

Summary
Endothelial cells are potent regulators of immune cell functions and have therefore been examined to determine their role in tumor-induced immune suppression. Previous studies by our laboratory showed that exposure to Lewis lung carcinoma (LLC)-secreted products induced endothelial cells to suppress T-cell functions in vitro. The current studies examined in vitro and in vivo the mechanism by which tumors induce the formation of suppressor endothelial cells and the means by which suppressor endothelial cells disrupt T-cell functions. In vitro studies demonstrated that inhibition of tumor-derived VEGF with neutralizing antibodies or treatment of endothelial cells with the VEGF receptor tyrosine kinase inhibitor, SU5416, prevented endothelial cells from being induced to suppress T-cell functions. Treatment of tumor-bearing mice with SU5416 blocked the development of endothelial cells that are suppressive to CD4+ and CD8+ T-cell functions. We next examined the role of suppressor endothelial cell-derived PGE2 in the inhibition of T-cell functions. Abrogation of endothelial cell PGE2 production in vitro with indomethacin prevented tumor-conditioned media from stimulating endothelial cell production of immune inhibitory activity toward T-cell functions. Similar treatment of endothelial cells from lungs of tumor-bearing mice blocked their capacity to produce T-cell-inhibitory mediators. These studies demonstrate that tumor-derived VEGF induces endothelial cells to upregulate production of PGE2 which, in turn, leads to suppression of T-cell functions.
Keywords: endothelial cell, T cells, tumor immunity, lung, cytokines
Patients with solid tumors, have defects in immune effector cell function which is recapitulated in mice bearing Lewis lung carcinoma (LLC) tumors. While numerous mechanisms by which tumors suppress immune functions have been identified, tumor-induced immune suppression remains a significant hurdle to the treatment of cancer. The steps leading to the development of tumor vasculature, one of the most crucial processes to the progression of solid tumors, can themselves be immune suppressive. Examples of tumor-derived factors that are both pro-angiogenenic and immune suppressive include vascular endothelial growth factor (VEGF), transforming growth factor-β (TGF-β) and prostaglandin E2 (PGE2).1 Elevated levels of VEGF can promote endothelial cell migration and proliferation, disrupt of dendritic cell function and suppress T-cell development.2,3 Tumor secretion of PGE2 promotes angiogenesis and can suppress T-cell proliferation, expression of Th1 cytokines and dendritic cell functions.4,5 Tumor-secretion of TGF-β also promotes angiogenesis and suppresses T-cell, NK cell, dendritic cell and macrophage functions.6,7 These and other studies suggest that many of the mechanisms involved in development of the tumor vasculature may also contribute to tumor-induced immune suppression.
In addition to direct suppression of immune cell functions, tumors can indirectly suppress immune competence by recruitment of normal host cells to inhibit immune functions. Examples of this include tumor-associated macrophages (TAM), Tregs, myeloid derived suppressor cells (MDSC) and CD34+ progenitor cells.8 For example, while macrophages have the capacity to eradicate tumor cells, in the tumor microenvironment they can be induced to downregulate production of the immune stimulatory cytokine IL-12. Furthermore, TAM have elevated expression of the immune suppressants IL-10 and TGF-β, resulting in diminished T-cell and NK cell functions.9,10 Tregs represent another cell population recruited by tumors to assist in the suppression of immune cell functions. Tregs are selectively recruited into the tumor microenvironment where they can inhibit functions of dendritic cells, NK cells and T cells through their expression of TGF-β and CTLA-4.11 In patients with hepatocellular carcinoma, elevated numbers of tumor-infiltrating Treg cells correlate with a significant reduction in disease-free survival.12 MDSC are another class of immune suppressive cells that have been shown to be increased in patients with various types of solid tumors. MDSC can inhibit both CD4+ T-cell and CD8+ T-cell responses through the depletion of L-arginine, and the production of nitric oxide and reactive oxygen species.13 CD34+ progenitor cells, another category of MDSC, have been shown to suppress T-cell functions through the production of TGF-β and nitric oxide.14 Furthermore, in the presence of tumor-derived products, CD34+ cells can be skewed to differentiate into endothelial cells that incorporate into the tumor vasculature.15,16
Endothelial cells play a critical structural role in blood and lymphatic vasculature, and can serve as regulators of immune cell functions.17 Thus, endothelial cells could potentially be induced by tumors to suppress immune functions through their capacity to secrete immune suppressive factors including VEGF, PGE2, TGF-β, IL-6 and IL-10.18–20 Previous studies by our laboratory have demonstrated that exposure of endothelial cells to Lewis lung carcinoma (LLC)-secreted products induces the formation of suppressor endothelial cells in vitro.21 In the present studies, we investigated in vitro and in vivo the mechanism by which tumors induce the formation of suppressor endothelial cells and the means by which suppressor endothelial cells disrupt T-cell functions. Tumor-derived VEGF was shown to be the mediator by which LLC tumors induce the formation of suppressor endothelial cells. PGE2 was, in turn, shown to be a contributor to the mechanism by which suppressor endothelial cells inhibit T-cell IFN-γ and IL-2 production. Together, the results of these studies demonstrate the role of endothelial cells in tumor-induced immune suppression and provide support for the use of VEGF and PGE2 targeting therapies as a means of interrupting endothelial cell suppression of T-cell functions.
MATERIALS AND METHODS
Cell Culture
Mouse bEnd.3 endothelial cells, MLE 12 epithelial cells, (ATCC, Manassas, VA) and a metastatic variant of Lewis lung carcinoma (LCC) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) culture medium containing 10% heat-inactivated fetal bovine serum, 200 U/mL penicillin G, 200 μg/mL streptomycin sulfate, 500 μg/mL amphotericin B and 5×10−5 M 2-mercaptoethanol (Sigma-Aldrich, St. Louis, MO). MLE 12 mouse lung epithelial cells were used as a control for LLC cells as both are of lung epithelial cell origin.
In vitro induction of immune suppressive endothelial cells was performed as previously described.21 Briefly, subconfluent cultures of endothelial cells were treated with medium alone or 40% conditioned medium from epithelial cells or LLC cells. After 24 hours, endothelial cells were washed and fresh DMEM culture medium was added for an additional 24 hours of incubation. After this time, endothelial cell-conditioned medium was collected and assayed by ELISA for the presence of immune regulatory products or used to treat freshly isolated immune cell populations.
