

Abstract
Background
Forkhead transcription factors belonging to the FOXO subfamily are negatively regulated by protein kinase B (PKB) in response to signaling by insulin and insulin-like growth factor in Caenorhabditis elegans and mammals. In Drosophila, the insulin-signaling pathway regulates the size of cells, organs, and the entire body in response to nutrient availability, by controlling both cell size and cell number. In this study, we present a genetic characterization of dFOXO, the only Drosophila FOXO ortholog.
Results
Ectopic expression of dFOXO and human FOXO3a induced organ-size reduction and cell death in a manner dependent on phosphoinositide (PI) 3-kinase and nutrient levels. Surprisingly, flies homozygous for dFOXO null alleles are viable and of normal size. They are, however, more sensitive to oxidative stress. Furthermore, dFOXO function is required for growth inhibition associated with reduced insulin signaling. Loss of dFOXO suppresses the reduction in cell number but not the cell-size reduction elicited by mutations in the insulin-signaling pathway. By microarray analysis and subsequent genetic validation, we have identified d4E-BP, which encodes a translation inhibitor, as a relevant dFOXO target gene.
Conclusion
Our results show that dFOXO is a crucial mediator of insulin signaling in Drosophila, mediating the reduction in cell number in insulin-signaling mutants. We propose that in response to cellular stresses, such as nutrient deprivation or increased levels of reactive oxygen species, dFOXO is activated and inhibits growth through the action of target genes such as d4E-BP.
Background
Receptors for insulin and insulin-like growth factors (IGFs) are central regulators of energy metabolism and organismal growth in vertebrates and invertebrates. In mammals, the insulin receptor regulates glucose homeostasis and embryonic growth [1], whereas the insulin-like growth factor 1 receptor (IGF1-R) regulates embryonic and postembryonic growth [2] and longevity [3]. In Caenorhabditis elegans, DAF-2 – the homolog of the mammalian insulin/IGF receptor – controls organismal growth in response to poor nutrient conditions indirectly by controlling formation of the long-lived, stress-resistant dauer stage during larval development, and lifespan in the adult [4]. In Drosophila, the insulin/IGF receptor homolog DInr controls organismal growth directly by regulating cell size and cell number [5]. Furthermore, reduced insulin signaling causes female sterility and independently increases lifespan [6,7]. The striking conservation of insulin receptor function is also reflected in the conservation of the intracellular signaling cascade. Binding of insulin-like peptides to their receptor tyrosine kinases leads to the activation of class IA phosphatidylinositol (PI) 3-kinases and increased intracellular concentrations of the lipid second messenger phosphatidylinositol (3,4,5)-trisphosphate (PIP3). This results in recruitment to the membrane, and activation, of the protein kinases phosphoinositide-dependent protein kinase 1 (PDK1) and protein kinase B (PKB/AKT), both of which contain pleckstrin homology (PH) domains and which in turn modulate the activity of downstream effector proteins [8]. The lipid phosphatase PTEN (phosphatase and tensin homolog on chromosome 10) catalyzes the 3-dephosphorylation of PIP3, thereby acting as a negative regulator of insulin signaling [9]. The demonstration that the lethality associated with loss of dPTEN in Drosophila is rescued by a mutant form of dPKB with impaired affinity for PIP3 indicates that PKB is a key effector of this pathway [10]. Genetic and biochemical studies have identified two critical targets of PKB, namely forkhead transcription factors of the FOXO subfamily and the Tuberous Sclerosis Complex 2 (TSC2) tumor suppressor protein.
In C. elegans, the only FOXO transcription factor is encoded by daf-16. Loss-of-function mutations in daf-16 completely suppress the dauer-constitutive and longevity phenotypes associated with reduced function of insulin-signaling components. On the basis of knowledge about DAF signaling in C. elegans, forkhead transcription factors belonging to the FOXO subfamily have been identified as direct targets of insulin/IGF signaling in mammals [11-13]. The mammalian DAF-16 homologs comprise the proteins FOXO1 (FKHR), FOXO3a (FKHRL1) and FOXO4 (AFX). Their phosphorylation by the insulin-activated kinases PKB and serum- and glucocorticoid-regulated protein kinase (SGK) creates binding sites for 14-3-3 proteins, and this leads to inactivation of FOXO proteins via cytoplasmic sequestration [12,14]. The result of this process is an insulin-induced transcriptional repression of FOXO target genes, which are involved in the response to DNA damage [15] and oxidative stress [16,17], apoptosis [12,18], cell-cycle control [19-21] and metabolism [22]. In addition to their transcriptional activation capabilities, FOXO proteins have recently been shown to induce cell-cycle arrest by repressing transcription of genes encoding D-type cyclins [23,24]. FOXO transcription factors mediate insulin resistance in diabetic mice [25], and have been proposed to be tumor suppressors, as several chromosomal translocations disrupting FOXO genes are found in cancers [26,27], and overexpressed FOXO proteins can inhibit tumor growth [23].
TSC2, the second target of PKB, forms a complex with TSC1 and acts as a negative regulator of growth in Drosophila, and as a tumor suppressor in mammals. Overexpressed activated PKB phosphorylates TSC2 and thereby disrupts the TSC1/2 complex in Drosophila and in mammalian cells [28,29]. In Drosophila, the TSC1/2 complex functions by negatively regulating two kinases, dTOR (homolog of the mammalian target of rapamycin) [30] and dS6K (homolog of the mammalian ribosomal protein S6 kinase) [31]. Recent genetic and biochemical evidence indicates that TSC1/2 regulates S6K activity by acting as a GTPase-activating protein (GAP) for the small GTPase Rheb [32-35]. Interestingly, flies lacking dS6K function are reduced in size because of a reduction in cell size but not in cell number [36]. The growth control pathways regulating cell size and cell number therefore bifurcate either at dPKB or between dPKB and dS6K.
In this study, we describe the identification of dFOXO, the single FOXO ortholog in Drosophila. Although dFOXO function is not essential for development and organismal growth control under normal culture conditions, it mediates the reduction in cell number associated with reduced insulin signaling. Our results show that dFOXO regulates expression of d4E-BP, which mediates part of the cell-number reduction in dPKB mutants. We propose that dFOXO upregulates d4E-BP transcription under conditions of low insulin signaling. Furthermore, our observations suggest that dFOXO is required for resistance against oxidative stress in adult flies.
Results
dFOXO is the only Drosophila homolog of FOXO and DAF-16
The Drosophila genome contains a single homolog of the DAF-16/FOXO family of transcription factors. This notion is supported by the phylogenetic tree diagram calculated from the multiple sequence alignment (Figure 1a). The dFOXO gene is more closely related to the mammalian FOXO subfamily and daf-16 than any other Drosophila forkhead gene. The amino-acid sequences of the predicted 613 amino-acid dFOXO protein and hFOXO3a are 27% identical over the full protein length, and 82% identical within the forkhead DNA-binding domain. Furthermore, dFOXO is the only Drosophila forkhead gene encoding a putative protein containing conserved PKB phosphorylation sites [37]. The orientation of the three PKB consensus sites relative to the forkhead domain (Figure 1b) is conserved among the mammalian FOXO proteins, DAF-16 and dFOXO. Figure 1c shows the high degree of sequence conservation between dFOXO and FOXO/DAF-16 proteins within the DNA-binding domain. Taken together, these observations strongly suggest that dFOXO is the only Drosophila homolog of the mammalian FOXO transcription factors and C. elegans DAF-16.
Figure 1.
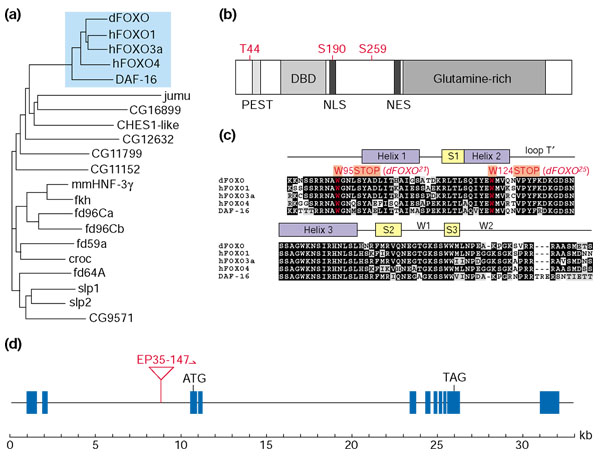
dFOXO is the only Drosophila FOXO/DAF-16 homolog. A TBLASTN search of the Drosophila genome for known and predicted genes encoding forkhead transcription factors retrieved 16 genes. (a) A phylogenetic tree calculated from a multiple sequence alignment of the forkhead domains of these 16 proteins and of the human FOXO proteins FOXO1 (FKHR), FOXO3a (FKHRL1) and FOXO4 (AFX), the C. elegans DAF-16 and mouse Foxa3 (HNF-3γ; protein names on the figure are from GenBank). The similarity of dFOXO to FOXO proteins is highlighted in blue. (b) dFOXO has three PKB phosphorylation sites in the same orientation as those of mammalian FOXO proteins. The sites are indicated above the protein; PEST (destruction), nuclear localization (NLS), nuclear export (NES) and DNA-binding sequences are also shown. (c) A multiple amino-acid sequence alignment of the dFOXO, human FOXO and DAF-16 forkhead domains illustrates the high degree of sequence conservation especially within the DNA-binding domain. The secondary structure is indicated above the alignment. Similar and identical amino-acid residues are shaded in gray and black, respectively. The region encoding helix 3 of the forkhead domain, which is the DNA-recognition helix contacting the major groove of the DNA double helix, is identical in the five proteins. Given the high structural similarity between the DNA-binding domains of FOXO4 (AFX) and HNF-3γ [85], it is likely that FOXO proteins contact insulin response elements through helix 3. Two EMS-induced point mutations described in this study are shown in red. (d) The dFOXO gene spans a genomic region of 31 kilobases (kb) and contains 11 exons (blue bars). The EP35-147 transposable element is inserted in the second intron upstream of the open reading frame, allowing GAL4-induced expression of endogenous dFOXO.
