muscle exhaustion or calf pain.
Figure 1.
Photograph of a subject using the custom-built MRI-compatible pedal ankle ergometer during 31P-MRS acquisition.
31P-MRS data acquisition
31P-MRS of the posterior calf muscles muscle was performed using a 3-Tesla MR system (General Electric Healthcare, Milwaukee, Wisconsin). A 7.5-cm circular 31P surface coil was centered on the maximum diameter of the calf (approximately 4 to 8 cm below the posterior knee crease). Magnetic field homogeneity was adjusted while observing the 31P signal. Spectra were acquired using a pulse-and-acquire free induction decay (FID) sequence. Localization was accomplished by the surface coil. The 31P-MRS acquisition parameters were: sweep width, 2048 Hz; number of complex points, 1024; TR, 5 s; signal averages, 2; time per acquisition, 10 s. Subjects were studied for: (1) one minute prior to calf exercise, (2) up to 7 minutes during exhaustive exercise, and (3) for 6 minutes following exercise during the recovery period. 31P spectra were acquired at 10 s intervals (Fig. 2).
Figure 2.
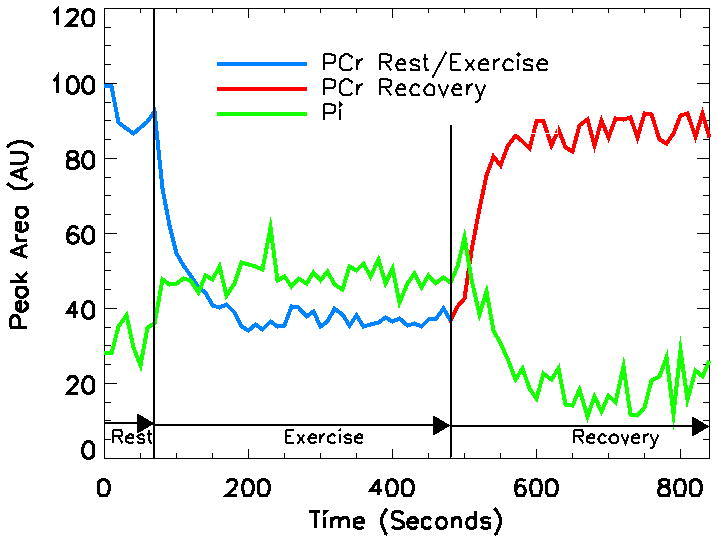
Graph showing the variability in the amounts of phosphocreatine (PCr) and inorganic phosphate (Pi) during the rest, exercise, and recovery phases of calf exercise as measured by 31P MR spectroscopy in a 44 year old white man prior to statin therapy.
31P-MRS data analysis
The 31P spectral data were processed using IDL software (Version 6.0. Research Systems, Inc. Boulder, CO). Each FID was processed with 3-Hz exponential line broadening without zero filling. Spectra were manually phased using zero and first-order phase corrections. The areas of the PCr peaks were calculated and normalized to the pre-exercise value. Monoexponential curve fitting was performed to compare the PCr recovery time between pre- and post-statin therapy. The model fitted is
where b is PCr depletion and k is the PCr metabolic recovery time constant.32, 33 We fitted this nonlinear model to each of the subjects pre- and post-statin therapy using the statistical nonlinear model fitting procedure NLIN of the SAS/STAT software version 9 (SAS Institute Inc., Cary NC). The procedure used nonlinear least squares solution via the Newton method. The two parameters b, and k and their standard errors were estimated for each subject. We were interested in testing the hypothesis that there was a mean difference in the parameter k between pre and post intervention. We calculated both unweighted and weighted mean data, with the weight computed from the inverse of the square of the standard error. With only 10 subjects, we did not have normality for the distribution of the estimates of parameter k, so we were not able to use a weighted paired t-test to obtain inference. Therefore, we used a nonparametric signed rank test on the paired differences.
