

Abstract
The TET family of FE(II) and 2-oxoglutarate-dependent enzymes (Tet1/2/3) promote DNA demethylation by converting 5-methylcytosine to 5-hydroxymethylcytosine (5hmC), which they further oxidize into 5-formylcytosine and 5-carboxylcytosine. Tet1 is robustly expressed in mouse embryonic stem cells (mESCs) and has been implicated in mESC maintenance. Here we demonstrate that, unlike genetic deletion, RNAi-mediated depletion of Tet1 in mESCs led to a significant reduction in 5hmC and loss of mESC identity. The differentiation phenotype due to Tet1 depletion positively correlated with the extent of 5hmC loss. Meta-analyses of genomic data sets suggested interaction between Tet1 and leukemia inhibitory factor (LIF) signaling. LIF signaling is known to promote self-renewal and pluripotency in mESCs partly by opposing MAPK/ERK-mediated differentiation. Withdrawal of LIF leads to differentiation of mESCs. We discovered that Tet1 depletion impaired LIF-dependent Stat3-mediated gene activation by affecting Stat3's ability to bind to its target sites on chromatin. Nanog overexpression or inhibition of MAPK/ERK signaling, both known to maintain mESCs in the absence of LIF, rescued Tet1 depletion, further supporting the dependence of LIF/Stat3 signaling on Tet1. These data support the conclusion that analysis of mESCs in the hours/days immediately following efficient Tet1 depletion reveals Tet1's normal physiological role in maintaining the pluripotent state that may be subject to homeostatic compensation in genetic models.
INTRODUCTION
Embryonic stem cells (ESCs) appear to have an unique epigenetic state that maintains the pluripotent genome in a stable program of self-renewal, while allowing rapid induction of alternate transcriptional programs to initiate differentiation (1–8). DNA methylation is one of the principle regulators of the epigenetic landscape that shapes and refines gene expression programs during embryogenesis and stem cell differentiation. While mechanisms of establishment and maintenance of DNA methylation by DNA methyltransferases are well characterized (9,10), it was less clear which enzymatic machinery is responsible for DNA demethylation or even which pathways lead to DNA demethylation (11,12). Recently, it was discovered that proteins of the Tet family of FE(II) and 2-oxoglutarate-dependent enzymes (Tet1/2/3) oxidize 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) (13–17), which they further oxidize into 5-formylcytosine and 5-carboxylcytosine, thereby promoting DNA demethylation (18–23). Tet proteins and 5hmC have been shown to have roles in cancer and stem cell biology (13,16,24,25). Mouse embryonic stem cells (mESCs) deficient in DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b lack 5mC as well as 5hmC implying that 5hmC marks are probably derived from pre-existing 5mC marks (26,27).
In mESCs, Tet1 is expressed at high levels, comparable to those of the master pluripotency factor Oct4 (16,28). Tet2, though well expressed in mESCs, is 5-fold less abundant than Tet1 (28). Tet3 is variably expressed in many tissues but not in mESCs (16). Though various tissues express one or more Tet proteins, the 5hmC modification is particularly abundant in mESCs and Purkinje neurons (13,14,16,29). In mESCs, Tet1 binding and 5hmC occupancy are correlated with CpG density, are enriched at the promoters and gene bodies of nearly two-thirds of all genes, and have been linked to both gene activation and repression (27,30–33). Tet1 has also been shown to be required for the recruitment of transcriptional repressors Ezh2 (30) and Sin3a (27) at CpG-rich promoters of developmental regulators. Though Tet1-mediated regulation is believed to be due to its catalytic activity, Tet1 might have other functions in addition to converting 5mC to 5hmC (27).
A recent study reported that RNAi-mediated knockdown (KD) of Tet1 in mESCs resulted in downregulation of pluripotency marker Nanog and loss of undifferentiated state implicating Tet1 in mESC maintenance (16). In contrast, subsequent studies found that Tet1 KD cells were morphologically indistinguishable from control mESCs with no changes in Nanog expression, modest reduction in total 5hmC levels, and a minor-to-moderate increase in 5mC levels (26–28,31). Discrepancy between these studies may be due to differences in KD efficiency, off-target effects, or homeostatic compensation masked by antibiotic selection. More recently, it was reported that Tet1−/− mice are viable, fertile and grossly normal, and that Tet1−/− mESCs maintained a normal undifferentiated mESC morphology with only ∼35% reduction in total 5hmC levels (34). Since only a modest reduction in 5hmC levels is observed in this genetic model, it is possible that maternal contribution or unaltered Tet2 in Tet1 null cells may be compensating Tet1's function, thereby obscuring the direct impact of Tet1 loss. The discrepancies observed in the various RNAi-mediated depletion experiments in addition to the puzzling retention of 5hmC in the Tet1 null mESCs suggest that unanswered questions remain regarding the role of Tet1 in the maintenance of the pluripotent state of mESCs.
To understand and clarify the role of Tet1 and 5hmC in epigenetic and transcriptional regulation of mESCs, we used RNAi to acutely deplete Tet1 in mESCs and performed expression and genome-wide 5hmC occupancy studies. We find that acute short-term depletion as opposed to genetic deletion of Tet1 results in a significant decrease in 5hmC levels, downregulation of pluripotency-associated factors, impairment of LIF-dependent Stat3-mediated gene activation, and loss of embryonic stem cell identity.
MATERIALS AND METHODS
Mouse ES cell culture, RNAi and alkaline phosphatase staining
Oct4GiP mESCs were kindly provided by Dr Austin Smith. E14Tg2a mESCs were obtained from Mutant Mouse Research Resource Centers, and J1 mESCs were obtained from ATCC. The cells were routinely maintained on gelatin-coated plates in the ESGRO complete plus clonal grade medium (Millipore), and were used at passage 20–35 for experiments. For siRNA transfections, mESCs were cultured on gelatin-coated plates in M15 medium: Dulbecco's Modified Eagle Medium (Invitrogen) supplemented with 15% Fetal Bovine Serum (FBS) (Invitrogen), 10 μM 2-mercaptoethanol, 0.1 mM non-essential amino acids (Invitrogen), 1× EmbryoMax nucleosides (Millipore), 1000 U of ESGRO (Millipore). For cells to be harvested or stained 96 h after transfection, 20–25 × 103 mESCs were transfected with siRNAs at 100 nM in M15 medium in one well of a 96-well plate. About 0.7 μl of Lipofectamine 2000 (Invitrogen) was pre-mixed with 10 μl of Opti-MEM (Invitrogen) and then mixed with 1 × 10−11 mol siRNAs. Dissociated mESCs were plated at 25 × 103 per well in 100 μl of M15 medium in gelatin-coated plates. siRNA–lipids complexes were incubated at room temperature for 15–30 min and then added to the cells. About 25–50% of the cells in each well was re-plated into one well of a 12-well plate the next day, and cultured in M15 medium for another 3 days. For cells to be harvested 48 h after transfection, 75 × 103 mESCs were transfected with siRNAs at 50 nM in M15 medium in one well of a 24-well plate, and cells were collected 2 days after transfection. RNAi experiments were performed using indicated individual siRNAs: Tet1 siRNA #1 (Invitrogen, MSS284895), Tet1 siRNA #2 (Invitrogen, MSS284897), Tet1 siRNA #3 (Dharmacon, D-062861-01), Tet1 siRNA #4 (Dharmacon, D-062861-02), Tet1 siRNA #5 (Dharmacon, D-062861-03), Tet1 siRNA #6 (Dharmacon, D-062861-04) and control siRNA duplex targeting firefly luciferase (Dharmacon, 5′-CGTACGCGGAATACTTCGA). For lineage marker [quantitative reverse-transcriptase PCR (qRT-PCR)] and microarray analysis, cells were harvested 48 and 96 h after transfection for RNA extraction. For alkaline phosphatase (AP) staining, cells were fixed, permeablized and stained for AP activity with the Alkaline Phosphatase Detection Kit (Millipore).