Overexpressed dFOXO is responsive to insulin signaling and nutrient levels, inducing organ-size reduction and cell death
To assess whether dFOXO has a key function in insulin signaling like that of DAF-16 in C. elegans, we tested whether overexpression of wild-type or mutant forms of hFOXO3a and dFOXO could antagonize insulin signaling. Elimination of the three PKB consensus phosphorylation sites in mammalian FOXO3a prevents its phosphorylation, subsequent binding to 14-3-3 proteins, and sequestration in the cytoplasm [12]. This leads to constitutive nuclear localization of the mutant FOXO3a and transcriptional activation of its target genes. Assuming that blocking the PKB signal would have the same activating effect on dFOXO, we overexpressed wild-type and triple PKB-phosphorylation-mutant variants of both dFOXO and human FOXO3a. Furthermore, we identified an EP transposable element insertion in the second dFOXO intron, which permits the GAL4-induced overexpression of endogenous dFOXO (Figure 1d). We used the GMR-Gal4 construct to drive UAS-dependent expression in postmitotic cells in the eye imaginal disc [38]. While expression of wild-type hFOXO3a in the developing eye did not result in a visible phenotype (Figure 2b), hFOXO3a-TM expression caused pupal lethality. Few escaper flies eclosed and displayed a strong necrotic eye phenotype (Figure 2c). A block of cell differentiation and necrosis was also observed when hFOXO3a-TM was expressed in cell clones in the developing eye (Figure 2d).
Figure 2.
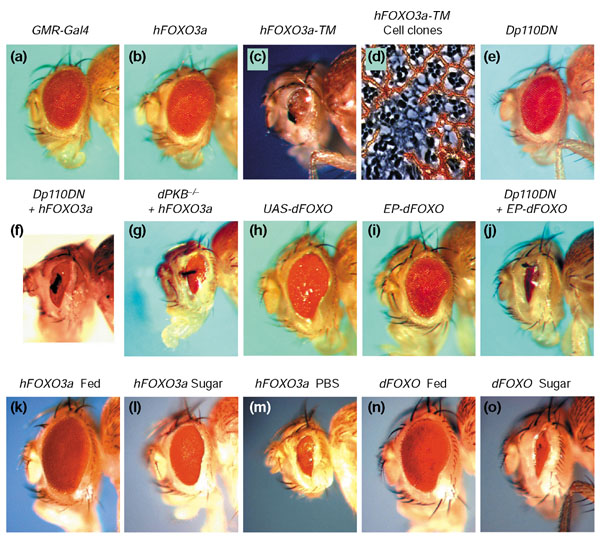
Targeted hFOXO3a and dFOXO expression in the developing Drosophila eye induces organ-size reduction and cell death, and the phenotypes are sensitive to insulin signaling and nutrient levels. (a) GMR-Gal4- expressing control fly. (b) No discernible phenotype results from hFOXO3a expression. (c) Expression of hFOXO3a-TM in the eye disc leads to pupal lethality; escapers at 18°C show a necrotic phenotype and severely disrupted cell specification. (d) Expression in w--marked clones of cells induces a similar phenotype at 25°C. (e) Dp110DN expression slightly reduces eye size, and (f) co-expression of wild-type hFOXO3a partially mimicks the hFOXO3a-TM escaper phenotype. (g) The same enhancement of hFOXO3a activity was observed in a dPKB-/- background. (h,i) Expression of transgenic or endogenous dFOXO results in a small-eye phenotype, which is also dramatically enhanced by (j) Dp110DN. (k-o) hFOXO3a and dFOXO phenotypes are progressively exacerbated by protein deprivation ('sugar') and complete starvation ('PBS'). Flies like the one shown in (m) die within one day, and complete starvation of dFOXO-expressing flies resulted in pupal lethality (not shown). Genotypes are: (a) y w; GMR-Gal4/+; (b) y w; GMR-Gal4/+; UAS-hFOXO3a/+; (c) y w; GMR-Gal4/+; UAS-hFOXO3a-TM/+; (d) y w hs-flp/y w; GMR >FRT-w+ STOP - FRT >Gal-4/+; UAS-hFOXO3a-TM/+; (e) y w; GMR-Gal4 UAS-Dp110DN/+; (f) y w; GMR-Gal4 UAS-Dp110DN/+; UAS-hFOXO3a/+; (g) y w; UAS-hFOXO3a/GMR-Gal4; dPKB3/dPKB1; (h) y w; UAS-dFOXO/GMR-Gal4; (i) y w; GMR-Gal4/+; EP-dFOXO/+; (j) y w; GMR-Gal4 UAS-Dp110DN/+; EP-dFOXO/+; (k-m) y w; GMR-Gal4/+; UAS-hFOXO3a/+; (n,o) y w; GMR-Gal4/+; EP-dFOXO/+.
Assuming that the lack of a phenotype observed upon UAS-hFOXO3a expression is due to hFOXO3a inactivation by endogenous DInr signaling in the eye disc, we performed the same experiment in a background of reduced insulin signaling. Indeed, in the presence of a dominant-negative (DN) form of Dp110 (encoding the PI 3-kinase catalytic subunit) [39], hFOXO3a expression induced a necrotic phenotype similar to the one observed with the hyperactive phosphorylation mutant (Figure 2f). To confirm that hFOXO3a is responsive to Drosophila insulin signaling and rule out artificial coexpression effects, we expressed hFOXO3a in flies mutant for either dPKB (Figure 2g) or Dp110 (not shown), and observed similar phenotypes to those seen upon coexpression of Dp110DN. Drosophila FOXO has qualitatively similar, but stronger effects. Expressing the wild-type form of dFOXO causes a weak eye-size reduction and disruption of the ommatidial pattern even in a wild-type background (Figure 2h,i), and the phenotype is strongly affected by Dp110DN as well (Figure 2j). The UAS-dFOXO-TM transgene appears to cause lethality even in the absence of a Gal4 driver, as we did not obtain viable transgenic lines with this construct. Furthermore, we examined the effects of nutrient deprivation on FOXO-expressing tissues. If nutrient availability is limited, FOXO should be more active in response to lowered insulin signaling. Indeed, we observed that the overexpression phenotypes of both hFOXO3a and dFOXO are enhanced under conditions of starvation. Drosophila larvae that are starved until 70 h after egg laying (AEL) die within a few days. But if the onset of nutrient deprivation occurs after they have surpassed the metabolic '70 h change' [40,41], they survive and develop into small adult flies. We therefore subjected larvae expressing hFOXO3a or dFOXO (under GMR control) to either protein starvation (sugar as the only energy source) or complete starvation, starting 80–90 h AEL, and analyzed the effect on the adult's eyes. Both phenotypes (Figure 2k,n) were progressively exacerbated by protein starvation (Figure 2l,o) and complete starvation (Figure 2m), the latter condition being accompanied by early adult or larval lethality, in the case of hFOXO3a or dFOXO, respectively. The resulting phenotypes are due to the FOXO transgenes, as wild-type control flies that have been starved during development display only a body-size reduction while maintaining normal proportions and normal eye structure.
