

Abstract
Few diseases exemplify the integration of research from bench to bedside as well as neonatal lupus, often referred to as a model of passively acquired autoimmunity. In essence, this disease encompasses two patients, both the mother and her child. The signature histologic lesion of autoimmune-associated congenital heart block is fibrosis of the conducting tissue, and in some cases the surrounding myocardium. It is astounding how rapid and, in most cases, irreversible is the fibrotic response to injury. The mechanism by which maternal anti-SSA/Ro–SSB/La antibodies initiate and perpetuate inflammation, and eventuate in scarring of the atrioventricular node, is not yet defined. In vitro and in vivo studies suggest that one pathologic cascade leading to scarring may be initiated via apoptosis, resulting in translocation of SSA/Ro–SSB/La antigens and subsequent surface binding by maternal autoantibodies. These opsonized cardiocytes are phagocytosed by macrophages, which secrete factors that transdifferentiate fibroblasts into myofibroblasts, a scarring phenotype. Dissecting the individual components in this fibrotic pathway should provide insights into the rarity of irreversible injury and should form the basis of rational approaches to prevention and treatment.
Keywords: anti-SSA/Ro and SSB/La antibodies, congenital heart block, myofibroblasts, neonatal lupus
Introduction
Following the seminal observation in the early 1980s that sera from nearly all mothers of children with isolated congenital heart block (CHB) contain specific autoantibodies [1,2], this disease, previously of interest only to the disciples of cardiology, became an important model of passively acquired autoimmunity. The identification of CHB in a fetus, particularly in the late second trimester and in the absence of structural abnormalities, currently predicts with at least 85% certainty that the mother will have autoantibodies to SSA/Ro alone or in conjunction with antibodies to SSB/La ribonucleoproteins [3]. Disease in the fetus is entirely independent of whether the mother has systemic lupus erythematosus or Sjögren's syndrome, or is totally asymptomatic [3,4].
One of the most intriguing aspects of CHB is that it is an injury unique to some phase(s) of development, since it has never been reported in the maternal heart despite the presence of identical antibodies in the maternal circulation. CHB carries a substantial mortality (approaching 20%) and morbidity, with over 60% of affected children requiring lifelong pacemakers [5]. To date, third-degree atrioventricular (AV) block is irreversible. With advances in fetal echocardiography, first-degree and second-degree blocks have also been detected in utero, an observation that suggests a window of opportunity with regard to treatment. Based on a review of records from the Research Registry for Neonatal Lupus [6], however, we have recently learned that incomplete AV block can progress postnatally despite the clearance of the candidate maternal antibodies from the neonatal circulation [7].
The present model of passively acquired autoimmunity offers an exceptional opportunity to examine the effector arm of immunity and to define the pathogenicity of an autoantibody in mediating tissue injury. The study of CHB exemplifies not only translational research, which inherently draws upon clinical observations and explores them in the laboratory, but also 'integrational' research, which attempts to fit together critical clinical and basic observations, even those seemingly at odds. The pathologic cascade that eventuates in irreversible fibrosis, characteristic of this autoimmune-associated cardiac disease, has been difficult to define at the molecular level. The challenge rests on integrating the initial antibody insult with the final cardiac injury, and on reconciling the facts that the target antigens are intracellular and that most infants born to mothers with the candidate antibodies do not have clinically detectable AV block [8]. Antibodies are therefore necessary but insufficient to cause CHB, and the final pathway leading to fibrosis may be variable, kept totally in check in most fetuses (normal sinus rhythm), being subclinical in others (first-degree block) and being fully executed in very few (advanced block).
Pathologic cascade from antibody insult to fibrosis
Accessibility of intracellular target antigen to the extracellular antibodies
In consideration of surface binding, one hypothesis is that apoptosis might result in translocation of intracellular antigens to the external leaflet of the membrane. Casciola-Rosen and colleagues first demonstrated, by confocal microscopy, the presence of SSA/Ro and SSB/La in surface blebs of apoptotic keratinocytes [9]. Applicability of apoptosis to the pathogenesis of CHB is supported by several observations. It is a selective process of physiological cell deletion in embryogenesis and normal tissue turnover, plays an important role in shaping morphological and functional maturity [10,11], and affects scattered single cells rather than tracts of contiguous cells [12]. Perhaps a novel view of apoptosis is that it facilitates the placing of target autoantigens in a position to be recognized by previously generated antibodies. In autoimmune-associated CHB, the newly accessible antigen is not 'inducing' an immune response (i.e. immunogenic), but rather becomes a target of cognate maternal autoantibodies already present in the fetal circulation (i.e. antigenic).