Nanog overexpression cells
Mouse Nanog was cloned into pcDNA-EF-HA/Flag vector and sequence verified (see Supplementary Material). The resulting plasmid, mNanog-pcDNA-EF-HA/Flag, was transfected into Oct4GiP cells with Lipofectamine 2000 (Invitrogen), and the cells were selected with G418 and plated at colonal density. Single clones were picked and expanded, and clone #2 was chosen for the experiment. The expression of the exogenous HA/Flag-tagged Nanog was estimated to be at the same level of the endogenous Nanog based on western blot.
Oct4GiP reporter assay
Oct4GiP cells were transfected in 96-well plates: 0.35 μl of Lipofectamine 2000 (Invitrogen) was pre-mixed with 10 μl of Opti-MEM (Invitrogen) and then mixed with 5 × 10−12 mol siRNAs. Dissociated cells were plated at 10 × 103 per well in 100 μl of M15 medium in gelatin-coated plates. siRNA–lipid complexes were incubated at room temperature for 15–30 min and then added to the cells. Cells were cultured in M15 medium with daily medium change. Four days after transfection, cells were lifted and dissociated by trypsinization, pipetted into single cell suspension in 10% FBS in Phosphate Buffered Saline (PBS), and fluorescence-activated cell sorting (FACS) analyzed on LSRII FACS analyzer (BD). Percentage of differentiation was determined by measuring the percentage of green fluorescent protein (GFP)-negative cells. For each experimental condition, 3–4 independent transfections were carried out, and data was plotted as mean ± SEM (standard error of the mean).
Western blot
Cells were directly lysed in SDS–PAGE sample buffer. Proteins were resolved by SDS–PAGE, transferred to nitrocellulose membrane using iBlot (Invitrogen), and probed with the indicated antibodies. Loading was normalized based on Ran or Tubulin. Primary antibodies used: Nanog (Millipore, AB9220), Stat3–Y705-phospho-specific (Cell Signaling, 9145), Stat3 (Cell Signaling, 9132), Dnmt3a (Santa Cruz, H-295), Dnmt3b (IMEGENEX, IMG-184A), Ran (BD, 610340) and Tubulin (Santa Cruz, sc-9104).
Quantitative RT-PCR
Total RNAs were prepared from cells using the RNeasy (Qiagen), and cDNAs were generated using the Omniscript RT kit (Qiagen) according to the manufacturer's instructions. Quantitative PCRs were performed using the Ssofast Evagreen Supermix (Bio-Rad) on the Bio-Rad CFX-384 or CFX-96 Real-Time PCR System. At least three biological repeats were carried out for each experiment. For every biological repeat, triplicate or quadruplicate PCR reactions were performed for each sample, and Actin was used for normalization. Data was plotted as mean ± SEM. Please refer to Supplementary Table S1 for gene specific primers used for RT-PCR analysis.
5hmC slot blot, and genome-wide mapping and analysis of 5hmC
Mouse E14Tg2a cells were transfected with control siRNA or an siRNA targeting Tet1 in 6-well plates. Cells were lysed 96 h after transfection in Trizol reagent, and both total RNA and genomic DNA were prepared according to manufacturer's instructions. Total RNA was used to determine the Tet1 KD efficiency. Genomic DNA was quantified with NanoDrop, and 100 or 500 ng was loaded on GE Hybond+ nylon membrane using the GE slot blot device according to manufacturer's instructions and blotted for 5hmC. Primary antibody used: 5hmC (Active Motif, 39769). Relative signal strengths of 5hmC bands were quantified using the Image-J software (http://imagej.nih.gov/ij/).
For genome-wide mapping of 5hmC, the genomic DNA obtained from control KD and Tet1 KD cells were processed using the QUEST 5hmC detection kit (Zymo Research) according to manufacturer's instructions. Briefly, the genomic DNA was treated with 5hmC glucosyltransferase enzyme, which specifically tags the 5hmC in DNA with a glucose moiety yielding a modified base, glucosyl-5hmC. Next the DNA was digested using glucosyl-5hmC sensitive restriction endonuclease MspI. MspI efficiently cleaves the DNA when cytosine, 5mC, or 5hmC but not glucysyl-5hmC is located at the inner C position within its recognition sequence (i.e. CCGG). In other words, the 5hmC sites in genomic DNA treated with 5-glycosyltransferase enzyme are protected from MspI digestion. By comparing the qPCR or next-generation sequencing results for treated versus untreated genomic DNA, 5hmC sites are detected (see below for details). Computational analysis indicate that digesting mouse genomic DNA with MspI, selecting 40–220 bp fragments, and performing 36-bp sequencing would cover ∼1 million distinct CpG dinucleotides (∼5% of all CpGs), with roughly half located within ‘CpG islands’ (including sequences from 90% of all CpG islands) and the rest distributed between other relatively CpG-poor sequence features (35). Every MspI cleaved sequence read will thus include at least one informative CpG position. Based on this information, MspI-digested DNA from treated and untreated samples derived from control and Tet1 KD mESCs were size-selected (40–220 bp) and sequenced using Illumina GAII.