The dFOXO overexpression phenotype (Figure 2i,j) does not appear to be caused by the activation of any of the known cell-death pathways. Expression of the caspase inhibitors p35 or DIAP1, or of p21, an inhibitor of p53-induced apoptosis [42], and loss of eiger, which encodes the Drosophila homolog of tumor necrosis factor (TNF) [43], did not suppress the eye phenotype (data not shown). In agreement with our results, it was observed in a parallel study that the GMR-dFOXO overexpression phenotype is insensitive to caspase inhibitors, and is not accompanied by increased acridine-orange-detectable apoptosis in the imaginal disc [44]. It therefore remains unclear whether high levels of nuclear dFOXO induce a specific caspase-independent cell-death program or whether nuclear accumulation of overexpressed dFOXO leads to secondary necrosis in a rather nonspecific fashion. Furthermore, the necrotic eye phenotype does not reflect the phenotype observed following a complete block in insulin signaling. Loss-of-function mutations in insulin-signaling components reduce cell size and cell number but do not increase cell death in larval tissues [45,46]. In summary, our overexpression experiments are consistent with a model in which, under normal conditions, excess FOXO transcription factor is phosphorylated by dPKB and kept inactive in the cytoplasm. Under conditions of reduced insulin-signaling activity or nutrient deprivation, dFOXO or hFOXO3a protein translocates to the nucleus and induces growth arrest and necrosis.
dFOXO loss-of-function mutants are viable, have no overgrowth phenotype and are hypersensitive to oxidative stress
Although the overexpression experiments described above did not reveal the physiological function of dFOXO, they provided the entry point for isolation of loss-of-function mutations. We made use of the EP35-147 element, which permits the generation of the necrotic eye phenotype (Figure 2j) by driving expression of endogenous dFOXO in the presence of Dp110DN. We mutagenized homozygous EP males, mated them to homozygous GMR-Gal4 UAS-Dp110DN females and then screened the F1 generation for reversion of the strong gain-of-function phenotype and its associated semilethality. Several loss-of-function alleles of dFOXO were isolated and molecularly characterized. Two such revertants are shown in Figure 3c (dFOXO21) and Figure 3d (dFOXO25). In dFOXO21 and dFOXO25, the codons for W95 and W124 within the forkhead domain are mutated to stop codons, respectively (Figure 1c), so they are assumed to be null alleles of dFOXO. We performed the subsequent phenotypic and epistasis analyses with these two lines.
Figure 3.
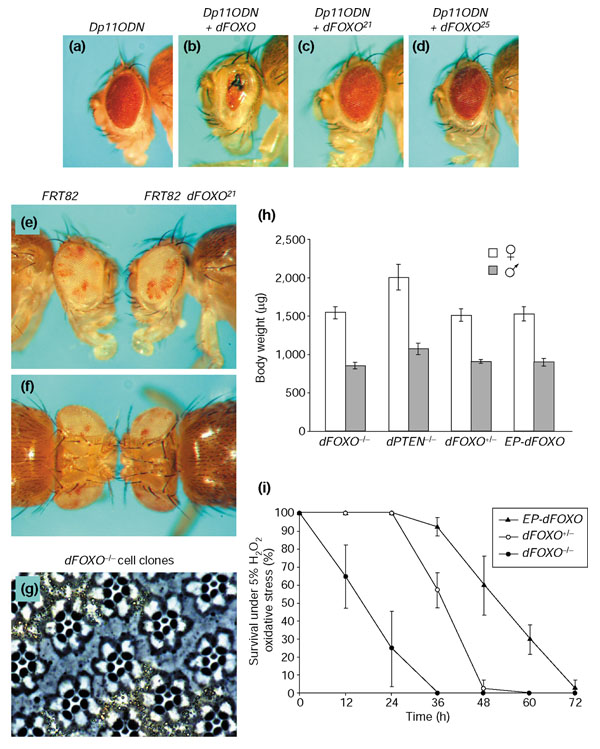
Null dFOXO mutants are viable, have no overgrowth phenotype and are hypersensitive to oxidative stress. (a) Dp110DN expressing control fly. (b) EP-driven coexpression of dFOXO elicits a necrotic eye phenotype. (c,d) EMS-induced mutations in dFOXO lead to a reversion of the overexpression phenotype. (e,f) Selective removal of dFOXO from the head (right) does not lead to an organ-size alteration compared to a control fly (left). (g) w--marked dFOXO-deficient photoreceptor cells are the same size as wild-type cells. (h) In contrast to dPTEN, dFOXO null mutants have no organismal growth phenotype. For each genotype, the left bar indicates the body weight of females and the right bar the weight of males. Values are shown ± standard deviation (SD). (i) dFOXO mutants are hypersensitive to oxidative stress. The graph shows a survival curve of male adult flies on PBS/sucrose gel containing 5% hydrogen peroxide. The observed hypersensitivity is more pronounced in males, but is also observed in females (not shown). The increased resistance of homozygous EP-dFOXO flies might be caused by low basal dFOXO overexpression from the EP element, which occurs due to leakiness of UAS enhancers in the absence of Gal4. Control flies placed on PBS/sucrose without oxidant survived during the time window shown. Genotypes are: (a) y w; GMR-Gal4 UAS-Dp110DN/+; (b) y w; GMR-Gal4 UAS-Dp110DN/+; EP-dFOXO/+; (c) y w; GMR-Gal4 UAS-Dp110DN/+; EP-dFOXO21/+; (d) y w; GMR-Gal4 UAS-Dp110DN/+; EP-dFOXO25/+; (e,f) y w ey-flp/y w; FRT82/FRT82 cl3R3 w+ (left); y w ey-flp/y w; FRT82 EP-dFOXO21/FRT82 cl3R3 w+ (right); (g) y w hs-flp/y w; FRT82 EP-dFOXO21/FRT82 w+.
Because FOXO transcription factors have been proposed to be the primary effectors of insulin signaling, on the basis of epistasis of daf-16 over daf-2 in C. elegans, it seemed reasonable to expect an overgrowth phenotype in dFOXO-/- flies as is observed in dPTEN loss-of-function mutants. To our surprise, dFOXO loss-of-function mutants are homozygous-viable and display no obvious phenotype under normal culturing conditions (Figure 3h). Thus, dFOXO is not essential for development. Only close inspection of the dFOXO mutants revealed that their wing size is significantly reduced (Figure 4i). But cellular and organismal growth are unaffected by dFOXO mutations. To assess whether dFOXO-mutant tissue grows to a different size than wild-type tissue, we recombined the dFOXO21 and dFOXO25 alleles onto the FRT82 chromosome and induced genetic mosaic flies with the ey-Flp/FRT system [47]. When the eye and head capsule were composed almost exclusively of dFOXO-/- tissue (w--marked in Figure 3e,f, on the right), no head-size difference was observed compared to the control fly with a head homozygous for the FRT82 chromosome without the dFOXO mutation (Figure 3e,f, left). This is consistent with experience from extensive genetic screens for recessive growth mutations carried out in our lab. An ey-Flp-screen on the right arm of chromosome 3 did not reveal any mutations in dFOXO based on an altered head-size phenotype (H.S. and E.H., unpublished observations).
Figure 4.
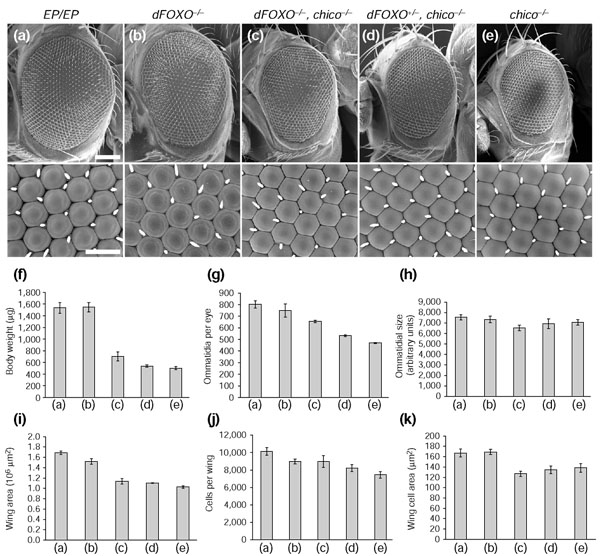
Loss of dFOXO suppresses the cell-number reduction in chico mutants. (a-e) Partial rescue of the chico phenotype by mutations in dFOXO. Bar sizes are 100 μm (low magnification) and 20 μm (high magnification). Each graph displays the variation of a single parameter between the five genotypes shown in (a-e): (f) body weight, (g) cell number in the eye, (h) cell size in the eye, (i) wing area, (j) cell number in the wing, and (k) cell size in the wing. (f) dFOXO-/-partially suppresses the low-body-weight phenotype of chico-/-. The suppression is less pronounced in the wing (i), because dFOXO-null mutants have significantly smaller wings than control flies, although their body weight is the same. In a chico-/- background, loss of dFOXO leads to increased cell numbers in the eye (g) and in the wing (j) compared to the chico single mutant. Although organ and tissue size is increased, cell size significantly decreases in the chico-dFOXO double mutant both in the eye (h) and in the wing (k). It seems that loss of dFOXO in a chico-/- background leads to increased proliferation rates. All values are shown ± SD. Genotypes are: (a) y w;; EP-dFOXO/EP-dFOXO; (b) y w;; EP-dFOXO21/EP-dFOXO25; (c) y w; chico1/chico2; EP-dFOXO21/+; (d) y w; chico1/chico2; EP-dFOXO21/ EP-dFOXO25; (e) y w; chico1/chico2.