Our laboratory initially addressed apoptosis in vitro using cultured human fetal cardiocytes. Incubation of 4-day cultured human fetal cardiocytes with 0.5 μM staurosporine or with 0.3 mM 2,3-dimethoxy-1,4-naphthoquinone induced the characteristic morphologic changes of apoptosis, the internucleosomal cleavage of DNA, and the signature 85 kDa cleavage fragment of poly ADP-ribose polymerase [13]. Apoptosis could also be induced by culturing the cells on poly(2-)hydroxyethylmethacrylate [14]. The surface expression of 48 kDa SSB/La and of 52 kDa and 60 kDa SSA/Ro was demonstrated by confocal microscopy and scanning electron microscopy using affinity-purified antibodies to each of the respective antigens [13,15].
Tran and colleagues have recently identified physiologic apoptosis, translocation of SSB/La, and binding of anti-SSB/La antibodies in the developing murine heart [16,17]. We have extended this in vivo work and examined cardiac sections from several available autopsies. As assessed by TUNEL (FITC and immunoperoxidase detection), apoptosis was increased in available sections including septal tissue (containing the conduction system), the right ventricle and left ventricle from two fetuses (20 and 22 weeks gestational age) dying with CHB, compared to age-matched normal abortuses from elective termination. Notably, apoptotic cardiocytes were not present in contiguous tracts, but rather were diffusely scattered between nonapoptotic cells (Fig. 1A). Apoptosis was most pronounced in the septal regions. Furthermore, human IgG was colocalized to the apoptotic cells (Fig. 1C). Although apoptosis had not been previously examined, earlier studies have shown deposition of IgG in the hearts from two infants dying of CHB and hydrops (29 and 30 weeks gestation, respectively) in several areas of the heart, including the conduction system [18,19]. Lee and colleagues reported that, in some areas, "IgG appeared to outline cells" [19].
Figure 1.
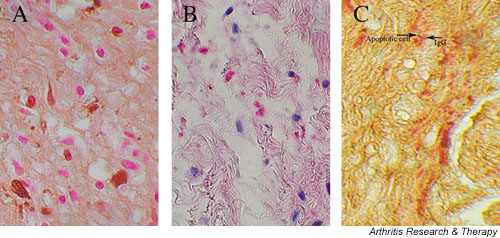
Histological evidence of increased apoptosis and deposition of human IgG in conduction tissue of a 20-week fetus with congenital heart block (CHB). (A) A longitudinal section through the septal tissue of a 20-week fetus with CHB, counterstained with hematoxylin and eosin. Apoptotic cells are identified by TUNEL peroxidase and are scattered among healthy cells. (B) The same tissue section stained with alkaline phosphatase conjugated to anti-human IgG. (C) The same tissue section double-stained with TUNEL peroxidase and alkaline phosphatase conjugated to anti-human IgG to demonstrate colocalization of apoptosis and of IgG, respectively.
What these experiments do not address is the specificity of the anti-SSA/Ro–SSB/La antibodies and the unique vulnerability of the fetal heart. Dieude and colleagues have recently reported (also confirmed in our laboratory) that lamin B1 is redistributed during apoptosis but, unlike SSA/Ro or SSB/La, is not bound by cognate antibodies [20]. These findings support discordance in the final cellular destination of translocated nuclear autoantigens during the process of apoptosis. In the case of lamin B1, physiologic noninflammatory clearance of apoptotic cells should proceed uneventfully even in the presence of circulating cognate antibodies. In CHB, however, the maternal anti-SSA/Ro–SSB/La antibodies result in opsonization and inflammatory/fibrotic sequelae. Even if it is found that SSA/Ro–SSB/La are not absolutely unique in this regard, there may be other factors such as complement binding of certain antigens or degradation of antigens that facilitate clearing without further sequelae. Establishing the fact that at least one other nuclear autoantigen is not surface bound during apoptosis of human fetal cardiomyocytes is a step forward.