Creatine kinase (CK)
CK serum levels were drawn on all subjects prior to and 4 weeks after the initiation of statin medications. The following are used as the upper limits of normal CK values at our institution: (Male/Female): Whites 322/201 IU/L; African-Americans 801/414 IU/L; Asians 641/313 IU/L.
Results
Patient Symptoms
Five of the 10 patients had taken statins previously, and 4 of the 5 patients had suspected statin-associated muscle pains that resolved with discontinuation of the medication. Of the 4 patients with a history of muscle pain while on statins, 3 had mild muscle pain while on treatment during the study. Half of the subjects were sedentary or performed light activities only; the other half engaged in moderate to vigorous exercise most or all days of the week. All subjects reported no changes in exercise habits or diets. The patient demographics are summarized in Table 1.
Table 1.
Subjects undergoing 31P-MRS before and after statin medications
| Pt# | Age (yrs) | G | Race | Statin, dose (mg/day) | Prior Statin Use/Pain | Pain during Study | CK Pre/Post (U/L) | Exercise Pre/Post Time (secs) | Recovery Time Pre-Statin Seconds (SE) | Recovery Time Post-Statin Seconds (SE) | Difference in Pre-Post Recovery Seconds (SE) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 47 | F | White | simvastatin, 40 | −/NA | − | 62/57 | 120/180 | 44.1 (4.8) | 116.9 (9.1) | −72.7 (8.4) |
| 2 | 67 | F | White | simvastatin, 20 | −/NA | − | 53/60 | 440/420 | 30.2 (6.3) | 147.5 (29.4) | −117.2 (27.4) |
| 3 | 35 | M | Asian | simvastatin, 40 | −/NA | − | 162/263 | 60/80 | 21.1 (4.3) | 15.0 (3.6) | 6.2 (4.3) |
| 4 | 47 | M | Asian | atorvastatin, 10 | +/− | − | 92/148 | 90/210 | 24.0 (4.3) | 18.3 (2.7) | 5.7 (4.0) |
| 5 | 47 | M | White | simvastatin, 20 | +/+ | + | 80/73 | 420/410 | 20.9 (16.8) | 36.5 (8.7) | −15.7 (15.4) |
| 6 | 44 | M | White | simvastatin, 40 | −/NA | − | 205/245 | 420/420 | 15.0 (5.6) | 35.9 (3.0) | −20.9 (5.2) |
| 7 | 56 | F | White | rosuvastatin, 5 | +/+ | + | 79/89 | 100/50 | 26.8 (8.6) | 61.6 (6.5) | −34.8 (8.4) |
| 8 | 69 | F | White | atorvastatin, 5 | +/+ | − | 81/92 | 160/220 | 34.2 (6.4) | 44.5 (5.1) | −10.3 (6.4) |
| 9 | 54 | M | White | atorvastatin, 10 | +/+ | + | 150/145 | 420/420 | 14.5 (5.6) | 18.0 (2.9) | −3.5 (5.2) |
| 10 | 50 | F | White | simvastatin, 20 | −/NA | + | 101/88 | 120/90 | 50.0 (2.7) | 59.6 (3.2) | −9.7 (3.3) |
NA-not available; CK-creatine kinase; SE-standard error
PCr Recovery
The mean metabolic recovery time constant in subjects increased from 28.1s (SE=6.5s) at baseline to 55.4s (SE=7.4s) following statin therapy (Fig. 3). The unweighted mean of the pre-post recovery time difference was −27.3s (SE=12.4s); (p-value = 0.02, using nonparametric signed rank test). The weighted mean was −8.9s (SE=6.0s). Eight out of 10 subjects had an increase in the metabolic recovery time constant, including all 4 patients with reported muscle symptoms during the study. The 2 patients with a small decrease in PCr metabolic recovery time constant did not experience pain while on statin therapy.
Figure 3.