We next asked whether cell size, like organ size, was not affected by the loss of dFOXO. For this purpose, we used a heat shock-inducible Flp construct to generate clones of homozygous dFOXO-/-photoreceptor cells and wild-type cells within one adult eye (Figure 3g). The cells lacking dFOXO are marked by the absence of pigment granules. Consistent with the absence of a 'bighead' phenotype, dFOXO-/- cells and wild-type cells have the same size. Similarly, no significant difference in the body weight of mutant and control flies was observed (Figure 3h). In contrast, flies with a viable heteroallelic combination of dPTEN loss-of-function alleles are significantly bigger than wild-type flies [48]. Taken together, these results argue that with the exception of the slight wing-size reduction, dFOXO is not required to control cellular, tissue, or organismal growth in a wild-type background.
A critical role has been reported for mammalian and C. elegans FOXO proteins in resistance against various cellular stresses, in particular oxidative stress [16,17,49], DNA damage [15] and cytokine withdrawal [50]. We tested the stress resistance of adult dFOXO mutant flies by measuring survival time following different challenges. Among starvation on water, oxidative-stress challenge, bacterial infection, heat shock, and heavy-metal stress, the only condition for which hypersensitivity was observed is oxidative stress. When placed on hydrogen-peroxide-containing food, dFOXO mutant flies display a significantly reduced survival time compared to control flies (Figure 3i). A very similar effect is elicited by paraquat feeding. These observations are consistent with the paraquat hypersensitivity of daf-16 mutants in C. elegans [51], suggesting that a role for FOXO proteins in protecting against oxidative stress is conserved across species.
The growth-deficient phenotypes of Dlnr, chico, Dp110 and dPKB mutants are significantly suppressed by loss of dFOXO
We performed genetic epistasis experiments to examine whether the growth phenotypes of DInr-signaling mutants are dependent on dFOXO function. For this purpose, we either generated double-mutant flies or investigated the double-mutant effect only in the head using the ey-Flp/FRT system. In contrast to the absence of a growth phenotype in single dFOXO mutant flies, lack of dFOXO significantly suppresses the growth-deficient phenotype observed in flies mutant for the insulin receptor substrate (IRS) homolog chico (Figure 4). Flies mutant for chico are smaller because they have fewer and smaller cells [45]. Loss of one dFOXO copy dominantly suppresses the cell-number reduction in chico mutant flies without affecting cell size. The suppression is more pronounced when both copies of dFOXO are removed in a chico mutant background. In this situation, the chico small body-size phenotype is partially suppressed. Homozygous chico-dFOXO double-mutant flies have more, and even slightly smaller, cells than homozygous chico single mutants. It seems that removal of dFOXO accelerates the cell cycle at the expense of cell size in a chico background.
We next asked whether dFOXO interacts with other components of the Drosophila insulin-signaling pathway. The ey-Flp/FRT system was used to generate heterozygous insulin-signaling mutant flies with heads homozygous for each mutation. Removal of DInr, Dp110 or dPKB leads to a characteristic 'pinhead' phenotype, which is substantially suppressed by the presence of a dFOXO loss-of-function allele on the same FRT chromosome as the insulin-signaling mutation. In all three cases, we observed a partial rather than a complete rescue of the tissue growth repression, consistent with the finding that dFOXO mutations affect only the cell-number aspect of the chico phenotype. Surprisingly, loss of dFOXO dramatically delays lethality in dPKB mutants. Complete loss of dPKB leads to larval lethality in the early third instar, but homozygous dPKB-dFOXO double mutants are able to develop into pharate adults of reduced size, most of which fail to eclose (Figure 5l). The lethality associated with the complete loss of dPKB is therefore largely due to hyperactivation of dFOXO.
Figure 5.
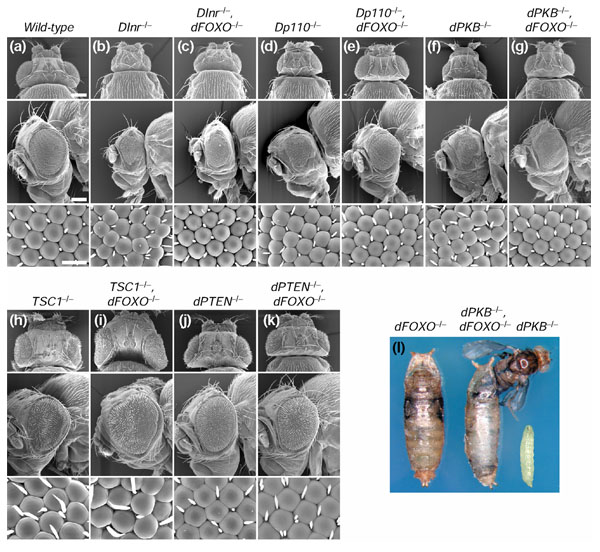
Growth-deficient phenotypes of DInr, Dp110 and dPKB mutants are suppressed by loss of dFOXO. (a) Control fly. (b) Selective removal of DInr from the head leads to a pinhead phenotype, which is partially suppressed by the loss of dFOXO (c). The same suppression is observed in Dp110-, and dPKB-pinheads (d-g). The TSC1-/- bighead phenotype (h) is enhanced by mutations in dFOXO (i), but the dPTEN-/- bighead (j) is slightly suppressed (k). (l) Living without PKB. In contrast to the larval lethality of dPKB null mutants, dPKB-dFOXO double mutants develop into small pharate adults, most of which fail to eclose. Bar sizes are 200 μm (low magnification) and 20 μm (high magnification). Genotypes are: (a) y w ey-flp/y w; FRT82/FRT82 cl3R3 w+; (b) y w ey-flp/y w; FRT82 DInr304/FRT82 cl3R3 w+; (c) y w ey-flp/y w; FRT82 DInr304 EP-dFOXO25/FRT82 cl3R3 w+; (d) y w ey-flp/y w; FRT82 Dp1105W3/FRT82 cl3R3 w+; (e) y w ey-flp/y w; FRT82 Dp1105W3 EP-dFOXO25/FRT82 cl3R3 w+; (f) y w ey-flp/y w; FRT82 dPKB1/FRT82 cl3R3 w+; (g) y w ey-flp/y w; FRT82 dPKB1EP-dFOXO25/FRT82 cl3R3 w+; (h) y w ey-flp/y w; FRT82 dTSC1Q87X/FRT82 cl3R3 w+; (i) y w ey-flp/y w; FRT82 dTSC1Q87X EP-dFOXO25/FRT82 cl3R3 w+; (j) y w ey-flp/y w; FRT40 dPTEN117-4/FRT40 cl2L3 w+; (k) y w ey-flp/y w; FRT40 dPTEN117-4/FRT40 cl2L3 w+; FRT82 EP-dFOXO25/FRT82 cl3R3 w+; (l) y w;; EP-dFOXO21/EPdFOXO25 (left), y w;; dPKB1EP-dFOXO21/dPKB1EP-dFOXO25 (middle), dPKB1/dPKB1 (right).
We also observed that dFOXO interacts with the tumor suppressors dTSC1 and dPTEN. Tissue-specific removal of either gene from the head leads to a bighead phenotype (Figure 5h,j). The dTSC1-/- bighead phenotype is enhanced by loss of dFOXO (Figure 5i). This observation is consistent with the recently reported negative feedback loop between dS6K and dPKB. Mutant dTSC1 larvae have elevated levels of dS6K activity, which in turn downregulates dPKB activity [31]. This reduction in dPKB activity probably leads to enhanced activation of dFOXO, which in turn partially mitigates the overgrowth phenotype by slowing down proliferation. The dTSC1 phenotype can therefore be enhanced by loss of the inhibitory function of dFOXO. Unexpectedly, the dPTEN-/- bighead phenotype was slightly suppressed by dFOXO mutations (Figure 5k). From the current model, it would be expected that in a dPTEN mutant dPKB activity is high and dFOXO is to a large extent inactive in the cytoplasm. Thus, removal of dFOXO function should have no effect on the dPTEN phenotype. At present, we can only speculate about possible explanations for this observation. In a parallel study, it has been shown that dFOXO can induce transcription of DInr [52]. It may be that in a dPTEN-mutant background dFOXO activates DInr expression in a negative-feedback loop. In this model, concomitant loss of dFOXO would alleviate the dPTEN overgrowth phenotype by lowering DInr levels. Another possible explanation is that dFOXO has additional functions when localized to the cytoplasm or during its nuclear export, such as interacting with other proteins. Loss of dFOXO might affect the function of interaction partners that have a role in dPTEN signaling.