Sequenced 36-bp reads were aligned to the reference genome (mouse NCBI36/mm8 assembly) using Bowtie 0.12.2 (36). Those reads that mapped to unique genomic locations with at most two mismatches were retained. Roughly ∼50–65% of all reads were mapped to unique genomic locations. To increase the mapping efficiency, we trimmed one base from the 3′-end of the unmapped reads, aligned the residual 36-bp reads to the reference genome and retained those that mapped to unique genomic locations. This procedure was repeated until the residual reads were of length 18, and, in each iteration, reads that mapped to unique genomic locations were retained for further analysis. The reads that mapped to unique genomic locations but whose corresponding 5′- to 3′-sequence in the reference genome neither started with CGG nor ended with GGC were considered noise and discarded. The rationale for this is because the MspI cleaves the DNA containing its recognition site CCGG at the inner C position (C/CGG) (37), the DNA fragments that is sequenced must all start with CGG or end with GGC (complementary strand). Reads that start or end with anything other than CGG or GGC, respectively, are likely to be byproducts of DNA degradation or sequencing errors. The resulting mapped reads for the four samples (control KD treated, control KD untreated, Tet1 KD treated and Tet1 KD untreated) were normalized by total reads in each sample so that the number of reads mapping to a specific site is directly comparable across all four samples. Sites with less than two reads in all four samples were excluded from the analysis. A site was defined as a 5hmC site only if the normalized read count at this site in the control untreated sample is at least 1.5-fold higher than that in the control treated sample. A 5hmC site was defined to have reduced 5hmC levels in Tet1 KD mESCs only if
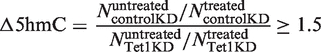 |
where 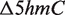 measures the reduction in 5hmC levels in Tet1 KD cells and
measures the reduction in 5hmC levels in Tet1 KD cells and 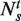 is the normalized read count in sample s with treatment t. Genes were considered to have 5hmC occupancy if they have one or more detected 5hmC sites within 5 kb upstream of the transcription start site (TSS) to the transcription end site (TES). Please refer to Supplementary Table S2 for PCR primers used for quantitative 5hmC analysis.
is the normalized read count in sample s with treatment t. Genes were considered to have 5hmC occupancy if they have one or more detected 5hmC sites within 5 kb upstream of the transcription start site (TSS) to the transcription end site (TES). Please refer to Supplementary Table S2 for PCR primers used for quantitative 5hmC analysis.
Chromatin immunoprecipitation
E14Tg2a mESCs were crosslinked with 1% formaldehyde in PBS for 10 min, and the resulting chromatin was sonicated using Diagenode Bioruptor for 15–18 cycles, 30 s per cycle with a 30 s rest between each cycle, to obtain ∼200–500 bp fragments. Chromatin immunoprecipitation (ChIP) was performed by incubating indicated antibodies with Dynabeads protein A magnetic beads (Invitrogen, 100-02D) for 4–6 h at 4°C, followed by overnight incubation with the chromatin at 4°C. Subsequent washing and reverse crosslinking steps were performed as described earlier (38). Each ChIP was performed using chromatin from ∼2 × 107 cells. Primary antibodies used: Stat3 (Santa Cruz, C-20), H3K9me3 (Abcam, ab8898) and IgG (Santa Cruz, H-270). Please refer to Supplementary Table S3 for ChIP primers used to analyze Stat3-target/binding sites.
ChIP-Seq factor binding data analysis
Published ChIP-Seq data sets for various transcription factors and chromatin remodelers (listed in Supplementary Table S4) were downloaded from the Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) or the BROAD Institute website. In cases where the data was mapped to mouse reference genome assembly mm9, the data was realigned to mm8 assembly using the liftOver tool (39). Where available for download, binding sites (peaks) as defined in the original publication were used. Otherwise SISSRs (40) was used for peak calling. Direct target genes of a particular factor (for example, Tet1) were defined as genes that have one or more detected factor binding sites within 5-kb upstream of the TSS to the TES.
Microarray gene expression data analysis
Raw CEL files (Affymetrix array type Mouse 430 2.0) generated from this study were processed using relevant R/Bioconductor packages (41). Specifically, RMA (42) was used for background correction and data normalization. Probe sets were defined using Entrez gene based custom chip definition files (CDFs), version 13 (http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/genomic_curated_CDF.asp) (43). This resulted in a single expression intensity measure per Entrez gene ID. All subsequent analyses were carried out on the log2 scale. To determine differentially expressed genes, a family-wise moderated t-test (44) (KD versus control) was performed followed by a multiple testing correction procedure to control the false discovery rate (FDR) (45). Unless otherwise noted, genes were considered differentially expressed if their FDR ≤ 0.1 and fold change ≥1.5. Since global gene expression changes due to Tet1 KDs using Tet1 siRNAs #1 and #2 were highly similar, we treated them as biological replicates in the differential expression analysis. Unsupervised hierarchical clustering was performed using the CLEAN software package (46). Functional enrichment analysis of differentially regulated lists of genes was performed using R packages CLEAN (46) and GO.db (41).
Published microarray data sets (listed in Supplementary Table S5) were downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo/). Where available, raw CEL files were re-processed using the above described microarray processing pipeline. Otherwise, the respective Series Matrix File was used to obtain preprocessed expression values and corresponding probe annotations. All data were transformed and analyzed on the log2 scale. Each probe identifier was mapped to an Entrez gene ID using relevant Bioconductor packages by continually matching annotations in the following order until an Entrez gene ID was found: Entrez gene ID, RefSeq ID, gene symbol or alias, gene name, unigene ID, Ensembl transcript ID. Probes mapping to the same Entrez gene ID were averaged (after the log-transformation). Fold changes and adjusted P-values (KD versus control) using moderated t-statistic described above were then computed for each data set independently.
Data availability
All microarray and 5hmC sequencing data generated for this study were deposited in NCBI GEO repository under the accession number GSE34267.
RESULTS
Acute depletion of Tet1 results in loss of embryonic stem cell identity
To elucidate the role of Tet1 and 5hmC in epigenetic and transcriptional regulation of mESC self-renewal and pluripotency, we used RNAi to acutely deplete Tet1 in Oct4GiP mESCs, which express the GFP under the control of the Oct4 promoter (47). Since Oct4 is exclusively expressed in ESCs, the GFP expression faithfully correlates with the ESC identity. Two small-interfering RNAs (siRNAs #1 and #2) targeting Tet1 were used to ensure that the effects were specific. Cells transfected with siRNAs were maintained in normal mESC culture conditions for 4 days, and subjected to FACS to determine the fraction of differentiated (GFP-negative) cells. Acute depletion of Tet1 resulted in small but significant increase in the percentage of differentiated cells (Figure 1A). Tet1 knockdown (KD) efficiency was confirmed using qRT-PCR and western blot (Figure 1B and Supplementary Figure S1A). Examination of Tet1 KD mESCs for colony morphology and AP staining revealed morphological changes and loss of AP staining consistent with differentiation (Figure 1C). The outcome remained similar even when E14Tg2a or J1 cell lines were used (Supplementary Figure S1B). To rule out the possibility that the observed changes were due to siRNA off-target effects, we used four additional siRNAs (#3–6), some of which were used in other studies that observed no obvious phenotype (28,31), and confirmed that acute depletion of Tet1 indeed induces morphological changes and loss of AP staining consistent with differentiation (Supplementary Figure S1B).