As indicated in specific experiments, 0.025 μg/mL of neutralizing antibodies against VEGF-A164 or goat IgG isotype control antibodies (R&D Systems, Minneapolis, MN) were added to tumor-conditioned medium immediately preceding addition to endothelial cells. In other studies, 10 μg/mL of neutralizing antibodies to VEGF-R1 or goat IgG isotype control antibodies (R&D Systems) were added to endothelial cells just prior to tumorconditioned medium treatment. As an additional method of VEGF inhibition, the VEGF-R1, -R2 and -R3 receptor tyrosine kinase inhibitor, SU5416 (Sigma-Aldrich), was added to endothelial cells at a dose of 1 μM in DMSO 20 minutes prior to the addition of tumor-conditioned medium. This dose of SU5416 was selected based on previously published in vitro studies using doses of 1 μM or higher.22–25 In addition, work published by other laboratories has shown that 1 μM of SU5416 did not affect other signaling pathway including AKT, ERK or STAT3.26
To examine the role of endothelial cell-derived PGE2 in disrupting T-cell functions, the cyclooxygenase inhibitor indomethacin (Sigma-Aldrich) was used to inhibit endothelial cell PGE2 production. Indomethacin or DMSO vehicle control was added to endothelial cells at a dose of 1 μM, 24 hours prior to the addition of tumor-conditioned medium. Indomethacin was again added at the time of the 24-hour treatment with tumor-conditioned media at the same final concentration to maintain inhibition of PGE2 production. After washing to remove tumor-conditioned medium and indomethacin, endothelial cells were cultured for an additional 24 hours in the absence of indomethacin for collection of endothelial cell-conditioned medium.
Mice, Tumor Model, and Treatments
Eight- to twelve-week-old female C57BL/6 mice were obtained from Charles River (Wilmington, MA). Mice were housed 5 to a cage with a 12/12 hour light dark cycle and fed ad libitum. Mice were humanely euthanized by CO2 asphyxiation followed by cervical dislocation. All procedures were conducted with Institutional Animal Care and Use Committee approval.
Spleens from normal mice were collected and used as a source of T cells. Endothelial cells were isolated from the lungs from normal mice or from mice bearing LLC lung tumors. LLC cells (1×106) or PBS alone were injected i.v. via the tail vein. After allowing tumors to develop for 21 days, mice were humanely euthanized by CO2 asphyxiation followed by cervical dislocation. Normal or tumor-bearing lungs were then collected and used as the source of endothelial cells. As indicated in specific in vivo experiments, VEGF signaling was inhibited using SU5416, administered subcutaneously at a dose of 25 mg/kg in 0.1mL of DMSO daily from days 17 through 20 post-tumor inoculation. This dose was selected as it is the same or lower than other previously published in vivo doses of SU5416.27–30 In vivo inhibition of PGE2 production was achieved by oral administration of indomethacin. On days 14–21 posttumor injection, mice were given indomethacin or DMSO vehicle control in their drinking water at a concentration of 16.5 μg/mL, resulting in a final dose of 0.5 mg/kg/day based on the average mouse consumption of 15mL of water per 100 gram body weight per day.
T-cell Isolation
CD90+ (Thy1.2+) T cells were immunomagnetically isolated from the spleens of normal female C57BL/6 mice. Spleens were homogenized into a single cell suspension using a Stomacher 80 Biomaster. ACK lysis buffer was then used to lyse red blood cells, after which cells were washed three times and resuspended in separation buffer. Labeling antibody was added per the manufacturer’s instructions (Miltenyi Biotech Inc.). Following isolation, T cells were washed and resuspended in RPMI culture medium with 10% heat-inactivated FBS, 200 U/mL penicillin G, 200 μg/mL streptomycin sulfate, 500 μg/mL amphotericin B, 5×10−5 M 2-mercaptoethanol (Sigma-Aldrich) and 10 units/mL of recombinant mouse IL-2 to maintain cell viability (R&D Systems). T cells were plated at a density of 2.5×105 cells per well on anti-CD3 antibody-coated plates. CD3 staining followed by flow cytometric analysis was used to confirm T-cell purity and was determined to be 94% or higher.
Immune Function Assays
T cells were treated with medium alone or 40% endothelial cell-conditioned medium for 24 hours. T cells were then washed, new medium was added and cells were allowed to incubate for an additional 24 hours. After this time, conditioned medium from T cells were collected for measurement of secreted immune regulatory products or else the T cells were used for functional assessments.
Secretion of immune modulatory products by endothelial cells and T cells was assessed by ELISA and included measurement of PGE2, VEGF, IL-6, IL-10, IL-12 (R&D Systems, Minneapolis, MN), and IFN-γ (BD Biosciences). All ELISAs were performed according to the manufacturers’ instructions. Since detectable quantities of PGE2 were present in FBS, endothelial cell culture medium alone was assayed for PGE2 alongside endothelial cell conditioned medium. The average amount of PGE2 detected in the medium alone control was subtracted from the levels detected in the endothelial cell conditioned medium to calculate the quantity of PGE2 secreted by endothelial cells.
T-cell intracellular cytokine production was measured by immunostaining followed by flow cytometric analysis. Prior to surface and intracellular staining, monensin (GolgiStop) was added to T cells for 2 hours according to the CytoStain Kit protocol. FcγII/III receptors were blocked with anti-CD16/CD32 monoclonal antibodies. Cell surface antigen staining was performed using anti-CD4 and anti-CD8 monoclonal antibodies. After staining, cells were washed twice, then fixed and permeabilized with Cytofix/Cytoperm. Intracellular cytokine staining was done by adding anti-IL-2 and anti-IFN-γ antibodies. Marker channels were set using the antibody isotype controls specific to each antibody. Four-color flow cytometric analysis was performed on a BD FACSCantos using FACSDiva flow cytometry analysis software. All reagents for immunostaining and subsequent flow cytometric analyses were obtained from BD Bioscience.
T-cell proliferation was assessed by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) analysis (Promega, Madison, WI). MTS is a tetrazolium compound that is reduced to formazan by metabolically active cells and was detected by measuring the absorbance at 492 nm. T-cell proliferation was assessed 24 hours after the removal of endothelial cell supernatants.
Endothelial Cell Isolation
Endothelial cells were isolated from the lungs of normal and LLC tumor-bearing mice. Once removed, lungs were rinsed twice in HBSS, finely minced and placed in an HBSS-based enzyme solution containing collagenase (type IV), DNase, and hyaluronidase (Sigma-Aldrich) for 90 minutes. Cells were then passed through a 70 μm strainer and washed twice. Red blood cells were lysed with ACK lysis buffer and the cells were washed twice more. FcγII/III receptors were then blocked with anti-CD16/CD32 monoclonal antibody to prevent non-specific antibody binding (BD Biosciences, San Jose, CA). Endothelial cells were then labeled with biotin-labeled anti-CD31 antibody per the manufacturer’s instructions (eBioscience, San Diego, CA). Cells were then washed to remove excess anti-CD31 antibody, resuspended in separation buffer and strepavidin-labeled microbeads were added followed by immunomagnetic separation conducted per the manufacturer’s instructions (Miltenyi Biotech Inc.). Endothelial cell purity was evaluated by immunostaining for the endothelial cell-selective adhesion molecule (ESAM) (eBioscience) and was determined to be at minimum 91%. Following isolation, endothelial cells were washed, counted, and plated in 96-well round-bottom tissue culture plates at a concentration 5×104 cells/well in phenol-free DMEM culture medium. Cells were allowed to incubate for 24 hours at 37°C with 5% CO2. Conditioned medium from isolated endothelial cells was collected and stored at −80°C until use.