In summary, our epistasis analysis provides strong genetic evidence that dFOXO is required to mediate the organismal growth arrest that is elicited in insulin-signaling mutants.
dFOXO upregulates transcription of the d4E-BP gene
We have shown previously that Drosophila embryonic Kc167 cells respond to insulin stimulation with upregulated activities of dPKB and dS6K [53,54]. We performed mRNA profiling experiments using the Affymetrix GeneChip system to measure on a genome-wide scale the transcriptional changes induced by insulin in these cells. On the basis of the currently held model that FOXO transcription factors are transcriptional activators that are negatively regulated by insulin, we expected potential dFOXO target genes to be repressed in Kc167 cells upon insulin stimulation. Figure 6a shows a selection of dFOXO target gene candidates that are transcriptionally downregulated by a factor of two or more upon insulin stimulation and whose promoter regions contain one or more conserved forkhead-response elements (FHREs) with the consensus sequence (G/A)TAAACAA [55]. Three of these candidate gene products are each involved in one of two biological processes known to be negatively regulated by insulin, namely gluconeogenesis (PEPCK) and lipid catabolism (CPTI and long-chain-fatty-acid-CoA-ligase). The remaining candidates are involved in stress responses (cytochrome P450 enzymes), DNA repair (DNA polymerase iota), transcription and translation control (d4E-BP and CDK8), and cell-cycle control (centaurin gamma and CG3799). Several of the insulin-repressed genes have been reported to be transcriptionally induced in Drosophila larvae under conditions of complete starvation (d4E-BP and PEPCK) or sugar-only diet (CPTI and long-chain-fatty-acid-CoA-ligase) [41,56].
Figure 6.
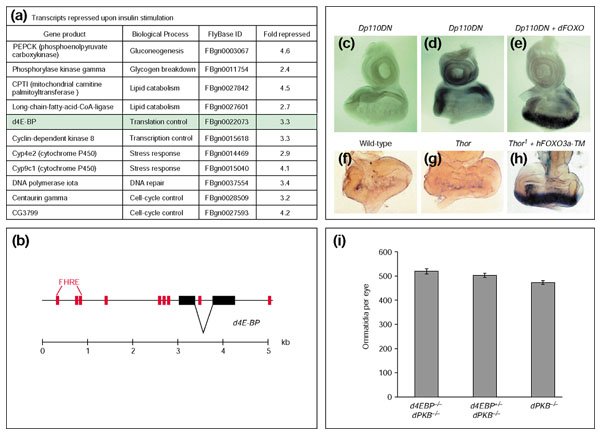
dFOXO regulates transcription of the d4E-BP gene. (a) A selection of microarray-identified genes that are transcriptionally downregulated after 2 h of insulin stimulation in Kc167 cells and contain forkhead response elements (FHREs) in their genomic upstream or intronic sequences. (b) FHREs (red) at the d4E-BP locus; black boxes are exons. (c,d) Overexpression of Dp110DN alone does not induce transcription of d4E-BP in imaginal discs, but (e) coexpression of dFOXO strongly upregulates the gene. (f-h) Expression of human FOXO3a-TM induces expression of the d4E-BP enhancer trap line Thor1. (i) d4E-BP and dPKB interact genetically. The Thor1 mutation increases the ommatidial number in dPKB-mutants by 9% without affecting cell size. Values are shown ± SD. Genotypes are: (c) y w; GMR-Gal4 UAS-Dp110DN/+; (d) y w; GMR-Gal4 UAS-Dp110DN/+; (e) y w; GMR-Gal4 UAS-Dp110DN/+; EP-dFOXO/+; (f) y w; (g) y w; Thor1/+; (h) y w; Thor1/GMR-Gal4; UAS-hFOXO3a-TM/+; (i) from right to left: y w;; dPKB3/dPKB1, y w; Thor1/+; dPKB3/dPKB1, y w; Thor1/Thor1; dPKB3/dPKB1.
We chose d4E-BP for further investigation, because it has previously been reported to be insulin-regulated at the level of protein phosphorylation, but not at the level of gene expression [57]. The d4E-BP gene encodes a translational repressor and was initially identified as the immune-compromised Thor mutant in a genetic screen for genes involved in the innate immune response to bacterial infection [58,59]. Figure 6b shows the presence of several FHREs in the genomic region around the d4E-BP locus. The d4E-BP protein is negatively regulated by insulin through LY294002- and rapamycin-sensitive phosphorylation [57], suggesting involvement of the Dp110 and dTOR signaling pathways. Phosphorylation of d4E-BP leads to the dissociation of d4E-BP from its binding partner, the translation initiation factor deIF4E, which then participates in the formation of a functional initiation complex. Positive transcriptional regulation of d4E-BP by dFOXO, which corresponds to negative transcriptional regulation by insulin, would be a complementary mechanism of regulation.
We then investigated whether overexpression of endogenous dFOXO could induce transcriptional upregulation of the d4E-BP gene. On the basis of our overexpression results, we chose the Dp110DN-dFOXO coexpression to efficiently activate dFOXO. Eye imaginal discs from Dp110DN-expressing third instar larvae display a low level of basal d4E-BP transcription throughout the disc, which is not induced by the driver construct alone (Figure 6d). Coexpression of dFOXO elicited a dramatic upregulation of d4E-BP transcription posterior to the morphogenetic furrow (Figure 6e). Consistent with this observation, we were able to induce expression of the d4E-BP enhancer trap line Thor1 with human FOXO3a-TM (Figure 6f-h). It remained unclear, however, whether regulation of d4E-BP expression by dFOXO is of physiological relevance.
It has been previously reported that overexpression of d4E-BP partially suppresses the dPKB overexpression phenotype [57], but as ectopic expression experiments have to be interpreted with some caution, we assessed whether loss of d4E-BP function suppresses the cell-number reduction in insulin-signaling mutants as does loss of dFOXO function. We generated double-mutant flies for dPKB and d4E-BP and observed that the Thor1 mutation slightly but significantly suppressed the reduced cell-number phenotype in a dose-dependent manner. The Thor1 mutation itself had no effect on ommatidial number compared to wild-type flies (data not shown), so we can rule out additive effects of d4E-BP and dPKB. These observations strongly argue that under conditions of reduced insulin-signaling activity the dFOXO-dependent reduction in cell number is in part mediated by the transcriptional upregulation of its target d4E-BP. Microarray studies in both mammalian [23] and Drosophila [52] cells imply that FOXO transcription factors exert their physiological functions by modulating expression of large sets of target genes.
Discussion
Forkhead transcription factors of the FOXO subfamily mediate insulin-regulated gene expression in C. elegans and mammals. In this study, we provide genetic evidence that the Drosophila FOXO/DAF-16 homolog dFOXO is an important downstream effector of Drosophila insulin signaling and a regulator of stress resistance.
dFOXO is a critical target of dPKB but mediates only part of its function
Genetic studies in C. elegans and Drosophila have led to two models regarding the output of the insulin pathway. First, the complete epistasis of daf-16 over the insulin pathway mutants daf-2, age-1, akt-1 and akt-2 suggests that the primary function of PKB is to inactivate FOXO transcription factors [60]. Second, it has been proposed that the TSC tumor suppressor complex is the major target of PKB [61,62] in the regulation of cell growth in Drosophila. Our analysis of Drosophila FOXO indicates that it is indeed a critical PKB target, but that it mediates only one aspect of PKB function. Several lines of evidence support this model. Firstly, the effects of ectopic overexpression of dFOXO and hFOXO3a in the developing Drosophila eye are altered by Dp110 and dPKB signaling as well as by nutrient levels. Under conditions of lowered insulin signaling, the phenotypes resulting from expression of dFOXO and hFOXO3a were dramatically enhanced. This situation was mimicked by expressing a dPKB-insensitive phosphorylation mutant, suggesting that endogenous dPKB signaling is required to mitigate the effects of ectopically expressed dFOXO and hFOXO3a. Secondly, the physiological relevance of dFOXO in dPKB signaling is most vividly demonstrated by our observation that the larval lethality associated with the complete loss of dPKB is rescued by dFOXO mutations to the extent that some flies develop to pharate adults. The lethality associated with loss of dPKB function is therefore to a large extent due to the hyperactivation of dFOXO. Thirdly, loss of dFOXO function suppresses the effects of insulin-signaling mutations only partially; dFOXO mediated a reduction in cell number but not in cell size in response to reduced insulin signaling.
dFOXO controls the reduction in cell number in body-size mutants
Genetic analysis of the control of body size in Drosophila has revealed two classes of mutations. Flies carrying mutations in chico or viable allelic combinations of DInr, Dp110, and dPKB are reduced in body size by up to 50% owing to a reduction in both cell size and cell number. Conversely, flies mutant for dS6K exhibit a more moderate reduction in body size, caused almost exclusively by a reduction in cell size [36]. This suggests that the pathways controlling cell number and cell size bifurcate at or below dPKB. Although dFOXO single mutants have no obvious size phenotype, loss of dFOXO substantially suppresses the cell-number reduction observed in insulin-signaling mutants. It appears that dFOXO mediates the repression of proliferation in flies mutant for DInr, chico, Dp110, and dPKB without being required for the reduction in cell size. Chico-dFOXO double mutant flies even have slightly smaller cells than chico mutants, suggesting that removal of dFOXO permits cell-cycle acceleration under conditions of impaired insulin signaling. The pathway controlling body size in response to insulin therefore bifurcates at the level of dPKB: dPKB controls cell number by inhibiting dFOXO function and dPKB controls cell size, at least under some conditions, by regulating S6K activity by phosphorylation of dTSC2 [29].