Figure 1.

Depletion of Tet1 and 5hmC levels results in loss of mouse embryonic stem identity. (A) Oct4GiP mESCs were transfected with indicated siRNAs in normal ESC medium and cultured for 96 h. The percentage of differentiated cells was determined by measuring the percentage of GFP-negative cells using FACS at 96 h after transfection (**P < 0.001; *P < 0.01). Error bars represent SEM of three experiments. (B) Relative Tet1 mRNA level in control and Tet1 KD Oct4GiP mESCs 48 h after transfection. Data are normalized to Actin. Error bars represent SEM of three experiments (**P < 0.001). Error bars represent SEM of three experiments. (C) AP staining of Oct4GiP mESCs transfected with control siRNA, and Tet1 siRNAs #1 and #2. Cells were cultured in normal ESC medium, and AP staining was performed 96 h after transfection. See Supplementary Figure S1B for AP staining results for siRNA #1 and four additional siRNAs on Oct4GiP, J1 and E14tg2a mESCs. (D) Expression fold changes of selected genes upon Tet1 KD in mESCs based on microarray analysis performed 48 and 96 h after transfection. Fold changes from data generated for this study [Tet1-KD (96 h) #1, #2 and Tet1-KD (48 h) #1] are presented alongside fold changes observed in recently published Tet1 KD (≥96 h) or knockout studies (color-coded on the top-right). Each column corresponds to fold changes obtained from an individual array computed in relation to its corresponding control array. Observed/reported morphological changes are symbolically indicated at the top, with columns ordered based on unsupervised hierarchical clustering. See Supplementary Figure S1C for a comprehensive heatmap. (E) Gene ontology analyses of up- and down-regulated genes in Tet1 KD cells compared to control cells. Only selected categories are shown. For complete lists, see Supplementary Table S6. (F) Relative mRNA levels of selected mESC pluripotency-associated genes and lineage marker genes in control and Tet1 KD mESCs at 96 h after transfection. The mRNA levels in control cells are set as one. Data are normalized to Actin. Error bars represent SEM of three experiments. (G) Scatter plot showing strong positive correlation among relative Tet1 mRNA levels, relative total 5hmC levels, morphogical changes, and AP staining in control and Tet1 KD E14Tg2a mESCs. Each data point corresponds to a siRNA that was used for transfection. The y-axis indicates relative Tet1 mRNA levels 96 h after KD, and the x-axis represents quantified intensity of 5hmC signal inferred from slot blot (96 h). Bottom panel show the corresponding representative morphological changes and AP staining for each siRNA. Error bars represent SEM of data from five replicates (different amounts of DNA were spotted) from two independent experiments.
ESC markers are downregulated and differentiation markers are upregulated upon Tet1 depletion
To determine whether Tet1 depletion has an effect on gene expression programs influencing mESC self-renewal, pluripotency and differentiation, we used microarrays to profile global gene expression in mESCs transfected with control and Tet1 siRNAs at 96 h after transfection. Pluripotency-associated genes such as Nanog, Esrrb, Tcl1, Tbx3, Klf2, Klf4, Lefty1, Lefty2, Tcfcp2l1 and Prdm14 were downregulated, and differentiation-associated genes including ectoderm and neuronal markers Fgf5, Pitx2, Nestin, Nefm, CD133 (Prom1), CD44, Lef1 and Zic1 and trophectoderm markers Eomes and Krt8/18/19 were upregulated in response to Tet1 KD (Figure 1D). Gene ontology analysis of transcripts downregulated at 96 h revealed enrichment of many terms related to stem cell maintenance and proliferation, whereas upregulated transcripts were enriched for terms related to development and differentiation (Figure 1E). To understand the kinetics of downstream effects of Tet1 KD, we also carried out microarray analysis at 48 h after siRNA transfection when Tet1-depleted cells (Figure 1B and Supplementary Figure S1A) were morphologically indistinguishable from control cells (Supplementary Figure S1D). Interestingly, our 48 h Tet1 KD data clustered tightly with those from shRNA-mediated Tet1 KD studies that did not observe morphological changes even past 96 h (Figure 1D and Supplementary Figure S1C), and away from our 96 h Tet1 KD data that accompanies morphological changes, suggesting a kinetic effect. The lack of phenotype in shRNA-mediated depletion studies could be due to the (i) differences in reaction rates in shRNA- versus siRNA-mediated knockdowns as a result of additional processing time necessary for the shRNA hairpin structure to be cleaved into siRNA or (ii) homeostatic compensation of Tet1's function masked by antibiotic selection.
We performed qRT-PCR and confirmed gene expression changes observed in the microarray analysis. Tet1 depletion decreased the expression of key pluripotency factors: Nanog by nearly 3-fold, and Oct4 and Sox2 by small, but reproducible and statistically significant, ∼1.25-fold. Other pluripotency-associated genes such as Klf4, Esrrb, Tcl1, Tbx3, Prdm14 and Nodal antagonists Lefty1 and Lefty2, which are among the earliest to be downregulated upon ESC differentiation (48), were downregulated by ∼2- to 4-fold (Figure 1F). Tet1 depletion also increased the expression of several differentiation genes including Pitx2, Nestin, Nefm, Lef1 and Zic1 that are largely associated with Ectoderm lineage (Figure 1F). Furthermore, these changes were consistent across Oct4GiP, E14Tg2a and J1 mESCs (Supplementary Figure S2A), and remained consistent even when using an siRNA that was used in two previous studies that observed no obvious phenotype (28,31) (Supplementary Figure S2B). Taken together, we concluded that acute RNAi-mediated depletion of Tet1 results in the loss of mESC identity.
Tet1 depletion results in significant reduction in total 5hmC levels
Examination of total 5hmC levels in Tet1-depleted mESCs using slot blot revealed a significant reduction in 5hmC compared to cells transfected with control siRNA (Supplementary Figure S2C). The severity of observed morphological changes positively correlated with the extent of 5hmC loss (Figure 1G). We suspected that the downregulation of Tet2 in Tet1-depleted cells (Figure 1D), also observed in one other report with findings consistent with ours (30), might also be a contributor to reduced 5hmC levels. Quantitative RT-PCR analysis of Tet2 in Tet1-depleted mESCs revealed a reproducible and statistically significant ∼1.5-fold reduction in Tet2 levels (Supplementary Figure S2D). This along with Tet1 occupancy at Tet2 promoter (Supplementary Figure S2E) suggested regulation of Tet2 by Tet1. Together, these data revealed that the acute reduction in 5hmC primarily due to Tet1 KD and also possibly due to indirect reduction in Tet2 levels is probably the cause for the loss of mESC identity.