Statistical Analysis
Statistical analyses were conducted using GraphPad Prism 5.01. Student t test was used to determine statistically significant differences between the various control and treatment groups as indicated in each figure legend and results section. Data points shown in scatter plots represent results from treatments using isolated endothelial cell-conditioned media from individual animals. In bar graphs, error bars represent standard deviation or standard error of the mean, as indicated in each figure legend.
RESULTS
Tumor Secretion of VEGF Induces Endothelial Cells to Alter Production of Select Immune Regulatory Products
Previous studies by our laboratory demonstrated that tumor-secreted products alter endothelial cell production of immune regulatory products and skew endothelial cells to disrupt T-cell functions in vitro.21 In the current study, tumor-derived VEGF was examined to determine whether it was mediating the formation of suppressor endothelial cells. VEGF was hypothesized to be the tumor-secreted factor responsible for the induction of suppressor endothelial cells due to its abundant secretion by tumor cells and its ability to modulate numerous endothelial cell functions.31 Furthermore, VEGF has been shown to increase endothelial cell production of PGE2, while diminishing production of IL-12.21,32
Therefore it was examined was whether neutralization of tumor-secreted VEGF could prevent tumor-conditioned medium from altering endothelial cell production of the immune mediators that we previously showed to be modulated by tumors: PGE2, VEGF, IL-6, and IL-1221 (Fig. 1). Consistent with the results of our previous studies, endothelial cells that were pre-treated with tumor-conditioned medium (EndoLLC–sup) were stimulated to produce increased levels of PGE2, VEGF, and IL-6 (Figs. 1A–C) compared to levels produced by endothelial cells that had been pretreated with medium (EndoMedia) or epithelial cell supernatants (EndoEpi–sup). Neutralization of VEGF in tumor-conditioned medium prior to addition to endothelial cells prevented the induction of endothelial cells to increase secretion of PGE2, VEGF, or IL-6. Endothelial cells treated with tumor-conditioned medium whose VEGF was neutralized secreted the same levels of PGE2 as the control endothelial cells.
FIGURE 1.

VEGF neutralization blocks tumor-induced alterations in endothelial cell secretion of immune modulatory products. VEGF neutralizing antibody or isotype control antibody was added to tumor-conditioned medium immediately before treatment of endothelial cells as indicated. Supernatants were then prepared from endothelial cell that were pretreated with tumor-conditioned medium (EndoLLC–sup), with medium (EndoMedia) or with epithelial cell supernatants (EndoEpi–sup). Endothelial cell-conditioned media were collected and assayed by ELISA for presence of immune modulatory products. Data shown are mean ±SEM with n ≥4 per treatment group. Statistics shown are Student t test results between indicated treatment groups. ELISA indicates enzyme-linked immunosorbent assay; NS, no statistically significant difference between treatments; VEGF, vascular endothelial growth factor.
Last, endothelial cell secretion of the immune stimulatory product IL-12 was examined (Fig. 1D). Exposure of endothelial cells to tumor-conditioned medium reduced their production of IL-12 compared to levels produced by the control groups of endothelial cells (EndoMedia or EndoEpi–sup). Neutralization of tumor-derived VEGF blocked the capacity of tumor-conditioned medium to downregulate endothelial cell production of IL-12. These results demonstrate that VEGF is the tumor-derived factor responsible for inducing endothelial cells to increase their secretion of IL-6, PGE2, and VEGF, while downregulating their production of IL-12.
Tumor-derived VEGF Skews Endothelial Cells to Disrupt T-cell Responses to Anti-CD3 Stimulation
We next tested whether tumor-derived VEGF could induce endothelial cells to disrupt T-cell IFN-γ production (Fig. 2). Examination of intracellular expression of IFN-γ demonstrated that supernatants from EndoLLC–sup endothelial cells that had been treated with tumor-conditioned media significantly reduced IFN-γ staining in CD8+ T cells compared to staining in T cells that were treated with supernatants of the control endothelial cell groups EndoMedia and EndoEpi–sup (Fig. 2A). This reduction in IFN-γ staining intensity in CD8+ T cells correlated with a reduced percentage of T cells that stained positive for IFN-γ+ (Fig. 2B). Neutralization of VEGF prevented the capacity of tumor-conditioned medium from inducing endothelial cells that reduced T-cell IFN-γ production (both the levels of IFN-γ within CD8+ T cells and the percentage of CD8+ T cells staining positive for IFN-γ).
FIGURE 2.

VEGF neutralization blocks endothelial cells from suppressing CD8+ T-cell IFN-γ production in response to anti-CD3. A, Mean fluorescent intensity ± SD of IFN-γ+ staining in CD8+ cells after exposure to treatments with endothelial cell-conditioned media. B, Percent of cells staining double positive for CD8 and IFN-γ ± SD. Statistics shown are Student t test results between indicated treatment groups. IFN indicates interferon; NS, no statistically significant difference between treatments; VEGF, vascular endothelial growth factor. Results presented in Figures A and B are representative results of at least 4 experiments.
Inhibition of endothelial cell VEGF receptor signaling was also examined as an alternate approach to demonstrate that tumor-derived VEGF was a contributor to skewing of endothelial cells to disrupt T-cell functions. Figure 3A demonstrates that while supernatants of endothelial cells that were exposed to tumor-conditioned medium in the presence or absence of isotype control antibodies inhibited CD8+ T-cell expression of IFN-γ, antibodies to VEGF-R1 prevented this induction of suppressive endothelial cells by tumor-conditioned medium. Treatment of endothelial cells with the VEGF receptor tyrosine kinase inhibitor, SU5416, confirmed the role of tumor-derived VEGF in stimulating immune suppressive endothelial cells (Fig. 3B). Treatment of endothelial cells with SU5416 blocked the capacity of tumor-conditioned medium from stimulating them to release mediators that inhibit T-cell IFN-γ expression. Collectively, these results demonstrate tumor secretion of VEGF induces endothelial cells to secrete products that disrupt T-cell functions in vitro.
FIGURE 3.

Attenuation of endothelial cell VEGF signaling prevents tumor-conditioned medium modulation of T-cell IFN-γ production. A, Neutralizing antibody to VEGF-R1 or isotype control antibody was added immediately before exposure of endothelial cells to tumor-conditioned medium. Data shown are mean ± SD with n = 4 per treatment group and are representative of at least 3 experiments. B, 1 μM SU5416 was added to endothelial cells 20 minutes before their exposure to tumor-conditioned medium. Data shown are mean ± SD with n ≥6 per treatment group and are representative of at least 3 experiments. Statistics shown are Student t test results between indicated treatment groups. IFN indicates interferon; NS, no statistically significant difference between treatments; VEGF, vascular endothelial growth factor.