The signaling systems controlling cell size and cell number are tightly interconnected. Genetic and biochemical analyses have revealed five different links between the dTSC-dTOR-dS6K pathway and the DInr-dPKB-dFOXO pathway. First, under conditions of unnaturally high insulin-signaling activity (that is, following the oncogenic activation of dPKB) dPKB phosphorylates and inactivates dTSC2, resulting in increased activation of dS6K [29]. Under normal culture conditions this regulation does not seem critical, however, loss of dPKB function does not lower dS6K activity in larval extracts [54]. Second, under physiological conditions, dPDK1 regulates dPKB as well as dS6K [63]. Third, dS6K itself downregulates dPKB activity in a negative feedback loop [31]. Fourth, under severe starvation conditions, nuclear dFOXO presumably activates target genes that reduce cell proliferation. One of these target genes is d4E-BP, which encodes an inhibitor of translation initiation. When conditions improve, the insulin and TOR signaling pathways can stimulate translation by disrupting the 4E-BP/eIF4E complex via phosphorylation of 4E-BP, and in parallel by repressing FOXO-dependent 4E-BP expression. Fifth, under even more severe starvation or stress conditions, full activation of dFOXO upregulates expression of the insulin receptor itself, thus rendering the cell hypersensitive to low insulin levels (see [52]). These multiple positive and negative interactions ensure a continuous fine adjustment of the growth rate to changing environmental conditions.
Evolutionary conservation of insulin signaling and FOXO function
Genetic dissection of signaling by insulin and its target DAF-16 has been pioneered in C. elegans and has helped to unravel the role of this pathway in dauer formation and longevity. Our analysis shows that the same pathway with the homologous nuclear targets operates in flies in the control of cell growth and proliferation, processes that do not involve insulin signaling in worms. Dauer formation and possibly longevity affect the entire organism and do not depend on cell-autonomous functions of the insulin signaling pathway [64]. The cell-growth phenotype in Drosophila, however, depends on the cell-autonomous functioning of the insulin-signaling cascade [45]. Insects enter diapause in response to diverse environmental cues (nutrients, day length or temperature) and arrest development or the aging process in a manner similar to dauer formation in worms [65]. Ageing, and possibly diapause, is also under the control of the insulin pathway in Drosophila [65,66]. It has recently been shown that heterozygous IGF-1R mutant mice also exhibit a prolonged lifespan [3]. It therefore appears that the function of the insulin pathway, its components, and possibly at least some of its targets, have been conserved throughout evolution.
dFOXO may integrate different forms of cellular stress
The longevity phenotype of IGF-1R-deficient mice is associated with enhanced resistance to oxidative stress [3]. It is likely that this phenomenon is due to hyperactivation of FOXO proteins, as several studies have shown that FOXO transcription factors play a role in the oxidative-stress response in mammalian cells [16,17] as well as in C. elegans [49]. Our observation that dFOXO mutant flies are hypersensitive to oxidative stress confirms that, in addition to their role in insulin signaling, the role of FOXO proteins in protecting against cellular stress is highly conserved. The mechanism by which dFOXO confers oxidative-stress resistance is not yet known. In our microarray experiment, we identified several genes encoding cytochrome P450 enzymes as dFOXO target gene candidates (Figure 6a). As it has been shown that cytochrome P450 enzymes reduce the toxic effects of paraquat in mice [67], they might partially mediate the protective effect of dFOXO. Furthermore, it remains to be established whether the regulation of dFOXO by insulin is required for dFOXO's protective properties. It is tempting to speculate that distinct stress-induced signaling pathways activate dFOXO under conditions of cellular stress, in addition to the negative input from the insulin cascade, as several stress-induced phosphorylation sites are conserved between hFOXO3a and dFOXO (A. Brunet and M.E. Greenberg, personal communication). This view is supported by our observation that overexpression of a FOXO variant that cannot be inactivated by PKB elicits cell death, a phenotype not observed in larval tissues lacking insulin-signaling components [45]. This result argues that dFOXO induces cellular responses that are independent of insulin.
The emerging model postulates that positive and negative inputs converge on FOXO proteins in response to different environmental conditions, making them central and important integrators controlling cellular (cell-cycle progression) and organismal adaptations (dauer formation, diapause and longevity; see Figure 7). Elucidating the positive inputs that converge on FOXO, by mutating conserved phosphorylation sites in the single Drosophila homolog of this class, should help us to better understand dFOXO's integrator function.
Figure 7.
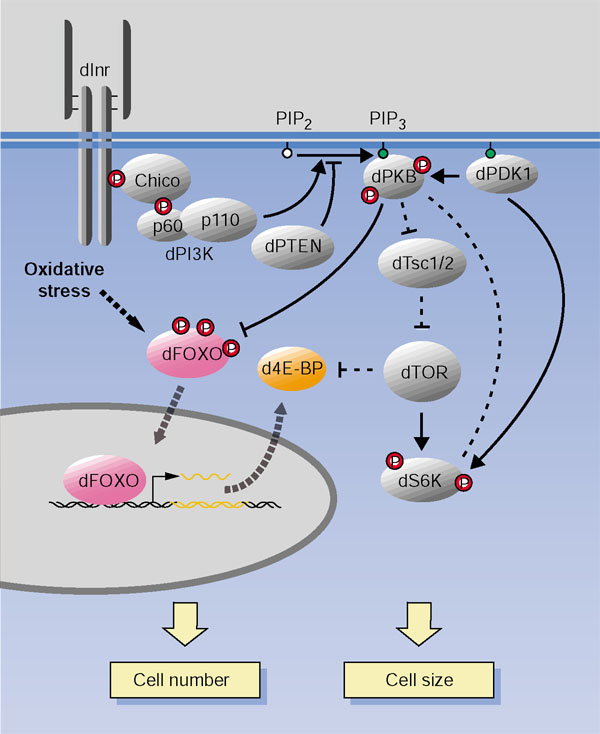
dFOXO may be an integrator of cellular stress. We propose a model in which dFOXO senses different forms of cellular stress (that is, nutrient deprivation or reactive oxygen species) and induces cellular responses, such as proliferation arrest, in part by repressing translation via upregulation of d4E-BP. The various signaling proteins shown in the figure are discussed in the text.
Materials and methods
Identification of dFOXO
We searched the Drosophila genome [68] using a TBLASTN algorithm for sequences with homology to the DNA-binding domain of human FOXO3a (amino acids 157–251). The resultant matches were further assessed for the presence of consensus PKB phosphorylation sites R-X-R-X-X-S/T [37].
We used a genomic DNA stretch flanking the only identified region fulfilling these criteria to search a collection of Drosophila expressed sequence tags [69], which eventually identified two clones (LD05569 and LD18492) containing identical full-length cDNA sequences of 3.7 kb length. The dFOXO gene is annotated in FlyBase [70] (FBgn0038197) under the name foxo.
Generation of plasmids and transgenic flies
The cDNA clone LD05569 contains the full-length dFOXO cDNA within the pBS-SK(+/-) vector (Stratagene [71]). To generate a triple PKB phosphorylation mutant of dFOXO, we used PCR-based site-directed mutagenesis (QuickChange, Stratagene) to introduce the three point mutations T44A, S190A and S259A. Primer sequences are available upon request. The mutated sequence was confirmed by double-stranded DNA sequencing. To generate UAS constructs, the cDNA inserts from both wild-type dFOXO and triple-mutant dFOXO were subcloned from pBS-SK(+/-) into the pUAST transformation vector [72] as EcoRI-Asp718 fragments. The corresponding UAS constructs containing the cDNA encoding wild-type and triple-mutant hFOXO3a [12] were generated by subcloning the inserts from pECE-HA-hFOXO3a and pECE-HA-hFOXO3a-TM (generous gifts of Anne Brunet) into pUAST as BglII-XbaI fragments. Fragments were excised from the pECE clones via complete digestion with XbaI followed by partial BglII digestion. All sequences were confirmed by double-stranded DNA sequencing. The four resultant UAS constructs are referred to as UAS-dFOXO, UAS-dFOXO-TM, UAS-hFOXO3a and UAS-hFOXO3a-TM. To generate transgenic Drosophila lines, P-element-mediated germline transformation was carried out as described previously [73]. Several independent transformant lines were recovered for each construct with the exception of UAS-dFOXO-TM, for which we did not obtain a viable transformant line.