Tet1-mediated gene regulation is context dependent
To investigate the role of Tet1 and 5hmC in the maintenance of mESC identity on the molecular level, we examined the genes that were differentially expressed upon Tet1 depletion in our microarray analysis. Using a reasonably stringent criterion (FDR < 0.1; fold ≥1.5), we identified 530 upregulated and 389 downregulated genes (Supplementary Table S7). Examination of published Tet1 occupancy data (27,30) revealed that Tet1 binds to 72% of upregulated and 63% of downregulated genes with no statistically significant preference for one group over the other, which is similar to the pattern observed with other factors known to play diverse regulatory roles in gene regulation such as the insulator binding transcription factor CTCF (Figure 2A and Supplementary Figure S3A). No correlation between Tet1 binding and gene expression changes was observed even when analyzing all genes with expression above detectable levels (Supplementary Figure S3B). These data indicate that Tet1 binding alone is not predictive of Tet1's function as an activator or a repressor.
Figure 2.

Meta-analysis of genomic data sets suggests interaction between Tet1 and LIF/Stat3 signaling. (A) Top: Transcription factor occupancy at 919 genes differentially expressed upon Tet1 KD (389 downregulated and 530 upregulated). Genes are represented along the y-axis and factor occupancy is denoted by blue bar. Target gene occupancy is defined as factor occupancy within 5-Kb upstream of the gene's TSS and/or within its gene body. Bottom: Log-fold enrichment of factor occupancy at up/downregulated genes. Red and green histograms denote enrichment in up- and down-regulated genes, respectively. *P < 0.01 after Bonferroni adjustment for multiple testing. (B) Top: Presence/absence of 5hmC sites (5hmC) in control mESCs, and 5hmC sites with ≥1.5-fold reduced hydroxymethylation levels (Δ5hmC) in Tet1 KD mESCs (96 h). Bottom: Log-fold enrichment at down- versus up-regulated genes, respectively. (C) Expression fold changes of 919 genes differentially expressed upon Tet1 KD (96 h; first column) presented alongside fold changes observed after KD or knockout (KO) of select other pluripotency factors in published reports. Genes are represented along the y-axis. Data sets along the x-axis have been ordered based on unsupervised hierarchical clustering of corresponding gene expression fold changes. Blue and yellow rectangles highlight Tet1-LIF and Polycomb clusters, respectively. Cells with no/missing data are colored in gray (DKO: double KO).
Tet1 has been reported to be required for the recruitment of transcriptional repressors Ezh2 (30) and Sin3a (27) at CpG-rich promoters of developmental regulators. We find that genes upregulated upon Tet1 KD are enriched for binding of polycomb members Suz12, Ezh2 and Ring1b (Figure 2A and Supplementary Figure S3A), whereas downregulated genes are enriched for binding of Stat3, Klf4, Oct4, Esrrb and Sin3a among others (Figure 2A). Since Tet1 binds to nearly two-thirds of all genes, its co-occupancy with other transcription factors is rather high, which could be misinterpreted for its functional interaction with these factors. When normalized by expected frequencies, Tet1's co-occupancy with other factors appears insignificant compared to that between factors that are known to function together (Supplementary Figure S3C). Like esBAF, a mESC specific chromatin remodeling complex (49) that facilitates both activation and repression (50,51), we propose that Tet1 probably plays a similar role as a general facilitator/recruiter of activating and repressing factors in a context dependent manner.
Tet1 depletion results in genome-wide reduction in 5hmC levels
To determine the extent of 5hmC loss in Tet1 KD cells on a genomic scale, we used a modified version of methylation sensitive restriction enzyme sequencing (52) and generated nucleotide resolution 5hmC maps covering ∼90% of sequences located within CpGs islands in control and Tet1 KD mESCs (see ‘Materials and Methods’ section for details). We identified 57 895 5hmC sites in control mESCs, of which 32 652 had at least 1.5-fold reduction in 5hmC levels in Tet1 KD cells (Supplementary Table S8). Quantitative PCR was performed to validate 5hmC sites and reduction in 5hmC levels (Supplementary Figure S3D). We find that genes that were upregulated or downregulated upon Tet1 depletion are equally likely to contain 5hmC sites and experience reduced 5hmC levels (Figure 2B), which is consistent with the established dual roles of Tet1 and 5hmC in both gene activation and repression (27,30,33). These data indicate that an acute depletion of Tet1 causes significant global reduction in 5hmC levels that were not specific to gene classes, and that the directionality of gene expression change (up/down) is probably determined by the gene context and local transcription factor occupancy (Figure 2A).
Meta-analysis of genomic data suggests interaction between Tet1 and LIF/Stat3 signaling
We next examined the fate of Tet1 regulated genes upon KD or knockout of other pluripotency factors. A meta-analysis of microarray data from published studies revealed that genes that are differentially expressed upon Tet1 KD underwent similar expression changes upon leukemia inhibitory factor (LIF) withdrawal or after depletion of key pluripotency-associated factors such as Esrrb, Tcl1, Tbx3 and Klf2/4/5, known to be associated with LIF/Stat3 signaling (8,53–55) (Figure 2C), suggesting Tet1's possible interaction with or co-regulation of transcriptional circuit regulated by LIF/Stat3 signaling (Figure 2A). In contrast, Tet1-regulated genes underwent largely divergent changes upon deletion/depletion of transcriptional repressors Tcf3 and Sall4 (Figure 2C).
LIF-dependent Stat3-mediated gene activation is dependent on Tet1
To examine Tet1's possible mechanistic interaction with LIF/Stat3 signaling (Figure 2A and C), we analyzed global gene expression changes upon Tet1 depletion against those after LIF withdrawal. Tet1-depleted and LIF-starved mESCs undergo similar changes in the global transcriptional profile (Figure 3A). The set of genes co-activated by Tet1 and LIF includes a number of factors essential for pluripotency, including Esrrb, Tcl1 and Tbx3 (56,57). This suggested that Tet1 or Tet1-dependent 5hmC levels might be required for LIF-dependent Stat3-mediated gene activation. To further explore this connection, we decided to study Tet1 KD mESCs 48 h after transfection, when Tet1 is depleted but the cells appeared normal compared to control transfected cells (Figure 1B, Supplementary Figure S1A and D). This time point was chosen so that we could clearly distinguish cause from effect. First, we asked whether LIF/Stat3 signal transduction was impaired in Tet1 KD mESCs. No significant changes in mRNA levels for Stat3 or LIF chimeric receptor consisting of gp130 and LIF receptor (LIFR) were observed at 48 h (Figure 3B and C). Tet1-depleted mESCs at 48 h after transfection had no significant changes in total Stat3 or Nanog levels but a modest reduction in phosphorylated/activated Stat3 (Figure 3D).
Figure 3.