Inhibition of VEGF Signaling In Vivo Prevents Tumors From Inducing Lung Endothelial Cells to Increase Production of Immune Suppressive Products
Based on our results thus far, the role of VEGF in inducing the formation of suppressor endothelial cells in vivo was examined. To accomplish this, tumor-bearing mice received no treatment, DMSO vehicle control or SU5416. Endothelial cells were then isolated from the lungs of these mice and cultured for 24 hours to generate endothelial cell-conditioned media.
First examined was endothelial cell production of IL-6, IL-10, and PGE2 (Fig. 4). Compared to endothelial cells isolated from the lungs of normal mice, those isolated from tumor-bearing lungs of mice receiving no treatment secreted elevated levels of IL-6, IL–10, and PGE2 (Figs. 4A–C). Lung endothelial cells from tumor-bearing mice treated with vehicle control also secreted high levels of these immune mediators. In contrast, treatment of tumor-bearing mice with SU5416 prevented their lung endothelial cells from secreting increased levels of IL-6, IL-10, and PGE2.
FIGURE 4.

Administration of SU5416 to tumor-bearing mice blocks endothelial cells from producing IL-6, IL-10, and PGE2. Mice were administered SU5416 daily, days 17 to 20 posttumor inoculation. Endothelial cells were isolated and cultured for 24 hours after which time conditioned medium was collected and assayed by ELISA for the presence of the indicated products. Data represent means ± SEM from 2 experiments for IL-6 (A) and 3 experiments for IL-10 (B) and PGE2 (C). Statistics shown are Student t test results between indicated treatment groups. ELISA indicates enzyme-linked immunosorbent assay; IL, interleukin; NS, no statistically significant difference between treatments; PGE2, prostaglandin E2; VEGF, vascular endothelial growth factor.
Inhibition of VEGF Signaling In Vivo Prevents Tumor-induced Alterations in Lung Endothelial Cells Modulation of T-cell Functions
Based on the above findings showing the role of VEGF in inducing endothelial cells to increase production of immune suppressive products, studies were designed to determine in vivo if inhibition of VEGF signaling could block endothelial cells from tumor-bearing lungs from disrupting T-cell functions (Fig. 5). Endothelial cell conditioned medium from lungs of tumor-bearing mice that were either untreated or treated with control vehicle inhibited CD8+ cell expression of IFN-γ (Fig. 5A). By comparison, T cells treated with conditioned medium of endothelial cells isolated from tumor-bearing mice receiving SU5416 had significantly higher levels of IFN-γ expression by CD8+ cells. In fact, there was no significant difference in IFN-γ expression by T cells that were exposed to conditioned medium from endothelial cells of normal mice or those treated with conditioned medium from endothelial cells isolated from tumor-bearing mice receiving SU5416.
FIGURE 5.

SU5416 blocks induction of immune suppressive endothelial cells capable of disrupting T-cell function. Analysis of intracellular cytokine expression by T cells treated with 24 hours conditioned medium from endothelial cells isolated from normal mice or tumor-bearing mice that received either no treatment, vehicle control or SU5416 treatment. A, Mean fluorescent intensity of IFN-γ+ staining in CD8+ cells after exposure to various treatments with endothelial cell-conditioned medium. B, Mean fluorescent intensity of IL-2 staining in CD4+ T cells. In A and B, data points represent results from individual mice and are representative of multiple experiments. Statistics shown are Student t test results between indicated treatment groups. Furthermore, in A and B there was no statistically significant difference in T-cell cytokine expression between treatment with normal endothelial cell-conditioned media and conditioned media from endothelial cells isolated from tumor-bearing mice receiving SU5416. IFN indicates interferon; IL, interleukin; NS, no statistically significant difference between treatments; VEGF, vascular endothelial growth factor.
The role of VEGF in modulating endothelial cell regulation of CD4+ T-cell IL-2 expression was also quantified as another indicator of T-cell function (Fig. 5B). Medium conditioned by endothelial cells from normal mice stimulated expression of IL-2 by CD4+ T cells compared to IL-2 expression by CD4+ T cells’ treated with fresh culture medium. In contrast, this T-cell-stimulatory activity of endothelial cells from tumor-bearing lungs of mice that were either untreated or treated with control vehicle was significantly diminished. The capacity of endothelial cells to stimulate CD4+ T-cell expression of IL-2 was restored by treating tumor-bearing mice with SU5416. In fact, there were no significant differences in the levels of IL-2 expression between CD4+ T cells that were treated with medium conditioned by endothelial cells from normal mice or tumor-bearing mice receiving SU5416 treatment. These results indicate that inhibition of VEGF signaling in vivo blocks the induction of suppressor endothelial cells capable of diminishing CD4+ and CD8+ T-cell functions.
Role of PGE2 in Mediating the Suppressive Activity of Tumor-exposed Endothelial Cells
To determine the identity of the product secreted by suppressor endothelial cells responsible for suppressing T-cell functions. To determine the contribution of PGE2 from suppressor endothelial cells for their suppression of T-cell functions, endothelial cells were treated with indomethacin prior to exposure to tumor-conditioned media. Measurement of PGE2 levels in medium conditioned by endothelial cells that had been pre-treated with indomethacin demonstrated the same low background levels as found in media alone (data not shown).
As detailed above, supernatants of endothelial cells that had been exposed to medium conditioned by LLC tumor (EndoLLC–sup) inhibited IFN-γ expression by CD8+ T cells as compared to IFN-γ expression by CD8+ T cells exposed to supernatants of control endothelial cells (EndoMedia or EndoEpi–sup) (Fig. 6). Pre-treatment of endothelial cells with vehicle control prior to exposure to tumor-conditioned medium had no effect on their suppression of IFN-γ expression by CD8+ T cells. However, treatment of endothelial cells with indomethacin prior to exposure to tumor-conditioned medium prevented them from producing a factor that inhibited expression of IFN-γ by CD8+ T cells. In fact, CD8+ T-cell expression of IFN-γ was similar for T cells that were treated with fresh culture medium, supernatants of control endothelial cell groups (EndoMedia or EndoEpi–sup) or supernatants of endothelial cells that had been treated with indomethacin prior to exposure to tumor-conditioned medium. These results identify PGE2 as the suppressor endothelial cells product that contributes to their inhibition of T-cell function.
FIGURE 6.