EMS reversion mutagenesis
To generate dFOXO loss-of-function mutants, homozygous y w;; EP35-147 males were mutagenized with 27 mM ethyl methanesulfonate (EMS) according to standard procedures [74]. Mutagenized males were mated to homozygous y w; GMR-Gal4 UAS-Dp110DN virgins. Roughly 60,000 F1 progeny were screened for suppression of semilethality and the eye phenotype shown in Figure 3b. F1 revertants were retested for transmission of the reversion to F2 and positive candidate lines were then balanced over TM3 Sb Ser. To characterize the mutations, the dFOXO open reading frame from each individual mutagenized chromosome was amplified by RT-PCR and sequenced. The cDNA derived from the unmutagenized EP35-147 chromosome was used as a reference sample to identify mutations. Promising mutations were verified by double peak analysis of PCR fragments amplified from genomic DNA using the Sequencher program (Gene Codes Corporation [75]).
Drosophila strains
The EP-35-147 line was kindly provided by Konrad Basler, the GMR-Gal4 driver was a gift from M. Freeman. The GMR-Gal4, UAS-Dp110DN line was obtained from Sally Leevers, the eiger mutants from Masayuki Miura, and the Thor1 line from Paul Lasko.
Phenotype analyses
All phenotypes were analyzed in females raised at 25°C unless indicated otherwise. Body weight, cell size and cell number were determined as described previously [5]. The body weight experiment was performed in duplicate, and male and female flies were measured separately (n = 12 for each gender and genotype; the highest and lowest values were excluded from the analysis). Flies were reared under identical, non-crowding conditions and were of identical age (2 d) at the time of the experiment. The sizes of ommatidia and rhabdomeres were quantified with the program NIH Image 1.61. [76].
Clonal analysis
To induce loss-of-function clones, we used the Flp/FRT and ey-Flp systems to generate mosaic flies by mitotic recombination [47,77]. Overexpression clones were generated as described [63].
In situ hybridizations
In situ hybridizations to eye imaginal discs was performed as described [78,79]. The d4E-BP cDNA was PCR-amplified with Pfu polymerase from Promega [80] from total double-stranded cDNA derived from adult y w flies and cloned into the pCAPs vector (PCR blunt-end cloning kit from Roche [81]). Insert orientation was determined by sequencing. Vector-specific PCR primers flanking the multiple cloning site (MCS) and containing either T7 or SP6 RNA polymerase promoters were used to synthesize double-stranded DNA templates for the labeling in vitro transcription reaction. The sense probe was transcribed with T7 and the antisense probe with SP6 RNA polymerase.
Cell culture
Drosophila embryonic Kc167 cells were maintained as described elsewhere [53]. Briefly, cells were grown at 25°C in Schneider's Drosophila medium (Gibco/Invitrogen [82]) supplemented with 10% heat-inactivated fetal calf serum, FCS. Cells were split and diluted to a density of 1 × 106 per ml twice a week. For the microarray experiment, cells were grown into the stationary phase for 7 d and then stimulated with 100 nM bovine insulin for 2 h.
Microarray experiment
The microarray experiment was performed at the Functional Genomics Center Zürich (FGCZ) using the Affymetrix GeneChip™ system [83]. Total RNA was extracted from untreated control cells and insulin-treated cells 2 h after stimulation using the RNeasy Mini kit (Qiagen [84]) according to the manufacturer's instructions. From each cell population, three independent samples were taken, processed in parallel and hybridized to three separate microarrays. Synthesis of cDNA and labeled cRNA, array hybridization and scanning were performed according to the standard Affymetrix protocols. The .chp files for the individual scanned microarrays were imported into the Affymetrix Data Mining Tool™ software for data analysis.
Stress treatments
Stress-resistance experiments were performed with 3-day-old adult flies, and males and females were assayed separately. For bacterial infection experiments, adult flies were pricked with a thin needle which had been dipped in a concentrated bacterial culture [85]. Bacterial strains tested were the Gram-negative Erwinia carotovora carotovora and the Gram-positive Micrococcus luteus. Heat shock was performed by continuous exposure to 37°C. Resistance to heavy metals during development was assayed by rearing flies on food containing either 2.5 mM copper, 6 mM zinc or 200 μM cadmium. For the starvation test, flies were transferred from normal food to empty vials closed with a wet foam stopper. For oxidative-stress challenge, flies were starved in empty vials for 6 h and then transferred to vials containing a gel of phosphate-buffered saline (PBS), 10% sucrose, 0.8% low-melt agarose and the respective oxidative agent (either 5% H2O2 or 20 mM paraquat). The oxidant was added to the solution after cooling to 40°C. A control population of flies was placed in vials containing the PBS-sucrose gel without oxidant. Dead flies were counted every 12 h (n = 80 for each gender and genotype). The hydrogen peroxide and paraquat experiments were each done in triplicate. Larval starvation was performed by rearing larvae on normal fly food until 80 h after egg deposition, then floating them in 30% glycerol, washing with water and transfering batches of 30–40 larvae to vials containing a gel of either PBS, 20% sucrose and 0.8% agarose (sugar condition) or PBS-agarose only (complete starvation).
Acknowledgments
Acknowledgements
We thank Konrad Basler, Sally Leevers, Paul Lasko and Masayuki Miura for fly stocks; Anne Brunet for the hFOXO3a constructs and critical reading of the manuscript; Christof Hugentobler and Dieter Egli for help with fly work and scanning electron microscopy; the DNA sequencing core facility at Children's Hospital, Boston, for DNA sequencing; Ruth Keist, Laura Huopaniemi and Andrea Patrignani for assistance in the microarray experiment; the FGCZ for financial support; members of the Hafen lab for helpful discussions; and Robert Tjian and Brian E. Staveley for exchanging information prior to publication. This work was supported by grants from the Swiss National Science Foundation and the Swiss Cancer League to E.H., by the Roche Research Foundation to T.R., and by the National Institutes of Health and the Mental Retardation Research Center to M.E.G. A research fellowship from Trinity College, Cambridge supported J.D.W. in part.
References
- Takahashi Y, Kadowaki H, Momomura K, Fukushima Y, Orban T, Okai T, Taketani Y, Akanuma Y, Yazaki Y, Kadowaki T. A homozygous kinase-defective mutation in the insulin receptor gene in a patient with leprechaunism. Diabetologia. 1997;40:412–420. doi: 10.1007/s001250050695. [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol. 2001;11:213–221. doi: 10.1016/s0960-9822(01)00068-9. [DOI] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Stocker H, Andjelkovic M, Oldham S, Laffargue M, Wymann MP, Hemmings BA, Hafen E. Living with lethal PIP3 levels: viability of flies lacking PTEN restored by a PH domain mutation in Akt/PKB. Science. 2002;295:2088–2091. doi: 10.1126/science.1068094. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27:352–360. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr, DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- Furukawa-Hibi Y, Yoshida-Araki K, Ohta T, Ikeda K, Motoyama N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J Biol Chem. 2002;277:26729–26732. doi: 10.1074/jbc.C200256200. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, et al. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1) Mol Cell Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez B, Martinez AC, Burgering BM, Carrera AC. Forkhead transcription factors contribute to execution of the mitotic programme in mammals. Nature. 2001;413:744–747. doi: 10.1038/35099574. [DOI] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Barthel A, Schmoll D, Kruger KD, Bahrenberg G, Walther R, Roth RA, Joost HG. Differential regulation of endogenous glucose-6-phosphatase and phosphoenolpyruvate carboxykinase gene expression by the forkhead transcription factor FKHR in H4IIE-hepatoma cells. Biochem Biophys Res Commun. 2001;285:897–902. doi: 10.1006/bbrc.2001.5261. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91. doi: 10.1016/s1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- Schmidt M, de Mattos SF, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002;22:7842–7852. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae J, Biggs WH, 3rd, Kitamura T, Cavenee WK, Wright CV, Arden KC, Accili D. Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet. 2002;32:245–253. doi: 10.1038/ng890. [DOI] [PubMed] [Google Scholar]
- Borkhardt A, Repp R, Haas OA, Leis T, Harbott J, Kreuder J, Hammermann J, Henn T, Lampert F. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11)(q13;q23) Oncogene. 1997;14:195–202. doi: 10.1038/sj.onc.1200814. [DOI] [PubMed] [Google Scholar]