Dependence of LIF/Stat3 signaling on Tet1. (A) Two-dimensional gene density heatmap depicting global gene expression changes 96 h after Tet1 KD against changes observed 48 h after LIF withdrawal (51). The intensity of each square represents the number of genes that fall within that square. Axes indicate degree of fold change, from nil (middle of axis) to >1.5-fold (outermost squares). (B) Gene expression fold changes observed in microarray analysis 48 and 96 h after Tet1 KD in mESCs, and 48 h after LIF withdrawal (51). (C) Relative Stat3 mRNA level in control and Tet1 KD mESCs. The mRNA level in control KD cells is set as one. Data are normalized to Actin. Error bars represent SEM of three experiments. (D) Western blot analysis showing protein levels in control and Tet1 KD (48 h) mESCs. Tubulin is used as a loading control. (E) Left: ChIP assay of select Stat3 target regions using an antibody against Stat3 in control KD and Tet1 KD mESCs (48 h). The y-axis represents enrichment over input normalized to a negative control region (Yipf2). Refer to Supplementary Figure S4 for data from control ChIP using a non-specific antibody against IgG. Error bars represent SEM of three experiments. Right: Gene expression fold changes observed in microarray analysis 48 and 96 h after Tet1 KD in mESCs, and 48 h after LIF withdrawal (51).
Next, we examined the ability of Stat3 to bind to its target sites on chromatin in the absence of Tet1. ChIP experiment using Stat3 antibody was carried out at 48 h after Tet1 KD. Stat3 binding was affected at many gene targets co-bound and co-activated by Tet1 and Stat3 (Figure 3E) indicating that Tet1 is required to facilitate LIF/Stat3 responsiveness in mESCs by enabling Stat3 to bind to its target sites on chromatin. Since the expression changes due to loss of Stat3 binding at Stat3-activated genes is not evident at 48 h but only at 96 after Tet1 KD (Figure 3E), we concluded that the loss of Stat3 binding precedes loss of expression. We do not rule out the possibility that a modest reduction in phosphorylated Stat3 levels (Figure 3D) might also have played a role in impaired Stat3 binding. Regardless, the LIF-dependent Stat3-mediated gene activation in mESCs is dependent on Tet1.
Nanog overexpression or inhibition of MAPK/ERK signaling rescues Tet1 KD phenotype
Since mESCs can be propagated without activation of Stat3 by imposing constitutive expression of core pluripotency factor Nanog (58), we checked if Nanog overexpression (Figure 4A) could rescue the differentiation phenotype exhibited by Tet1 KD cells. As expected, exogenous Nanog largely rescued the morphological changes and AP activity in Tet1 KD cells (Figure 4B). Also, since mESCs can be propagated without Stat3 activation by suppressing prodifferentiative MAPK/ERK signaling (59), we tested if mESCs transfected with Tet1 siRNA in 2i medium (which inhibits MAPK/ERK and Gsk-3b signaling) blocks differentiation. We found that while Tet1 depletion slightly decreased cell viability in 2i medium (data not shown), the expression of pluripotency-associated factors Nanog, Tcl1, Klf4 and Lefty2 were largely rescued (Figure 4C). In addition, Tet1 KD in 2i medium also suppressed upregulation of differentiation genes such as Pitx2, CD133, Nefm and Lef1. Together, these data provide evidence to further support the dependence of LIF/Stat3 signaling on Tet1.
Figure 4.

Nanog overexpression or suppression of MAPK/ERK signaling rescues Tet1 KD phenotype. (A) Western blot analysis showing Nanog overexpression with HA-tag. (B) AP staining of E14Tg2a mESCs, with and without Nanog overexpression, transfected with control siRNA and Tet1 siRNA #1. Cells were cultured in normal ESC medium, and AP staining was performed 96 h after transfection. (C) Relative mRNA levels of selected mESC pluripotency genes and differentiation marker genes in control and Tet1 KD mESCs in 2i medium. Oct4GiP cells were transfected with control siRNA or Tet1 siRNA #1 at 50 nM in 24-well plates in 2i-medium (which inhibits MAPK/ERK and Gsk-3b signaling) and cells were harvested 96 h after transfection. The mRNA levels in control cells are set as one. Data are normalized to Actin. Error bars represent SEM of three experiments.
Tet1 negatively regulates de novo DNA methyltransferase Dnmt3b
Given that genes downregulated in Tet1 KD cells undergo a minor-to-moderate increase in 5mC levels (26–28,31), and that repressive histone mark H3K9me3 has been known to recruit DNA methyltransferases and be associated with DNA methylation (35,60), we hypothesized that the loss of Stat3 binding and Stat3-mediated activation of its targets accompanies an increase in H3K9me3. ChIP using an antibody against H3K9me3 revealed that this was indeed the case (Figure 5A). It is not clear, however, whether Tet proteins alleviate/prevent gene silencing at Stat3 targets by preventing deposition of H3K9me3, or by maintaining a DNA-hypomethylated state. Interestingly, we noticed a 2-fold increase in Dnmt3b levels in Tet1 KD cells in microarray analysis (Figure 1D). Quantitative RT-PCR and western blot confirmed the increase in Dnmt3b at the mRNA and protein levels (Figure 5B and C). Since Tet1 targets Dnmt3b (Figure 5D), we examined the 5hmC levels at the Dnmt3b locus and found >3-fold reduction in 5hmC in Tet1 KD cells (Figure 5E). Based on this evidence, it is possible that Tet1's regulation of DNA methylation might also involve its negative regulation, direct or indirect, of de novo DNA methyltransferase Dnmt3b.
Figure 5.

Tet1 negatively regulates de novo DNA methyltransferase Dnmt3b. (A) ChIP assay of select Stat3 target regions using an antibody against H3K9me3 in control and Tet1 KD mESCs (48 h). The y-axis represents enrichment over input normalized to a positive control region (Myod1) for H3K9me3. Error bars represent SEM of three experiments. (B) Relative mRNA levels of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b in control and Tet1 KD mESCs 96 h after transfection. The mRNA levels in control cells are set as 1. Data are normalized to Actin. Error bars represent SEM of three experiments. (C) Western blot analysis showing protein levels of Nanog, Dnmt3a and Dnmt3b in control and Tet1 KD mESCs 96 h after transfection. Ran is used as a loading control. (D) Genome browser shot showing a region containing the Dnmt3b gene and results from Tet1 and Sin3a ChIP-Seq experiments by various groups (GSM numbers denote GEO accession). The red open rectangle highlights Tet1 occupancy at the promoter region of Dnmt3b, where a CpG island is present (green-filled rectangle). (E) Relative 5hmC levels at Dnmt3b locus in control and Tet1 KD (96 h) mESCs. Error bars represent SEM of three experiments.