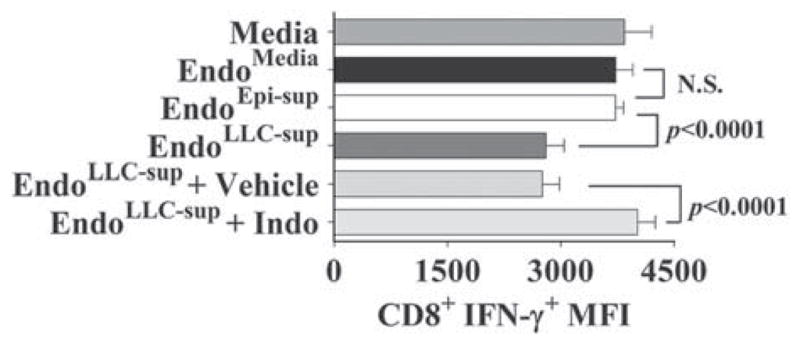
Inhibition of endothelial cell production of PGE2 in vitro blocks their suppressor phenotype. Endothelial cells were treated with indomethacin (Indo) to block the production of PGE2 before and during treatment with tumor-conditioned media. Mean fluorescent intensity ± SD (n≥5) of CD8+ T-cell immunostaining for IFN-γ+ after exposure to the indicated endothelial cell conditioned medium. Statistics shown are Student t test results between indicated treatment groups. IFN indicates interferon; NS, no statistically significant difference between treatments; PGE2, prostaglandin E2. Results presented are representative of data obtained from at least 4 experiments.
Treatment of Tumor-bearing Mice With Indomethacin Blocks the Ability of Their Endothelial Cells to Suppress T-cell Functions
We next tested the effect of inhibiting PGE2 in vivo on the ability of endothelial cells from tumor-bearing lungs to disrupt T-cell function. Prior to immunological studies, the effectiveness of in vivo administration of indomethacin on reducing endothelial cell PGE2 secretion was examined. Conditioned medium from endothelial cells isolated from the lungs of tumor-bearing mice receiving indomethacin secreted PGE2 at levels 95% lower than cells from mice receiving no treatment and 96% lower than those receiving vehicle control (data not shown). Immunological analyses showed that conditioned medium from endothelial cells from lungs of tumor-bearing mice that were either untreated or treated with vehicle control inhibited expression of IFN-γ by CD8+ cells as compared to expression by CD8+ T cells that were treated with media conditioned by endothelial cells from lungs of normal mice (Fig. 7A). Treatment of tumor-bearing mice with indomethacin blocked the production of immune suppressive activity by their lung endothelial cells. Moreover, there was no statistically significant difference in the levels of IFN-γ expressed by CD8+ T cells treated with conditioned medium from endothelial cells of normal mice or from tumor-bearing mice receiving indomethacin.
FIGURE 7.

Inhibition of PGE2 production in tumor-bearing mice blocks endothelial cells from disrupting T-cell function. Studies measured intracellular cytokine expression by T cells treated with 24 hours of conditioned media from endothelial cells isolated from normal mice or tumor-bearing mice receiving either no treatment, vehicle control, or indomethacin treatment. A, Mean fluorescent intensity of CD8+ T-cell immunostaining for IFN-γ+. B, Mean fluorescent intensity of CD4+ T-cell immunostaining for IL-2+. In A and B, data points are results from individual mice and are representative of 2 separate experiments. Statistics shown are Student t test results between indicated treatment groups. In both figures, there are no statistically significant differences in T-cell cytokine production between treatment with normal endothelial cell-conditioned media and conditioned media from endothelial cells isolated from tumor-bearing mice receiving indomethacin. IFN indicates interferon; IL, interleukin; INDO, indomethacin; PGE2, prostaglandin E2.
The ability of indomethacin treatment to prevent suppressor activity by endothelial cells from lungs of tumor-bearing mice toward CD4+ T-cell expression of IL-2 was examined (Fig. 7B). T cells treated with conditioned medium from endothelial cells from lungs of normal mice had increased CD4+ cell expression of IL-2. In comparison, CD4+ T-cell expression of IL-2 was significantly less after treatment with media conditioned by endothelial cells of tumor-bearing mice receiving no treatment or treatment with control vehicle. Conditioned medium of endothelial cells from tumor-bearing mice that received indomethacin treatment stimulated IL-2 expression by CD4+ T cells to the same level as did medium conditioned by endothelial cells from normal mice. Taken together, these results demonstrate that inhibition of endothelial cell PGE2 production in vitro and in vivo prevents tumor-exposed endothelial cells from disrupting T-cell function.
Administration of SU5416 or Indomethacin In Vivo Blocks Tumor-induced Alterations in Endothelial Cells’ Ability to Stimulate T-cell Proliferation
Previously it was demonstrated that in vitro-induced suppressor endothelial cells were capable of disrupting T-cell proliferation.21 This observation was extended by determining if endothelial cells isolated from tumor-bearing lungs can also alter T-cell proliferation (Fig. 8). Compared to T cells treated with media alone, T cells exposed to normal endothelial cell-conditioned medium had an increased proliferative response to anti-CD3 stimulation (P<0.0001). However, supernatants from endothelial cells isolated from tumor-bearing lungs lacked the ability to stimulate T-cell proliferation compared to normal endothelial cells (P=0.0004). Compared to endothelial cells isolated from mice treated with vehicle alone, endothelial cells isolated from mice receiving indomethacin possessed a heightened ability to stimulate T-cell proliferation (P=0.002). Administration of SU5416 in vivo also restored endothelial cells’ ability to stimulate T-cell proliferation compared to that of endothelial cells from mice that were treated with vehicle alone (P=0.0002). In summary, these results demonstrate that endothelial cells isolated from tumor-bearing lungs have a reduced ability to heighten T-cell proliferation in responses to anti-CD3 and that in vivo treatment with indomethacin or SU5416 restores their capacity to stimulate T-cell proliferation.
FIGURE 8.
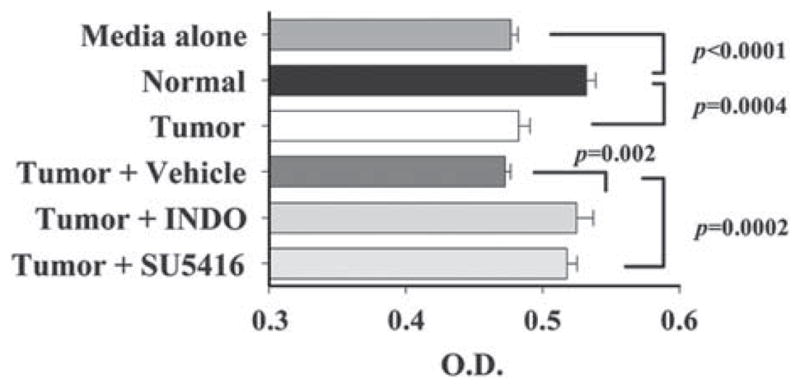
SU5416 and indomethacin are both capable of blocking suppressor endothelial cell-mediated disruption of T-cell proliferation. T-cell proliferation responses to immobilized anti-CD3 in the presence of endothelial cell-conditioned media were measured by MTS. Statistics shown are Student t test results between indicated treatment groups. Data shown are mean ± SD with n ≥6 mice per treatment group from 2 separate experiments. MTS indicates 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium. INDO, indomethacin.