The inflammatory component
The consequences of antibody-bound (opsonized) apoptotic cardiocytes were initially explored in vitro using a coculturing system [15]. Macrophages coincubated with these opsonized cells secreted increased levels of tumor necrosis factor alpha over basal conditions or coculture with apoptotic cardiocytes incubated with IgG from a healthy control. Other investigators have also demonstrated that phagocytosis of opsonized apoptotic cells is proinflammatory [21,22]; for example, the observation that ingestion of apoptotic cells bound by anticardiolipin antibodies results in the release of tumor necrosis factor alpha from cocultured macrophages [22]. Histologic studies confirmed the in vitro coculturing model. Giant cells and macrophages (frequently seen proximal to IgG) were present in septal regions as well as in areas of thickened fibrous subendocardium, most apparent in the two fetuses dying before 23 weeks. These studies extend previous reports of a mononuclear cell infiltration in the myocardium of a fetus dying in utero at 18 weeks of gestation [23] and the demonstration of patchy lymphoid aggregates throughout the myocardium of an infant delivered at 30 weeks and dying in the immediate postnatal period [19].
Macrophages potentially contribute to several aspects of the pathologic process mediated by maternal autoantibodies. Although the pathways of clearance and cytokine secretion may vary, these scavengers phagocytose both nonopsonized and opsonized apoptotic cells. Concomitantly or alternatively, macrophages may present antigen to lymphocytes (perhaps those of either maternal or fetal origin), further contributing to an inflammatory process. Moreover, macrophages may provide a critical link between inflammation and ultimate scarring by secretion of alkaline phosphatases, resulting in increased calcification [24]. In fact, macrophages could be seen contained in areas of calcification, particularly in the early cases. In a full-term neonate with CHB who died at birth, however, macrophages were less abundant and not associated with calcified areas, suggesting a diminished role in inflammation as the pathologic process evolves.
The fibrosing end of the line
Perhaps CHB occurs as a consequence of unresolved scarring of the AV node secondary to the transdifferentiation of cardiac fibroblasts to unchecked proliferating myofibroblasts (scarring phenotype in which smooth muscle actin is expressed). Histologic support for this hypothesis was provided by the detection of myofibroblasts in all the affected CHB fetuses regardless of the timing of death relative to detection. As expected, myofibroblasts were located in areas of fibrosis. In the 20-week CHB fetus, clusters of macrophages in close proximity to myofibroblasts were present in scar tissue near the AV groove as well as the thickened fibrous subendocardium (Fig. 2). Sections from the septum of the 22-week CHB fetus showed myofibroblasts associated with the extensive fibrous matrix, and showed marked calcification in the inferior portion of the atrial wall where the AV node is likely to reside. In the full-term neonate dying at birth of CHB, myofibroblasts were observed in areas of scar. In contrast, these cells were not observed in either septal or ventricular tissue from the control 22-week and 23-week abortuses and from a term neonate dying of noncardiac causes. For the 20-week and 22-week CHB hearts, there was a strong positive correlation between the absolute number of macrophages and the content of myofibroblasts.
Figure 2.
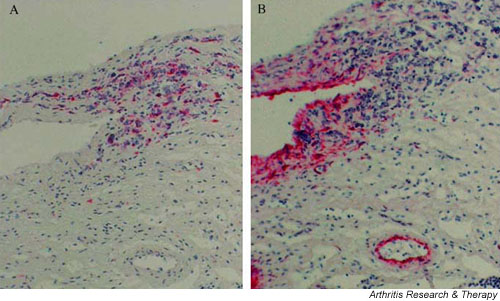
Proximity of macrophages and myofibroblasts in the septum of the heart from a 20-week fetus with congenital heart block (CHB). Longitudinal sections through the septum of a 20-week CHB heart. Tissue was first incubated with (A) anti-CD68 (macrophage) or (B) anti-smooth muscle actin (myofibroblast and blood vessel smooth muscle cells), and then stained with alkaline phosphatase.