Graph comparing the fitted cumulative PCr metabolic recovery time constant in 10 subjects before and after statin therapy. There is a prolonged and blunted cumulative recovery curve following 4 weeks of statin therapy when compared to baseline.
Creatine kinase (CK)
CK levels were not significantly different (p-value = 0.50) following 4 weeks of statin medication. The average CK was 107 U/L prior to initiating statin medication and 126 U/L after 4 weeks of treatment.
Discussion
To our knowledge, PCr recovery kinetics in patients with hyperlipidemia before and after statin medication use have not been studied previously. The recovery of PCr concentration in skeletal muscle following exercise relies on the availability of substrates and oxygen as well as the capacity for the mitochondria to carry out oxidative metabolism within the myocytes. Following statin therapy, participants in this study had a significant drop in the recovery of PCr in their calf muscles when compared to baseline. This suggests impairment of mitochondrial oxidative metabolism as a cause of statin-associated myopathy. Our study had too few participants to compare whether PCr recovery was different among those with and without suspected statin-associated muscle pain. Further investigations are of great interest, as PCr exercise recovery kinetics measured by 31P-MRS may be an effective study tool for diagnosing and evaluating statin-associated myopathy.
Two prior studies have used 31P-MRS to investigate the effect of statins on resting muscle.34, 35 Slade et al. found a 57% elevation in the phosphodiesterase (PDE), a breakdown product of cell membrane phospholipids, in 10 patients taking statins when compared to normals.35 Since increased levels of PDE are seen in active tumors, increased age, muscular dystrophies, and non-specific muscle pain syndromes, the authors hypothesize that statin use increases muscle membrane turnover.35 However, the patients studied did not have muscle symptoms, and MRS was performed at rest. Guis et al performed PCr exercise recovery kinetics in the forearm on 7 patients with a history of statin-associated myalgia and elevated CK.34 They saw no difference in the PCr recovery between normal subjects and patients with a prior history of statin myopathy.34 Moreover, the recovery of pH was slower in the patients with a history of statin myopathy versus normal subjects. This suggested reduced proton efflux, which the authors suggest is linked to failure of calcium homeostasis.34 It is unclear why Guis et al, did not see a blunted PCr recovery in their patients, however, this discrepancy may be because their patients had discontinued statin medications prior to 31P-MRS. Moreover, they performed 31P-MRS in the forearm, a less common site for statin-associated muscle pain in our experience.
The CK did not increase significantly in our patients during the 4 week course of therapy, which was not unexpected. Past studies have shown that CK levels do not correlate well with the degree of muscle pain. Statin-associated muscle symptoms confirmed on blinded statin rechallenge may occur in patients with normal CK levels,18 while muscle symptoms may be absent or mild, when severe muscle toxicity (i.e., rhabdomyolysis) is present.36, 37 Damage to the muscle can be seen histologically even when patients are asymptomatic or have normal CK levels. Draeger et al showed that ultrastructural damage (breakdown of the T-tubular system and subsarcolemmal rupture) occurs in skeletal muscle in asymptomatic patients on statin therapy without myalgia.38 Histologic analysis of patients with statin-associated muscle symptoms following blinded statin rechallenge without elevated serum CK showed microscopic changes of altered myofiber architecture and necrosis, along with signs of mitochondrial dysfunction which are reversible with drug cessation.18
A few limitations deserve mention. As with any pilot study, the small number of subjects precludes making firm conclusions, and additional studies with larger patient groups are needed to investigate the ability of 31P-MRS to elucidate the effects of statins on skeletal muscle. Four of our 5 subjects with prior statin use experienced mild-to-moderate muscle pain on a prior statin, which is higher than the general population. Thus there maybe a higher likelihood of a prolong PCr recovery time in our statin subject group when compared to a more normal patient distribution. Another potential limitation is that changes in nutritional status or exercise habits may alter PCr recovery kinetics, however, the subjects did not alter their diet or exercise habits during the 4 week statin course. Moreover, PCr exercise recovery kinetics with 31P-MRS can be difficult to perform, requiring 31P-MRS specific coils, pedal ergometer, and technical expertise in curve fitting and MR spectra data processing. Additional studies to show the reproducibility of the technique are necessary. Moreover, we performed unlocalized 31P-MRS, thus the spectra are believe to be the result of a summation of muscle tissue. Newer techniques that incorporate localized spectroscopy can be of benefit and should be evaluated.