- Sublett JE, Jeon IS, Shapiro DN. The alveolar rhabdomyosarcoma PAX3/FKHR fusion protein is a transcriptional activator. Oncogene. 1995;11:545–552. [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS, Ru B, Pan D. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol. 2002;4:699–704. doi: 10.1038/ncb847. [DOI] [PubMed] [Google Scholar]
- Radimerski T, Montagne J, Hemmings-Mieszczak M, Thomas G. Lethality of Drosophila lacking TSC tumor suppressor function rescued by reducing dS6K signaling. Genes Dev. 2002;16:2627–2632. doi: 10.1101/gad.239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003;5:559–566. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5:578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003;5:566–571. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–1466. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: a regulator of cell size. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 1996;399:333–338. doi: 10.1016/s0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wassarman DA, Rubin GM. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell. 1995;83:1253–1262. doi: 10.1016/0092-8674(95)90150-7. [DOI] [PubMed] [Google Scholar]
- Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD. The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J. 1996;15:6584–6594. [PMC free article] [PubMed] [Google Scholar]
- Beadle G, Tatum E, Clancy C. Food level in relation to rate of development and eye pigmentation in Drosophila melanogaster. Biol Bull. 1938;75:447–462. [Google Scholar]
- Zinke I, Schutz CS, Katzenberger JD, Bauer M, Pankratz MJ. Nutrient control of gene expression in Drosophila: microarray analysis of starvation and sugar-dependent response. EMBO J. 2002;21:6162–6173. doi: 10.1093/emboj/cdf600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollmann M, Young LM, Di Como CJ, Karim F, Belvin M, Robertson S, Whittaker K, Demsky M, Fisher WW, Buchman A, et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell. 2000;101:91–101. doi: 10.1016/S0092-8674(00)80626-1. [DOI] [PubMed] [Google Scholar]
- Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, Miura M. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002;21:3009–3018. doi: 10.1093/emboj/cdf306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JM, Davidge JT, Lockyer JM, Staveley BE. Expression of Drosophila FOXO regulates growth and can phenocopy starvation. BMC Dev Biol. 2003;3:5. doi: 10.1186/1471-213X-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss BF, Beckingham K, Hafen E. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell. 1999;97:865–875. doi: 10.1016/s0092-8674(00)80799-0. [DOI] [PubMed] [Google Scholar]
- Weinkove D, Neufeld TP, Twardzik T, Waterfield MD, Leevers SJ. Regulation of imaginal disc cell size, cell number and organ size by Drosophila class I(A) phosphoinositide 3-kinase and its adaptor. Curr Biol. 1999;9:1019–1029. doi: 10.1016/s0960-9822(99)80450-3. [DOI] [PubMed] [Google Scholar]
- Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- Oldham S, Stocker H, Laffargue M, Wittwer F, Wymann M, Hafen E. The Drosophila insulin/IGF receptor controls growth and size by modulating PtdInsP(3) levels. Development. 2002;129:4103–4109. doi: 10.1242/dev.129.17.4103. [DOI] [PubMed] [Google Scholar]
- Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999;13:1385–1393. [PubMed] [Google Scholar]
- Dijkers PF, Birkenkamp KU, Lam EW, Thomas NS, Lammers JW, Koenderman L, Coffer PJ. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J Cell Biol. 2002;156:531–542. doi: 10.1083/jcb.200108084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanase S, Yasuda K, Ishii N. Adaptive responses to oxidative damage in three mutants of Caenorhabditis elegans (age-1, mev-1 and daf-16) that affect life span. Mech Ageing Dev. 2002;123:1579–87. doi: 10.1016/s0047-6374(02)00093-3. [DOI] [PubMed] [Google Scholar]
- Puig O, Marr MT, Ruhf ML, Tjian R. Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev. 2003. [DOI] [PMC free article] [PubMed]
- Radimerski T, Mini T, Schneider U, Wettenhall RE, Thomas G, Jeno P. Identification of insulin-induced sites of ribosomal protein S6 phosphorylation in Drosophila melanogaster. Biochemistry. 2000;39:5766–5774. doi: 10.1021/bi9927484. [DOI] [PubMed] [Google Scholar]
- Radimerski T, Montagne J, Rintelen F, Stocker H, van der Kaay J, Downes CP, Hafen E, Thomas G. dS6K-regulated cell growth is dPKB/dPI(3)K-independent, but requires dPDK1. Nat Cell Biol. 2002;4:251–255. doi: 10.1038/ncb763. [DOI] [PubMed] [Google Scholar]
- Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J. 2000;349:629–634. doi: 10.1042/0264-6021:3490629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinke I, Kirchner C, Chao LC, Tetzlaff MT, Pankratz MJ. Suppression of food intake and growth by amino acids in Drosophila : the role of pumpless, a fat body expressed gene with homology to vertebrate glycine cleavage system. Development. 1999;126:5275–5284. doi: 10.1242/dev.126.23.5275. [DOI] [PubMed] [Google Scholar]
- Miron M, Verdu J, Lachance PE, Birnbaum MJ, Lasko PF, Sonenberg N. The translational inhibitor 4E-BP is an effector of PI(3)K/Akt signalling and cell growth in Drosophila. Nat Cell Biol. 2001;3:596–601. doi: 10.1038/35078571. [DOI] [PubMed] [Google Scholar]
- Bernal A, Kimbrell DA. Drosophila Thor participates in host immune defense and connects a translational regulator with innate immunity. Proc Natl Acad Sci USA. 2000;97:6019–6024. doi: 10.1073/pnas.100391597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Zhou Z, Tang ML, Meller S, Chen J, Bellen H, Kimbrell DA. Identification of immune system and response genes, and novel mutations causing melanotic tumor formation in Drosophila melanogaster. Genetics. 1996;143:929–940. doi: 10.1093/genetics/143.2.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Ruvkun G. Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 1998;12:2488–2498. doi: 10.1101/gad.12.16.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CJ, Huang H, Xu T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001;105:357–368. doi: 10.1016/s0092-8674(01)00333-6. [DOI] [PubMed] [Google Scholar]
- Gao X, Pan D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev. 2001;15:1383–1392. doi: 10.1101/gad.901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintelen F, Stocker H, Thomas G, Hafen E. PDK1 regulates growth through Akt and S6K in Drosophila. Proc Natl Acad Sci USA. 2001;98:15020–15025. doi: 10.1073/pnas.011318098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfeld J, Kenyon C. Cell nonautonomy of C. elegans daf-2 function in the regulation of diapause and life span. Cell. 1998;95:199–210. doi: 10.1016/s0092-8674(00)81751-1. [DOI] [PubMed] [Google Scholar]
- Tatar M, Yin C. Slow aging during insect reproductive diapause: why butterflies, grasshoppers and flies are like worms. Exp Gerontol. 2001;36:723–738. doi: 10.1016/s0531-5565(00)00238-2. [DOI] [PubMed] [Google Scholar]
- Partridge L, Gems D. Mechanisms of ageing: public or private? Nat Rev Genet. 2002;3:165–175. doi: 10.1038/nrg753. [DOI] [PubMed] [Google Scholar]
- Shimada H, Furuno H, Hirai K, Koyama J, Ariyama J, Simamura E. Paraquat detoxicative system in the mouse liver postmitochondrial fraction. Arch Biochem Biophys. 2002;402:149–157. doi: 10.1016/S0003-9861(02)00059-0. [DOI] [PubMed] [Google Scholar]
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Hong L, Brokstein P, Evans-Holm M, Frise E, Stapleton M, Harvey DA. A Drosophila complementary DNA resource. Science. 2000;287:2222–2224. doi: 10.1126/science.287.5461.2222. [DOI] [PubMed] [Google Scholar]
- FlyBase http://flybase.org
- Stratagene http://www.stratagene.com
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Basler K, Christen B, Hafen E. Ligand-independent activation of the sevenless receptor tyrosine kinase changes the fate of cells in the developing Drosophila eye. Cell. 1991;64:1069–1081. doi: 10.1016/0092-8674(91)90262-w. [DOI] [PubMed] [Google Scholar]
- Ashburner M. Drosophila. Cold Spring Harbor Laboratory Press: Cold Spring Harbor; 1989. [Google Scholar]
- Gene Codes Corporation http://www.genecodes.com
- NIH Image http://rsb.info.nih.gov/nih-image
- Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- Lehmann R, Tautz D. In situ hybridization to RNA. Methods Cell Biol. 1994;44:575–598. doi: 10.1016/s0091-679x(08)60933-4. [DOI] [PubMed] [Google Scholar]
- O'Neill JW, Bier E. Double-label in situ hybridization using biotin and digoxigenin-tagged RNA probes. Biotechniques. 1994;17:870–875. [PubMed] [Google Scholar]
- Promega http://www.promega.com
- Roche http://www.roche-applied-science.com
- Invitrogen life technologies http://www.invitrogen.com
- Affymetrix http://www.affymetrix.com
- Qiagen http://www.qiagen.com
- Lemaitre B, Reichhart JM, Hoffmann JA. Drosophila host defense: differential induction of antimicrobial peptide genes after infection by various classes of microorganisms. Proc Natl Acad Sci USA. 1997;94:14614–14619. doi: 10.1073/pnas.94.26.14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt J, Climent I, Dahlman-Wright K, Wikstrom M. Solution structure of the DNA binding domain of the human forkhead transcription factor AFX (FOXO4) Biochemistry. 2001;40:5861–5869. doi: 10.1021/bi001663w. [DOI] [PubMed] [Google Scholar]