DISCUSSION
Endothelial cells have long been studied as regulators of immune function. However, limited work has focused on the role of endothelial cells in regulating immune function in the context of the tumor microenvironment. Prior studies in other models have shown that endothelial cells are capable of suppressing immune cell function. For example, in vitro studies by Onoe et al demonstrated that endothelial cells can induce T-cell tolerance in a contact-dependent manner.33 In addition, endothelial cells in a murine kidney allograft rejection model were shown to be capable of inducing and activating Tregs that induce T-cell tolerance. 34 Our work is consistent with these findings defining the immune regulatory role of endothelial cells in that it demonstrates that endothelial cells can suppress T-cell function based on cues received from within their environment. The significance of the results presented here is that they support a role for endothelial cells in tumor-induced immune suppression.
Previously we have shown that products secreted by LLC cells could induce endothelial cells to be suppressive to T-cell activities.21 The focus of the present study was to ascertain the mechanism by which this was occurring in order to identify potential therapeutic targets to block suppressor endothelial cells. VEGF was hypothesized to be responsible for inducing endothelial cells to become suppressive based on its abundant secretion by tumors, its capacity to modulate endothelial cell functions and to increase endothelial cell production of PGE2.32,35,36 While results of these studies showed that tumor-derived VEGF induced suppressor endothelial cells, it is important to note that the ability of VEGF to induce suppressor endothelial cells is dependent on the quantity of VEGF, not just its mere presence. Endothelial cells treated with epithelial cellconditioned medium which contained over 300 pg/mL of VEGF were incapable of inducing suppressor endothelial cells. Only tumor-conditioned media, which contained over 1000 pg/mL of VEGF, was able to induce endothelial cells to suppress T-cell function. That tumor induction of suppressor endothelial cells was due to the increased amount of VEGF that they secreted and not to another contaminating mediator was suggested by the complete blockage of endothelial cell suppressor-inducing activity when tumor-secreted VEGF was neutralized with antibody. These results are consistent with the findings of Tamura et al demonstrating that VEGF induction of endothelial cell PGE2 production was dose-dependent.32 The results of these studies were also similar those demonstrating that primary human oral squamous cell carcinoma cell production of VEGF was capable of inducing endothelial cells in vitro to alter T-cell functions including production of the cytotoxic mediators granzyme B and perforin.37
The present studies also identified that suppressor endothelial cell production of PGE2 was responsible for disrupting T-cell functions. These findings are consistent with prior demonstrations that PGE2 can suppress T-cell IFN-γ and IL-2 production.5 Support for the role of PGE2 as the mediator by which suppressor endothelial cells disrupt T-cell function came from studies showing that tumor-exposed endothelial cells produced increased levels of PGE2 and from studies demonstrating that reducing endothelial cell PGE2 production, either by blockage with indomethacin or by neutralizing VEGF in tumor-conditioned medium, prevented the suppressive activity of tumor-exposed endothelial cells. While inhibition of the cyclooxygenase pathway inhibited PGE2 production and blocked suppressor endothelial cells’ ability to disrupt T-cell functions, the contribution of other products regulated by the cyclooxygenase pathway remains to be explored. For example, Cox-2 regulates the production of nitric oxide, which can induce immune suppression in patients with hepatocellular carcinoma.38 However, our examination of nitric oxide production revealed no different between endothelial cells exposed in vitro to tumor-conditioned media or control treatments (data not shown). This does not eliminate the possibility that other immune suppressive products regulated by PGE2 such as arginase may be involved in the disruption of T-cell functions.39
The results of these studies may apply to other murine and human tumors besides the one examined here. Numerous studies in a variety of solid tumor types have demonstrated progressive increases in VEGF secretion beginning in early stages of dysplasia and continuing throughout tumor progression.31,36,40 Endothelial cells also have access to most infiltrating immune cells that must pass between endothelial cells to enter the tumor. Thus, while tumors can directly suppress T-cell function through their own production of PGE2, upregulated endothelial cell production of PGE2 may further contribute to suppression of T-cell function.
One aspect of these studies that remains to be tested is their clinical implications. Currently, three VEGF targeting therapies aimed at disrupting the tumor vasculature have gained FDA approval for the treatment of cancer. While VEGF targeting therapies were not originally designed to improve immune cell functions, there is growing evidence that they may have some use as an immunotherapeutic agent. To date, three clinical trials have examined the use of VEGF targeting therapies as immunotherapeutic agents. Fricke et al, observed in a phase I clinical study examining 15 patients that treatment with VEGF-Trap, a fusion protein that binds all isoforms of VEGF-A, significantly increased the proportion of circulating mature dendritic cells.41 Similarly Osada et al examined of 41 patients with lung, breast or colorectal carcinoma and reveled that anti- VEGF antibody therapy enhanced the capacity of dendritic cells to stimulate T cell proliferation against recall antigens.42 Lastly, studies by Ko et al, demonstrated in patients with metastatic renal cell carcinoma that the VEGF and c-kit inhibitor, Sunitinib, was capable of reducing the levels of CD3+CD4+CD25hiFoxp3+ Treg cells and MDCS.43 The results of these studies and the findings in the present report provide support for the use of VEGF targeting therapies as immunotherapeutic agents.
Cyclooxygenase inhibitors, including indomethacin, have been shown to block angiogenesis, suppress solid tumor metastases and slow tumor growth in vitro and have been shown clinically to reduce the incidence and mortality of select malignancies.44,45 In addition, cyclooxygenase inhibitors have been explored as an immunotherapeutic agent. In patients with oral squamous cell carcinoma, treatment with indomethacin improved immune function and increased immune infiltrate into tumors.46 Indomethacin was also found to improve T-cell cell responses to antigen stimulation in patient with melanoma or lung cancer.47,48 Studies by Lang et al, have shown that use of the selective Cox-2 inhibitor, Rofecoxib, improved monocyte migration and function to levels seen in healthy controls.49 Other studies have shown that use of selective Cox-2 inhibitors as chemopreventative agents reduces the expansion of immature myeloid suppressor cells.50 Future studies may include clinical investigation into the efficacy of cyclooxygenase-targeting therapies in blocking the inhibitory effects of suppressor endothelial cells.