In conclusion, PCr metabolic recovery time constant in the calf is prolonged in patients following statin use. This supports the hypothesis that statins impair mitochondrial oxidative function. This initial pilot study shows that 31P-MRS is a potential tool in the study of statin-associated myopathy and may be useful for testing of effective treatment options.
Acknowledgments
This study was supported by grants from the Beth Israel Deaconess Radiologic Foundation and the Harvard Clinical and Translational Science Center, from the National Center for Research Resources (Grant Number 1 UL1 RR025758-01). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Abbreviations
- MRS
magnetic resonance spectroscopy
- HMG-CoA
3-hydroxy-3-methylglutaryl coenzyme A reductase
- PCr
phosphocreatine
- CK
creatine kinase
Footnotes
This study was presented as a poster presentation at the American Heart Association annual meeting in San Francisco, CA. March 2010.
References
- 1.Miller JA. Statins--challenges and provocations. Curr Opin Neurol. 2005;18:494–6. doi: 10.1097/01.wco.0000180661.65425.05. [DOI] [PubMed] [Google Scholar]
- 2.Graham DJ, Staffa JA, Shatin D, Andrade SE, Schech SD, La Grenade L, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. Jama. 2004;292:2585–90. doi: 10.1001/jama.292.21.2585. [DOI] [PubMed] [Google Scholar]
- 3.Lau TK, Leachman DR, Lufschanowski R. Severe rhabdomyolysis associated with the cerivastin-gemfibrozil combination therapy: report of a case. Tex Heart Inst J. 2001;28:142–5. [PMC free article] [PubMed] [Google Scholar]
- 4.Baker SK, Tarnopolsky MA. Statin myopathies: pathophysiologic and clinical perspectives. Clin Invest Med. 2001;24:258–72. [PubMed] [Google Scholar]
- 5.Bernini F, Poli A, Paoletti R. Safety of HMG-CoA reductase inhibitors: focus on atorvastatin. Cardiovasc Drugs Ther. 2001;15:211–8. doi: 10.1023/a:1011908004965. [DOI] [PubMed] [Google Scholar]
- 6.Chapman MJ, Carrie A. Mechanisms of statin-induced myopathy: a role for the ubiquitin-proteasome pathway? Arterioscler Thromb Vasc Biol. 2005;25:2441–4. doi: 10.1161/10.1161/01.ATV.0000194548.11901.a4. [DOI] [PubMed] [Google Scholar]
- 7.Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. Jama. 2003;289:1681–90. doi: 10.1001/jama.289.13.1681. [DOI] [PubMed] [Google Scholar]
- 8.Ucar M, Mjorndal T, Dahlqvist R. HMG-CoA reductase inhibitors and myotoxicity. Drug Saf. 2000;22:441–57. doi: 10.2165/00002018-200022060-00003. [DOI] [PubMed] [Google Scholar]
- 9.Sacks FM. Adherence to statin therapy: why aren’t we doing better? Am J Med. 2002;113:685–6. doi: 10.1016/s0002-9343(02)01428-6. [DOI] [PubMed] [Google Scholar]
- 10.Bouchard MH, Dragomir A, Blais L, Berard A, Pilon D, Perreault S. Impact of adherence to statins on coronary artery disease in primary prevention. Br J Clin Pharmacol. 2007;63:698–708. doi: 10.1111/j.1365-2125.2006.02828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker SK. Molecular clues into the pathogenesis of statin-mediated muscle toxicity. Muscle Nerve. 2005;31:572–80. doi: 10.1002/mus.20291. [DOI] [PubMed] [Google Scholar]
- 12.Evans M, Rees A. Effects of HMG-CoA reductase inhibitors on skeletal muscle: are all statins the same? Drug Saf. 2002;25:649–63. doi: 10.2165/00002018-200225090-00004. [DOI] [PubMed] [Google Scholar]