The functional implication of the cellular colocalization demonstrated on the histologic sections was examined by in vitro studies in which cultured human fetal cardiac fibroblasts, exposed to supernatants obtained from macrophages incubated with opsonized apoptotic cardiocytes, markedly increased the expression of the myofibroblast marker smooth muscle actin (scarring phenotype) [14]. The addition of neutralizing antitransforming growth factor beta antibodies to the 'opsonized' supernatant blocked expression of smooth muscle actin, supporting a potential role of transforming growth factor beta in the final pathologic cascade to scarring. Of relevance, preliminary genotyping data suggest that children with CHB have a higher frequency of the fibrosis-promoting polymorphism at codon 10 of transforming growth factor beta than do unaffected siblings [25].
While it is often assumed that fibrosis is simply the end result of an inflammatory insult, a recently published pathologic description of Lyme carditis associated with second-degree heart block prompts a reappraisal of the elements of tissue injury, response, and ultimate repair or scar in the human heart. Right ventricular biopsy revealed mono-nuclear cells around the myocardial microvasculature and within the endocardium [26]. Despite prolonged inflammation (second-degree heart block was present for 8 weeks), the cascade to fibrosis was not irrevocably programmed since the block resolved following antibiotic therapy. This absence of permanent injury stands in strong contrast to the rapid progression to scarring seen in autoantibody-associated CHB. The expression of specific combinations of cytokines may ultimately provide the explanation.
Conclusions
In summary, immunohistological analyses of available cardiac sections from several cases of CHB/myocarditis with varying degrees of pathology parallel the results obtained exploiting in vitro coculturing systems. Physiologic apoptosis may initiate an inflammatory process via antibody binding and ingestion by macrophages that not only fuels continued apoptosis, but contributes to the transdifferentiation of cardiac fibroblasts to a scarring phenotype.
The heart block of neonatal lupus is not only progressive (second to third degree) but also characteristically irreversible, despite brief exposure to autoantibodies and a limited period of inflammation. This is underscored by the finding of extensive fibrosis even in the earliest deaths. Furthermore, persistence of this phenotype even after birth may be related to the progression of block seen post-partum in some infants [7]. Moreover, fibrosis of the AV node contradicts the paradigm that fetal wounds heal without scarring [27].
Disruption of healing may involve the continued presence of myofibroblasts, a consequence of protracted stimulation from the macrophages. Irreversible fibrotic replacement of normal tissue may be unique to heart block acquired in utero following autoantibody-initiated inflammation. Other inflammatory stimuli, as in Lyme disease, induce transient block [26], arguing against the assumption that fibroblast transdifferentiation is merely a common final pathway of inflammation.
It seems reasonable to predict that there are both susceptibility and regulatory factors, such as fetal polymorphisms of Fc receptors and cytokines, each of which could influence the extent of the proposed pathologic cascade to result in permanent third-degree heart block. Dissecting the individual components in this fibrotic pathway should elucidate the pathogenesis of antibody-associated CHB and the rarity of irreversible injury, and may provide a rationale for therapy.
Competing interests
None declared.
Abbreviations
AV = atrioventricular; CHB = congenital heart block; Fc = crystallizable fragment; FITC = fluorescein isothiocyanate; TUNEL = terminal deoxynucleotidyl transferase-mediated 2'-deoxyuridine 5'-triphosphate (dUTP) nick end-labeling.
Acknowledgments
Acknowledgements
The original work described herein was supported by US National Institutes of Health Grant No AR42455 (JPB), Grant No AR48409 (RMC), and Contract No AR42220 (Research Registry for Neonatal Lupus).