DISCUSSION
Our studies indicate that acute short-term depletion as opposed to long-term genetic deletion of Tet1 in mESCs results in the loss of the mESC identity. Tet1 depletion, in our hands, reduced total 5hmC levels by roughly 2- to 3-fold, Nanog expression by ∼3-fold and Tet2 expression by ∼1.5-fold. The extent of 5hmC reduction positively correlated with morphological changes and extent of loss of AP staining (Figure 1) suggesting that it is the loss of 5hmC and not necessarily Tet1 that caused differentiation. Our findings are consistent with those by another group that used shRNAs to deplete Tet1 and observed similar changes in morphology, global gene expression, Nanog, Tet2 and total 5hmC levels (16,30). Our findings, however, are in contrast to Tet1 depletion studies by others (26–28,31), who observed only ∼1.5-fold reduction in total 5hmC levels, no reduction in Nanog or Tet2 levels and no distinguishable morphological changes. Most remarkably, our results were reproducible even when using a siRNA that was employed in two previous studies that observed no obvious phenotype (28,31). Interestingly, the molecular changes we observe after Tet1 depletion are consistent with those from siRNA-mediated KD but not shRNA-mediated KD with puromycin selection from a previous study (31) (data from Xu et al in Figure 1D). It should be noted that the only other study that reported a Tet1 KD phenotype used shRNA-mediated depletion of Tet1 with no antibiotic selection. Since the siRNAs we used do not lead to greater KD efficiency than the shRNAs used by studies with no phenotype, it is possible that the lack of phenotype observed by others could be the result of homeostatic compensation of Tet1's function masked by antibiotic selection.
At present, although we have no ready explanation for the discrepancy between our results and those from Tet1 null mice (34), a modest reduction in 5hmC levels in the genetic model raises an intriguing possibility of maternal contribution or unaltered Tet2 expression compensating for Tet1's function after prolonged loss of Tet1, therefore masking the direct effect of Tet1 loss that we observe hours/days after RNAi-mediated acute Tet1 KD. Our findings suggest that an acute as opposed to long-term chronic reduction in Tet1 and thus 5hmC levels is probably difficult for mESCs to overcome or compensate, thereby resulting in the loss of the pluripotent state. This is consistent with Tet2's critical role in regulating self-renewal, proliferation, and differentiation in hematopoietic stem cells (61–63), and Tet3's requirement for paternal genome conversion of 5mC into 5hmC (64–66).
The sharp reduction in Tet1 and 5hmC impaired LIF-dependent Stat3-mediated gene activation by affecting Stat3's ability to bind to its target sites on chromatin, indicating dependence of LIF/Stat3 signaling on Tet1 and/or 5hmC. Suppression of prodifferentiative MAPK/ERK signaling or Nanog overexpression, known to maintain mESCs in the absence of LIF or Stat3 activation, rescued Tet1 KD phenotype further supporting the dependence of LIF/Stat3 signaling on Tet1. Collectively, our results support a model in which the Tet1-mediated regulation of 5hmC levels is critical to the maintenance of the pluripotent state in mESCs (Figure 6). Tet1 aids LIF-mediated Stat3 gene activation by facilitating Stat3 binding to chromatin by a yet to be determined mechanism. Tet1 regulates key pluripotency-associated factors including Nanog, Klf2/4, Tcl1, Tbx3, Esrrb, Lefty1/2, Prdm14 and Tcfcp2l1. Regulation of DNA methylation by Tet1-mediated conversion of 5mC to 5hmC also involves Tet1's regulation of Tet2 and Dnmt3b. Our findings suggest that studying mESCs in the hours/days following depletion of Tet1, before any compensatory alterations could take place, reveals Tet1's normal physiological role in maintaining the pluripotent state, which may be subject to homeostatic compensation in genetic models. Tet1/2 double knockout may shed light into whether Tet2 is in fact compensating for Tet1's loss in Tet1 null cells. Further investigations are needed to better understand the mechanistic role of Tet1 and 5hmC in facilitating pluripotency, and LIF/Stat3 signaling.
Figure 6.

Proposed model for Tet1-mediated epigenetic and transcriptional regulation of mESC self-renewal and pluripotency. Red arrows denote regulatory interactions inferred from data generated for this study. Tet1, regulated by Oct4 (28), regulates DNA methylation (5mC) by converting 5mC to 5hmC (16,28). Tet1 regulates LIF/Stat3 signaling by facilitating Stat3 binding by an yet to be determined mechanism, and regulates the transcriptional regulatory module comprising Nanog, Esrrb, Tcl1, Tbx3, Klf2/4, Prdm14 and Lefty1/2. Tet1's regulation of Tet2 confers tight regulation of 5mC to 5hmC conversion. Tet1's negative regulation of de novo DNA methyltransferase Dnmt3b may provide an additional layer of Tet1-mediated regulation of 5mC. Silent and active promoters on the chromatin are denoted by broad red and green arrows, respectively.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–4, Supplementary Tables 1–8, Supplementary Methods and Supplementary References [67–79].
FUNDING
Funding for open access charge: The Intramural Research Program of the NIH, National Institute of Environmental Health Sciences [1ZIAES102625-03 (to R.J.), 1ZIAES102745-02 (to G.H.) and 1ZIAES101965-07 (to P.A.W.)].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Lena Ho (A-Star) for helpful discussions on ChIP experiments and critical comments on the article, NIEHS/NISC sequencing facility for their advice and help in mapping 5hmC sites, and the NIEHS Microarray Core for microarray data generation. We also thank Karen Adelman, Georgette Charles, Senthilkumar Cinghu, Anne Lai, Xiaoling Li, Thomas Kunkel, Larry Lazarus, Daniel Menendez and Sailesh Surapureddi for critical comments on the article. R.J. conceived the study, and wrote the article. G.H. and R.J. designed the experiments with contributions from S.C. and P.A.W. G.H. directed the experiments. S.G., B.L., X.Z., R.L. and G.H. executed the experiments. J.M.F. and R.J. analyzed the data with contributions from S.Y. J.M.F., G.H. and R.J. interpreted the data. All the authors read and approved the article.