- 13.Paiva H, Thelen KM, Van Coster R, Smet J, De Paepe B, Mattila KM, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther. 2005;78:60–8. doi: 10.1016/j.clpt.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Schick BA, Laaksonen R, Frohlich JJ, Paiva H, Lehtimaki T, Humphries KH, et al. Decreased skeletal muscle mitochondrial DNA in patients treated with high-dose simvastatin. Clin Pharmacol Ther. 2007;81:650–3. doi: 10.1038/sj.clpt.6100124. [DOI] [PubMed] [Google Scholar]
- 15.Cao P, Hanai J, Tanksale P, Imamura S, Sukhatme VP, Lecker SH. Statin-induced muscle damage and atrogin-1 induction is the result of a geranylgeranylation defect. Faseb J. 2009;23:2844–54. doi: 10.1096/fj.08-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanai J, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, et al. The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J Clin Invest. 2007;117:3940–51. doi: 10.1172/JCI32741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohaupt MG, Karas RH, Babiychuk EB, Sanchez-Freire V, Monastyrskaya K, Iyer L, et al. Association between statin-associated myopathy and skeletal muscle damage. Cmaj. 2009;181:E11–8. doi: 10.1503/cmaj.081785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips PS, Haas RH, Bannykh S, Hathaway S, Gray NL, Kimura BJ, et al. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med. 2002;137:581–5. doi: 10.7326/0003-4819-137-7-200210010-00009. [DOI] [PubMed] [Google Scholar]
- 19.Lupattelli G, Palumbo B, Sinzinger H. Statin induced myopathy does not show up in MIBI scintigraphy. Nucl Med Commun. 2001;22:575–8. doi: 10.1097/00006231-200105000-00017. [DOI] [PubMed] [Google Scholar]
- 20.Sheehy N, Israel DA. Findings on (18)FDG-PET imaging in statin-induced rhabdomyolysis. Clin Radiol. 2007;62:1012–4. doi: 10.1016/j.crad.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Trieu J, Emmett L, Perera C, Thanakrishnan K, Van Der Wall H. Rhabdomyolysis resulting from interaction of simvastatin and clarithromycin demonstrated by Tc-99m MDP scintigraphy. Clin Nucl Med. 2004;29:803–4. doi: 10.1097/00003072-200412000-00008. [DOI] [PubMed] [Google Scholar]
- 22.Chance B, Im J, Nioka S, Kushmerick M. Skeletal muscle energetics with PNMR: personal views and historic perspectives. NMR Biomed. 2006;19:904–26. doi: 10.1002/nbm.1109. [DOI] [PubMed] [Google Scholar]
- 23.Arnold DL, Matthews PM, Radda GK. Metabolic recovery after exercise and the assessment of mitochondrial function in vivo in human skeletal muscle by means of 31P NMR. Magn Reson Med. 1984;1:307–15. doi: 10.1002/mrm.1910010303. [DOI] [PubMed] [Google Scholar]
- 24.Isbell DC, Berr SS, Toledano AY, Epstein FH, Meyer CH, Rogers WJ, et al. Delayed calf muscle phosphocreatine recovery after exercise identifies peripheral arterial disease. J Am Coll Cardiol. 2006;47:2289–95. doi: 10.1016/j.jacc.2005.12.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kent-Braun JA, Sharma KR, Miller RG, Weiner MW. Postexercise phosphocreatine resynthesis is slowed in multiple sclerosis. Muscle Nerve. 1994;17:835–41. doi: 10.1002/mus.880170802. [DOI] [PubMed] [Google Scholar]