References
- Scott JS, Maddison PJ, Taylor PV, Esscher E, Scott O, Skinner RP. Connective-tissue disease, antibodies to ribonucleoprotein, and congenital heart block. N Engl J Med. 1983;309:209–212. doi: 10.1056/NEJM198307283090403. [DOI] [PubMed] [Google Scholar]
- Lee LA, Reed BR, Harmon C. Autoantibodies to SS-A/Ro in congenital heart block [abstract] Arthritis Rheum. 1983;20(suppl):S24. [Google Scholar]
- Buyon JP. Neonatal lupus syndromes. In: Lahita RG, editor. In Systemic Lupus Erythematosus. 3. San Diego, CA: Academic Press; 1999. pp. 337–359. [Google Scholar]
- Lee LA. Neonatal lupus erythematosus. J Invest Derm. 1993;100:9s–13s. doi: 10.1111/1523-1747.ep12355173. [DOI] [PubMed] [Google Scholar]
- Waltuck J, Buyon JP. Autoantibody-associated congenital heart block: outcome in mothers and children. Ann Intern Med. 1994;120:544–551. doi: 10.7326/0003-4819-120-7-199404010-00003. [DOI] [PubMed] [Google Scholar]
- Buyon JP, Hiebert R, Copel J, Craft J, Friedman D, Katholi M, Lee L, Marston K, Provost T, Reichlin M, Rider L, Rupel A, Saleeb S, Weston W, Skovron ML. Autoimmune-associated congenital heart block: mortality, morbidity, and recurrence rates obtained from a national neonatal lupus registry. J Am Coll Cardiol. 1998;31:1658–1666. doi: 10.1016/S0735-1097(98)00161-2. [DOI] [PubMed] [Google Scholar]
- Askanase AD, Friedman DM, Dische MR, Dubin A, Starc T, Katholi MC, Buyon JP. Spectrum and progression of conduction abnormalities in infants born to mothers with anti-SSA/Ro–SSB/La antibodies. Lupus. 2002;11:145–151. doi: 10.1191/0961203302lu173oa. [DOI] [PubMed] [Google Scholar]
- Brucato A, Frassi M, Franceschini F, Cimaz R, Faden D, Pisoni MP, Muscara M, Vignati G, Stramba-Badiale M, Catelli L, Lojacono A, Cavazzana I, Ghirardello A, Vescovi F, Gambari PF, Doria A, Meroni PL, Tincani A. Risk of congenital congenital heart block in newborns of mothers with anti-Ro/SSA antibodies detected by counterimmunoelectrophoresis. Arthritis Rheum. 2001;44:1832–1835. doi: 10.1002/1529-0131(200108)44:8<1832::AID-ART320>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ucker DS. Death by suicide: one way to go in mammalian cellular development? New Biol. 1991;3:103–109. [PubMed] [Google Scholar]
- Hale AJ, Smith CA, Sutherland LC, Stoneman VEA, Longthorne VL, Culhane AC, Williams GT. Apoptosis: molecular regulation of cell death. Eur J Biochem. 1996;236:1–26. doi: 10.1111/j.1432-1033.1996.00001.x. [DOI] [PubMed] [Google Scholar]
- Takeda K, Yu ZX, Nishikawa T, Tanaka M, Hosoda S, Ferrans VJ, Kasajima T. Apoptosis and DNA fragmentation in the bulbus cordis of the developing rat heart. J Mol Cell Cardiol. 1996;28:209–215. doi: 10.1006/jmcc.1996.0020. [DOI] [PubMed] [Google Scholar]
- Miranda-Carus ME, Tseng CE, Rashbaum W, Ochs RL, Casiano CA, DiDonato F, Chan EKL, Buyon JP. Accessibility of SSA/Ro and SSB/La antigens to maternal autoantibodies in apoptotic human fetal cardiac myocytes. J Immunol. 1998;161:5061–5069. [PubMed] [Google Scholar]
- Clancy RM, Askanase AD, Kapur RP, Chiopelas E, Azar N, Miranda-Carus ME, Buyon JP. Transdifferentiation of cardiac fibroblasts, a fetal factor in anti-SSA/Ro–SSB/La antibody-mediated congenital heart block. J Immunol. 2002;169:2156–2163. doi: 10.4049/jimmunol.169.4.2156. [DOI] [PubMed] [Google Scholar]