REFERENCES
- 1.Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Macarthur BD, Ma'ayan A, Lemischka IR. Systems biology of stem cell fate and cellular reprogramming. Nat. Rev. Mol. Cell Biol. 2009;10:672–681. doi: 10.1038/nrm2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nichols J, Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 4.Lessard JA, Crabtree GR. Chromatin regulatory mechanisms in pluripotency. Annu. Rev. Cell Dev. Biol. 2010;26:503–532. doi: 10.1146/annurev-cellbio-051809-102012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loh YH, Yang L, Yang JC, Li H, Collins JJ, Daley GQ. Genomic approaches to deconstruct pluripotency. Annu. Rev. Genomics Hum. Genet. 2011;12:165–185. doi: 10.1146/annurev-genom-082410-101506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng HH, Surani MA. The transcriptional and signalling networks of pluripotency. Nat. Cell Biol. 2011;13:490–496. doi: 10.1038/ncb0511-490. [DOI] [PubMed] [Google Scholar]
- 7.Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 10.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 11.Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell. 2008;133:1145–1148. doi: 10.1016/j.cell.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 12.Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2011;11:607–620. doi: 10.1038/nrm2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698–1710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veron N, Peters AH. Epigenetics: Tet proteins in the limelight. Nature. 2011;473:293–294. doi: 10.1038/473293a. [DOI] [PubMed] [Google Scholar]
- 18.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cimmino L, Abdel-Wahab O, Levine RL, Aifantis I. TET family proteins and their role in stem cell differentiation and transformation. Cell Stem Cell. 2011;9:193–204. doi: 10.1016/j.stem.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 25.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 27.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X, Li Y, Chen CH, Zhang W, Jian X, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu H, D'Alessio AC, Ito S, Xia K, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J, Barbera AJ, Zheng L, Zhang H, Huang S, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol. Cell. 2011;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu H, Zhang Y. Tet1 and 5-hydroxymethylation: a genome-wide view in mouse embryonic stem cells. Cell Cycle. 2011;10:2428–2436. doi: 10.4161/cc.10.15.16930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu H, D'Alessio AC, Ito S, Wang Z, Cui K, Zhao K, Sun YE, Zhang Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dawlaty MM, Ganz K, Powell BE, Hu YC, Markoulaki S, Cheng AW, Gao Q, Kim J, Choi SW, Page DC, et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell. 2011;9:166–175. doi: 10.1016/j.stem.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waalwijk C, Flavell RA. MspI, an isoschizomer of hpaII which cleaves both unmethylated and methylated hpaII sites. Nucleic Acids Res. 1978;5:3231–3236. doi: 10.1093/nar/5.9.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 43.Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smyth GK. In: Bioinformatics and Computational Biology Solutions using R and Bioconductor. Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- 45.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. J. Royal Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 46.Freudenberg JM, Joshi VK, Hu Z, Medvedovic M. CLEAN: CLustering Enrichment ANalysis. BMC Bioinformatics. 2009;10:234. doi: 10.1186/1471-2105-10-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu G, Kim J, Xu Q, Leng Y, Orkin SH, Elledge SJ. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009;23:837–848. doi: 10.1101/gad.1769609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glover CH, Marin M, Eaves CJ, Helgason CD, Piret JM, Bryan J. Meta-analysis of differentiating mouse embryonic stem cell gene expression kinetics reveals early change of a small gene set. PLoS Comput. Biol. 2006;2:e158. doi: 10.1371/journal.pcbi.0020158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho L, Ronan JL, Wu J, Staahl BT, Chen L, Kuo A, Lessard J, Nesvizhskii AI, Ranish J, Crabtree GR. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl Acad. Sci. USA. 2009;106:5181–5186. doi: 10.1073/pnas.0812889106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho L, Jothi R, Ronan JL, Cui K, Zhao K, Crabtree GR. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc. Natl Acad. Sci. USA. 2009;106:5187–5191. doi: 10.1073/pnas.0812888106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ho L, Miller EL, Ronan JL, Ho WQ, Jothi R, Crabtree GR. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat. Cell Biol. 2011;13:903–913. doi: 10.1038/ncb2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, Johnson BE, Fouse SD, Delaney A, Zhao Y, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010;28:1097–1105. doi: 10.1038/nbt.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hall J, Guo G, Wray J, Eyres I, Nichols J, Grotewold L, Morfopoulou S, Humphreys P, Mansfield W, Walker R, et al. Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell. 2009;5:597–609. doi: 10.1016/j.stem.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 54.Trouillas M, Saucourt C, Guillotin B, Gauthereau X, Ding L, Buchholz F, Doss MX, Sachinidis A, Hescheler J, Hummel O, et al. Three LIF-dependent signatures and gene clusters with atypical expression profiles, identified by transcriptome studies in mouse ES cells and early derivatives. BMC Genomics. 2009;10:73. doi: 10.1186/1471-2164-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Niwa H, Ogawa K, Shimosato D, Adachi K. A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature. 2009;460:118–122. doi: 10.1038/nature08113. [DOI] [PubMed] [Google Scholar]
- 56.Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, Schafer X, Lun Y, Lemischka IR. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 58.Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 59.Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR, Carey MF, Pradhan S. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20:3089–3103. doi: 10.1101/gad.1463706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, Tsangaratou A, Rajewsky K, Koralov SB, Rao A. Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl Acad. Sci. USA. 2011;108:14566–14571. doi: 10.1073/pnas.1112317108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, Masse A, Le Couedic JP, Pendino F, Carbonne B, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood. 2011;118:2551–2555. doi: 10.1182/blood-2010-12-324707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- 65.Iqbal K, Jin SG, Pfeifer GP, Szabo PE. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl Acad. Sci. USA. 2011;108:3642–3647. doi: 10.1073/pnas.1014033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334:194. doi: 10.1126/science.1212483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nishiyama A, Xin L, Sharov AA, Thomas M, Mowrer G, Meyers E, Piao Y, Mehta S, Yee S, Nakatake Y, et al. Uncovering early response of gene regulatory networks in ESCs by systematic induction of transcription factors. Cell Stem Cell. 2009;5:420–433. doi: 10.1016/j.stem.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 70.Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 72.Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell Biol. 2007;27:3769–3779. doi: 10.1128/MCB.01432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, Robson P, Zhong S, Ng HH. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat. Cell Biol. 2008;10:353–360. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- 74.Fazzio TG, Huff JT, Panning B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell. 2008;134:162–174. doi: 10.1016/j.cell.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lim CY, Tam WL, Zhang J, Ang HS, Jia H, Lipovich L, Ng HH, Wei CL, Sung WK, Robson P, et al. Sall4 regulates distinct transcription circuitries in different blastocyst-derived stem cell lineages. Cell Stem Cell. 2008;3: 543–554. doi: 10.1016/j.stem.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 76.Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell. 2008;32:491–502. doi: 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ding L, Paszkowski-Rogacz M, Nitzsche A, Slabicki MM, Heninger AK, de Vries I, Kittler R, Junqueira M, Shevchenko A, Schulz H, et al. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for embryonic stem cell identity. Cell Stem Cell. 2009;4:403–415. doi: 10.1016/j.stem.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 78.Leeb M, Pasini D, Novatchkova M, Jaritz M, Helin K, Wutz A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 2010;24:265–276. doi: 10.1101/gad.544410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yi F, Pereira L, Hoffman JA, Shy BR, Yuen CM, Liu DR, Merrill BJ. Opposing effects of Tcf3 and Tcf1 control Wnt stimulation of embryonic stem cell self-renewal. Nat. Cell Biol. 2011;13:762–770. doi: 10.1038/ncb2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All microarray and 5hmC sequencing data generated for this study were deposited in NCBI GEO repository under the accession number GSE34267.