- 26.Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, et al. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation. 2003;107:3040–6. doi: 10.1161/01.CIR.0000072789.89096.10. [DOI] [PubMed] [Google Scholar]
- 27.Nightingale AK, Crilley JG, Pegge NC, Boehm EA, Mumford C, Taylor DJ, et al. Chronic oral ascorbic acid therapy worsens skeletal muscle metabolism in patients with chronic heart failure. Eur J Heart Fail. 2007;9:287–91. doi: 10.1016/j.ejheart.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 28.Greenman RL, Panasyuk S, Wang X, Lyons TE, Dinh T, Longoria L, et al. Early changes in the skin microcirculation and muscle metabolism of the diabetic foot. Lancet. 2005;366:1711–7. doi: 10.1016/S0140-6736(05)67696-9. [DOI] [PubMed] [Google Scholar]
- 29.Greiner A, Esterhammer R, Messner H, Biebl M, Muhlthaler H, Fraedrich G, et al. High-energy phosphate metabolism during incremental calf exercise in patients with unilaterally symptomatic peripheral arterial disease measured by phosphor 31 magnetic resonance spectroscopy. J Vasc Surg. 2006;43:978–86. doi: 10.1016/j.jvs.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 30.Hug F, Bendahan D, Le Fur Y, Cozzone PJ, Grelot L. Metabolic recovery in professional road cyclists: a 31P-MRS study. Med Sci Sports Exerc. 2005;37:846–52. doi: 10.1249/01.mss.0000162616.20085.b4. [DOI] [PubMed] [Google Scholar]
- 31.Wagner BK, Kitami T, Gilbert TJ, Peck D, Ramanathan A, Schreiber SL, et al. Large-scale chemical dissection of mitochondrial function. Nat Biotechnol. 2008;26:343–51. doi: 10.1038/nbt1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kemp GJ, Thompson CH, Stratton JR, Brunotte F, Conway M, Adamopoulos S, et al. Abnormalities in exercising skeletal muscle in congestive heart failure can be explained in terms of decreased mitochondrial ATP synthesis, reduced metabolic efficiency, and increased glycogenolysis. Heart. 1996;76:35–41. doi: 10.1136/hrt.76.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor DJ, Bore PJ, Styles P, Gadian DG, Radda GK. Bioenergetics of intact human muscle. A 31P nuclear magnetic resonance study. Mol Biol Med. 1983;1:77–94. [PubMed] [Google Scholar]
- 34.Guis S, Figarella-Branger D, Mattei JP, Nicoli F, Le Fur Y, Kozak-Ribbens G, et al. In vivo and in vitro characterization of skeletal muscle metabolism in patients with statin-induced adverse effects. Arthritis Rheum. 2006;55:551–7. doi: 10.1002/art.22100. [DOI] [PubMed] [Google Scholar]
- 35.Slade JM, Delano MC, Meyer RA. Elevated skeletal muscle phosphodiesters in adults using statin medications. Muscle Nerve. 2006;34:782–4. doi: 10.1002/mus.20619. [DOI] [PubMed] [Google Scholar]
- 36.Schindler C, Thorns M, Matschke K, Tugtekin SM, Kirch W. Asymptomatic statin-induced rhabdomyolysis after long-term therapy with the hydrophilic drug pravastatin. Clin Ther. 2007;29:172–6. doi: 10.1016/j.clinthera.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Walsh RJ, Amato AA. Toxic myopathies. Neurol Clin. 2005;23:397–428. doi: 10.1016/j.ncl.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 38.Draeger A, Monastyrskaya K, Mohaupt M, Hoppeler H, Savolainen H, Allemann C, et al. Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. J Pathol. 2006;210:94–102. doi: 10.1002/path.2018. [DOI] [PubMed] [Google Scholar]