- Miranda-Carús ME, Dinu Askanase A, Clancy RM, Di Donato F, Chou TM, Libera MR, Chan EKL, Buyon JP. Anti-SSA/Ro and anti-SSB/La autoantibodies bind the surface of apoptotic fetal cardiocytes and promote secretion of tumor necrosis factor α by macrophages. J Immunol. 2000;165:5345–5351. doi: 10.4049/jimmunol.165.9.5345. [DOI] [PubMed] [Google Scholar]
- Tran HB, Ohlsson M, Beroukas D, Hiscock J, Bradley J, Buyon JP, Gordon TP. Subcellular redistribution of La(SS-B) autoantigen during physiologic apoptosis in the fetal mouse heart and conduction system: a clue to the pathogenesis of congenital heart block. Arthritis Rheum. 2002;46:202–208. doi: 10.1002/1529-0131(200201)46:1<202::AID-ART10062>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Tran HB, Macardle PJ, Hiscock J, Cavill D, Bradley J, Buyon JP, Gordon TP. Anti-La (SS-B) antibodies transported across the placenta bind apoptotic cells in fetal organs targeted in neonatal lupus. Arthritis Rheum. 2002;46:1572–1579. doi: 10.1002/art.10316. [DOI] [PubMed] [Google Scholar]
- Litsey SE, Noonan JA, O'Connor WN, Cottrill CM, Mitchell B. Maternal connective tissue disease and congenital heart block. Demonstration of immunoglobulin in cardiac tissue. N Engl J Med. 1985;312:98–100. doi: 10.1056/NEJM198501103120206. [DOI] [PubMed] [Google Scholar]
- Lee LA, Coulter S, Erner S, Chu H. Cardiac immunoglobulin deposition in congenital heart block associated with maternal anti-Ro antibody. Am J Med. 1987;83:793–796. doi: 10.1016/0002-9343(87)90918-1. [DOI] [PubMed] [Google Scholar]
- Dieude M, Senecal JL, Rauch J, Hanly JG, Fortin P, Brassard N, Raymond Y. Association of autoantibodies to nuclear lamin B1 with thromboprotection in systemic lupus erythematosus: lack of evidence for a direct role of lamin B1 in apoptotic blebs. Arthritis Rheum. 2002;46:2695–2707. doi: 10.1002/art.10552. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DA, Konowal A, Freed P, Westcott JY, Henson P. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2 and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi AA, Rovere P, Galati G, Heltai S, Bozzolo E, Soldini L, Davoust J, Balestrieri G, Tincani A, Sabbadini MG. Apoptotic cell clearance in systemic lupus erythematosus. I. Opsonization by antiphospholipid antibodies. Arthritis Rheum. 1998;41:205–214. doi: 10.1002/1529-0131(199802)41:2<205::AID-ART4>3.3.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Herreman G, Galezewski N. Maternal connective tissue disease and congenital heart block [letter] N Engl J Med. 1985;312:1329. doi: 10.1056/NEJM198505163122016. [DOI] [PubMed] [Google Scholar]
- Tintut Y, Patel J, Territo M, Saini T, Parhami F, Demer LL. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation. 2002;105:650–655. doi: 10.1161/hc0502.102969. [DOI] [PubMed] [Google Scholar]
- Backer C, Loma-Sanner I, Yin X, Chandrashekhar S, Sullivan K, Clancy R, Buyon J. Increased frequency of the high produce genotype TT (codon 10) of TGF beta in congenital heart block and TNF2 allele in families with neonatal lupus [abstract] Arthritis Rheum. 2002;46(suppl):S261. [Google Scholar]
- Hajjar RJ, Kradin RL. Case records of the Massachusetts General Hospital: case 17-2002 – a 55-year-old man with second-degree atrioventricular block and chest pain. N Engl J Med. 2002;346:1732–1738. doi: 10.1056/NEJMcpc020017. [DOI] [PubMed] [Google Scholar]
- Moulin V, Tam BY, Castilloux G, Auger FA, O'Connor-McCourt MD, Philip A, Germain L. Fetal and adult human skin fibroblasts display intrinsic differences in contractile capacity. J Cell Physiol. 2001;188:211–222. doi: 10.1002/jcp.1110. [DOI] [PubMed] [Google Scholar]