Several questions remain to be answered that were not addressed in these studies. While these studies demonstrated the mechanism by which suppressor endothelial cells are induced and how they disrupt T-cell function, they did not address the in vivo contribution of endothelial cells to tumor-induced immune suppression. Previous studies by our laboratory demonstrated that suppressor endothelial cells can inhibit not only T-cell function, but the function of NK cells and macrophages as well. However, the mechanisms by which tumor-exposed endothelial cells become suppressive toward NK and macrophage function have yet to be determined. Characterization of the phenotype of suppressor endothelial cells also remains. Expression of CD11b, CD11c, CD34 and Gr-1 by the endothelial cell line used in our prior study and endothelial cells isolated from normal or tumor-bearing lungs was examined to determine if suppressor endothelial cells were phenotypically similar to MDSC. Expression of these markers was found to occur in extremely low levels on the cultured bEnd.3 endothelial cell line and was not altered by treatment with tumor-conditioned medium or controls (data not shown). Furthermore, examination of these markers on endothelial cells isolated from normal or tumor-bearing lungs were examined. As in the in vitro studies, expression of these markers was very low and there was no statistically significant difference in the percent or intensity of expression in these makers between control and tumor-derived endothelial cells (data not shown). These results suggest that suppressor endothelial cells are phenotypically distinct from MDSC’s, even though they share similar precursor origins.
Acknowledgments
Supported by Research Services of the Department of Veterans Affairs and by grants R01CA85266, R01DE018168, and 1R01CA128837 from the National Institutes of Health to MRIY.
References
- 1.Walsh JE, Lathers DM, Chi AC, et al. Mechanisms of tumor growth and metastasis in head and neck squamous cell carcinoma. Curr Treat Options Oncol. 2007;8:227–238. doi: 10.1007/s11864-007-0032-2. [DOI] [PubMed] [Google Scholar]
- 2.Ohm JE, Gabrilovich DI, Sempowski GD, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101:4878–4886. doi: 10.1182/blood-2002-07-1956. [DOI] [PubMed] [Google Scholar]
- 3.Mimura K, Kono K, Takahashi A, et al. Vascular endothelial growth factor inhibits the function of human mature dendritic cells mediated by VEGF receptor-2. Cancer Immunol Immunother. 2007;56:761–770. doi: 10.1007/s00262-006-0234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alfranca A, Lopez-Oliva JM, Genis L, et al. PGE2 induces angiogenesis via MT1-MMP-mediated activation of the TGFβ/Alk5 signaling pathway. Blood. 2008;112:1120–1128. doi: 10.1182/blood-2007-09-112268. [DOI] [PubMed] [Google Scholar]
- 5.Pockaj BA, Basu GD, Pathangey LB, et al. Reduced T-cell and dendritic cell function is related to cyclooxygenase-2 overexpression and prostaglandin E2 secretion in patients with breast cancer. Ann Surg Oncol. 2004;11:328–339. doi: 10.1245/aso.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 6.Vinals F, Pouyssegur J. Transforming growth factor β1 (TGF-β1) promotes endothelial cell survival during in vitro angiogenesis via an autocrine mechanism implicating TGF-α signaling. Mol Cell Biol. 2001;21:7218–7230. doi: 10.1128/MCB.21.21.7218-7230.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor-β and the immune response: implications for anticancer therapy. Clin Cancer Res. 2007;13:5262–5270. doi: 10.1158/1078-0432.CCR-07-1157. [DOI] [PubMed] [Google Scholar]
- 8.Young MR. Protective mechanisms of head and neck squamous cell carcinomas from immune assault. Head Neck. 2006;28:462–470. doi: 10.1002/hed.20331. [DOI] [PubMed] [Google Scholar]
- 9.Elgert KD, Alleva DG, Mullins DW. Tumor-induced immune dysfunction: the macrophage connection. J Leukoc Biol. 1998;64:275–290. doi: 10.1002/jlb.64.3.275. [DOI] [PubMed] [Google Scholar]
- 10.Mantovani A, Schioppa T, Porta C, et al. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25:315–322. doi: 10.1007/s10555-006-9001-7. [DOI] [PubMed] [Google Scholar]
- 11.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sasaki A, Tanaka F, Mimori K, et al. Prognostic value of tumor-infiltrating FOXP3+ regulatory T cells in patients with hepatocellular carcinoma. Eur J Surg Oncol. 2008;34:173–179. doi: 10.1016/j.ejso.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res. 2007;13(18 Pt 1):5243–5248. doi: 10.1158/1078-0432.CCR-07-0182. [DOI] [PubMed] [Google Scholar]
- 14.Young MR, Wright MA, Matthews JP, et al. Suppression of T cell proliferation by tumor-induced granulocyte-macrophage progenitor cells producing transforming growth factor-β and nitric oxide. J Immunol. 1996;156:1916–1922. [PubMed] [Google Scholar]
- 15.Young MR. Tumor skewing of CD34+ progenitor cell differentiation into endothelial cells. Int J Cancer. 2004;109:516–524. doi: 10.1002/ijc.20003. [DOI] [PubMed] [Google Scholar]
- 16.Young MR, Cigal M. Tumor skewing of CD34+ cell differentiation from a dendritic cell pathway into endothelial cells. Cancer Immunol Immunother. 2006;55:558–568. doi: 10.1007/s00262-005-0036-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danese S, Dejana E, Fiocchi C. Immune regulation by microvascular endothelial cells: directing innate and adaptive immunity, coagulation and inflammation. J Immunol. 2007;178:6017–6022. doi: 10.4049/jimmunol.178.10.6017. [DOI] [PubMed] [Google Scholar]
- 18.Lefkowitz DL, Roberts E, Grattendick K, et al. The endothelium and cytokine secretion: the role of peroxidases as immunoregulators. Cell Immunol. 2000;202:23–30. doi: 10.1006/cimm.2000.1638. [DOI] [PubMed] [Google Scholar]
- 19.Ohira H, Abe K, Yokokawa J, et al. Adhesion molecules and CXC chemokines in endotoxin-induced liver injury. Fukushima J Med Sci. 2003;49:1–13. doi: 10.5387/fms.49.1. [DOI] [PubMed] [Google Scholar]
- 20.Pirtskhalaishvili G, Nelson JB. Endothelium-derived factors as paracrine mediators of prostate cancer progression. Prostate. 2000;44:77–87. doi: 10.1002/1097-0045(20000615)44:1<77::aid-pros10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 21.Mulligan J, Lathers D, Young M. Tumors skew endothelial cells to disrupt NK cell, T-cell and macrophage functions. Cancer Immunol Immunother. 2008;57:951–961. doi: 10.1007/s00262-007-0425-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abdollahi A, Lipson KE, Han X, et al. SU5416 and SU6668 Attenuate the Angiogenic Effects of Radiation-induced Tumor Cell Growth Factor Production and Amplify the Direct Anti-endothelial Action of Radiation in vitro. Cancer Res. 2003;63:3755–3763. [PubMed] [Google Scholar]
- 23.Bischof M, Abdollahi A, Gong P, et al. Triple combination of irradiation, chemotherapy (pemetrexed), and VEGFR inhibition (SU5416) in human endothelial and tumor cells. Int J Radiat Oncol Biol Phys. 2004;60:1220–1232. doi: 10.1016/j.ijrobp.2004.07.689. [DOI] [PubMed] [Google Scholar]
- 24.Kosmider O, Denis N, Dubreuil P, et al. Semaxinib (SU5416) as a therapeutic agent targeting oncogenic Kit mutants resistant to imatinib mesylate. Oncogene. 2006;26:3904–3908. doi: 10.1038/sj.onc.1210159. [DOI] [PubMed] [Google Scholar]
- 25.Zhong X-S, Zheng JZ, Reed E, et al. SU5416 inhibited VEGF and HIF-1[alpha] expression through the PI3K/AKT/p70S6K1 signaling pathway. Biochemical and Biophysical Research Communications. 2004;324:471–480. doi: 10.1016/j.bbrc.2004.09.082. [DOI] [PubMed] [Google Scholar]
- 26.Wang SY, Chen B, Zhan YQ, et al. SU5416 is a potent inhibitor of hepatocyte growth factor receptor (c-Met) and blocks HGF-induced invasiveness of human HepG2 hepatoma cells. Journal of Hepatology. 2004;41:267–273. doi: 10.1016/j.jhep.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 27.Cuneo KC, Fu A, Osusky KL, et al. Effects of vascular endothelial growth factor receptor inhibitor SU5416 and prostacyclin on murine lung metastasis. Anticancer Drugs. 2007;18:349–355. doi: 10.1097/CAD.0b013e328011fdab. [DOI] [PubMed] [Google Scholar]
- 28.Laschke MW, Elitzsch A, Vollmar B, et al. Combined inhibition of vascular endothelial growth factor (VEGF), fibroblast growth factor and platelet-derived growth factor, but not inhibition of VEGF alone, effectively suppresses angiogenesis and vessel maturation in endometriotic lesions. Hum Reprod. 2006;21:262–268. doi: 10.1093/humrep/dei308. [DOI] [PubMed] [Google Scholar]
- 29.Ou X-M, Li W-C, Liu D-S, et al. VEGFR-2 antagonist SU5416 attenuates bleomycin-induced pulmonary fibrosis in mice. Int Immunopharmacol. 2009;9:70–79. doi: 10.1016/j.intimp.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Timke C, Zieher H, Roth A, et al. Combination of Vascular Endothelial Growth Factor Receptor/Platelet-Derived Growth Factor Receptor Inhibition Markedly Improves Radiation Tumor Therapy. Clin Cancer Res. 2008;14:2210–2219. doi: 10.1158/1078-0432.CCR-07-1893. [DOI] [PubMed] [Google Scholar]
- 31.Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF) J Cell Mol Med. 2005;9:777–794. doi: 10.1111/j.1582-4934.2005.tb00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamura K, Sakurai T, Kogo H. Relationship between prostaglandin E2 and vascular endothelial growth factor (VEGF) in angiogenesis in human vascular endothelial cells. Vascul Pharmacol. 2006;44:411–416. doi: 10.1016/j.vph.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 33.Onoe T, Ohdan H, Tokita D, et al. Liver sinusoidal endothelial cells tolerize T-cells across MHC barriers in mice. J Immunol. 2005;175:139–146. doi: 10.4049/jimmunol.175.1.139. [DOI] [PubMed] [Google Scholar]
- 34.Krupnick AS, Gelman AE, Barchet W, et al. Murine vascular endothelium activates and induces the generation of allogeneic CD4+25+Foxp3+ regulatory T-cells. J Immunol. 2005;175:6265–6270. doi: 10.4049/jimmunol.175.10.6265. [DOI] [PubMed] [Google Scholar]
- 35.Gerber H-P, Ferrara N. The role of VEGF in normal and neoplastic hematopoiesis. J Mol Med. 2003;81:20–31. doi: 10.1007/s00109-002-0397-4. [DOI] [PubMed] [Google Scholar]
- 36.Johnstone S, Logan RM. Expression of vascular endothelial growth factor (VEGF) in normal oral mucosa, oral dysplasia and oral squamous cell carcinoma. Int J Oral Maxillofac Surg. 2007;36:263–266. doi: 10.1016/j.ijom.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 37.Mulligan JK, Day TA, Gillespie MB, et al. Secretion of vascular endothelial growth factor by oral squamous cell carcinoma cells skews endothelial cells to suppress T-cell functions. Hum Immunol. 2009;70:375–382. doi: 10.1016/j.humimm.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu T. Cyclooxygenase-2 in hepatocellular carcinoma. Cancer Treat Rev. 2006;32:28–44. doi: 10.1016/j.ctrv.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez PC, Hernandez CP, Quiceno D, et al. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med. 2005;202:931–939. doi: 10.1084/jem.20050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 41.Fricke I, Mirza N, Dupont J, et al. Vascular endothelial growth factor-trap overcomes defects in dendritic cell differentiation but does not improve antigen-specific immune responses. Clin Cancer Res. 2007;13:4840–4848. doi: 10.1158/1078-0432.CCR-07-0409. [DOI] [PubMed] [Google Scholar]
- 42.Osada T, Chong G, Tansik R, et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunol Immunother. 2008;57:1115–1124. doi: 10.1007/s00262-007-0441-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148–2157. doi: 10.1158/1078-0432.CCR-08-1332. [DOI] [PubMed] [Google Scholar]
- 44.Cervello M, Montalto G. Cyclooxygenases in hepatocellular carcinoma. World J Gastroenterol. 2006;12:5113–5121. doi: 10.3748/wjg.v12.i32.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blanke CD. Celecoxib with chemotherapy in colorectal cancer. Oncology (Williston Park) 2002;16(4 Suppl 3):17–21. [PubMed] [Google Scholar]
- 46.Cross DS, Platt JL, Juhn SK, et al. Administration of a prostaglandin synthetase inhibitor associated with an increased immune cell infiltrate in squamous cell carcinoma of the head and neck. Arch Otolaryngol Head Neck Surg. 1992;118:526–528. doi: 10.1001/archotol.1992.01880050080019. [DOI] [PubMed] [Google Scholar]
- 47.Tilden AB, Balch CM. Indomethacin enhancement of immunocompetence in melanoma patients. Surgery. 1981;90:77–84. [PubMed] [Google Scholar]
- 48.Han T, Takita H. Indomethacin-mediated enhancement of lymphocyte response to mitogens in healthy subjects and lung cancer patients. Cancer. 1980;46:2416–2420. doi: 10.1002/1097-0142(19801201)46:11<2416::aid-cncr2820461120>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 49.Lang S, Lauffer L, Clausen C, et al. Impaired monocyte function in cancer patients: restoration with a cyclooxygenase-2 inhibitor. FASEB Journal. 2002:02fje–0417fje. doi: 10.1096/fj.02-0417fje. [DOI] [PubMed] [Google Scholar]
- 50.Talmadge JE, Hood KC, Zobel LC, et al. Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int J Immunopharmacol. 2007;7:140–151. doi: 10.1016/j.intimp.2006.09.021. [DOI] [PubMed] [Google Scholar]