

Abstract
Ultraviolet-visible spectroscopy readily discerns the two types of melanin pigments (eumelanin and pheomelanin) although fundamental details regarding the optical properties and pigment heterogeneity are more difficult to disentangle via analysis of the broad featureless absorption spectrum alone. We employed nonlinear transient absorption spectroscopy to study different melanin pigments at near infrared wavelengths. Excited state absorption, ground state depletion and stimulated emission signal contributions were distinguished for natural and synthetic eumelanins and pheomelanins. A starker contrast amongst the pigments is observed in the nonlinear excitation regime because they all exhibit distinct transient absorptive amplitudes, phase shifts and nonexponential population dynamics spanning the femtosecond – nanosecond range. In this manner, different pigments within the pheomelanin subclass were distinguished in synthetic and human hair samples. These results highlight the potential of nonlinear spectroscopies to deliver an in situ analysis of natural melanins in tissue that are otherwise difficult to extract and purify.
Keywords: Melanin, Excited State Dynamics, Human Hair
1. Introduction
Ultraviolet (UV) photo-protection is naturally afforded by melanin pigments in the skin. Melanins in the skin come in two chemical forms: eumelanin (dark pigment) and pheomelanin (red/brown pigment). Important similarities and differences exist between these two types of pigments. First of all, both melanins are packaged in microscopic melanosomes which effectively absorb and scatter UV light. Typically, absorption of light is immediately followed by a rapid nonradiative down conversion to heat.1 Both melanins are chemically derived from the oxidation of tyrosine although the biochemical pathways diverge in the presence of cysteine.2 Pheomelanins are produced when cysteine reacts with dopaquinone to produce benzothiazine intermediates which subsequently polymerize.3 In the absence of cysteine, dopaquinone reacts to form indoles, which polymerize to form the dark eumelanin pigments. Further divergence between the pigments occurs in the microscopic assembly of the melanosomes. Eumelanosomes are elliptical in shape and approximately one micron in size while pheomelanosomes are observed to be smaller and spherical.4 Additionally, there exist varying amounts of protein and metal ions in both types of melanosomes.4,5 While the biochemical differences are notable, the low solubilities of melanins have steered chemical assays towards degradation studies whereby reaction products are analyzed to assess pigment content.6 These assays suffer from low sensitivities (low product yields) and are invasive in that they require the pigments to be isolated and subsequently chemically degraded. Other methods are required to characterize melanin pigments in situ such as in hair or tissue where they may be present in low concentrations.
Optical techniques are capable of interrogating melanins in tissue because of their high sensitivity. The sensitivity arises from the exceptional optical detectors available that benefit from experimental observables and/or arrangements with suppressed background contributions. For biological applications, fluorescence is typically implemented because the emission may be readily filtered from the excitation light and background fluorescence may be minimized at near infrared (near-IR) wavelengths. It has been used to map the fluorescence of eumelanins although the specific quantum yields are extremely small.7,8 This property makes it difficult to study the fluorescent properties of melanins in tissue. Alternatively, absorption may be used to study melanins albeit with lower sensitivity because measurements are made with the background excitation source present. For instance, diffuse reflectance studies have reported changes in the melanin spectrum for different pigmented skin lesions9 although the reason for the changes is subject to debate until the linear absorption spectrum is well understood.
Melanins exhibit broad linear absorption spectra that extend from the ultraviolet to the near IR with pheomelanin exhibiting a sharper increase in absorption at shorter wavelengths giving rise to its reddish color. The broad absorption spectrum which is uncharacteristic of organic molecules is similar in appearance to that of an amorphous semiconductor with a smeared band edge.10 Such an electronic structure may occur in extended conjugated polymers with delocalized molecular orbitals. Due to the disordered nature of melanin polymerization, it is very likely that melanosomes contain a pigment distribution that gives rise to the absorption spectrum. In this case, shorter oligomers whose absorption spectra superpose may also account for the broad spectral features.11 This scenario is consistent with structural models for eumelanin12 whereby oligomers may stack to potentially form the nanoscopic structures observed in high resolution AFM studies.13,14 The interpretations illustrate the difficulty in modeling a relatively featureless absorption spectrum. Higher order optical measurements may be used to extend molecular signatures to higher dimensions where different pigments either within an ensemble or in a mixture (such as eumelanin and pheomelanin) may be more clearly resolved.
The simplest variants of nonlinear optical spectroscopies involve focusing a single pulsed laser on a sample to observe nonlinear intensity modulations (such as two-photon absorption15) or the generation of new light frequencies (such as two-photon fluorescence,16 second harmonic generation,17 self-phase modulation18 etc.). While extremely useful for imaging applications, they also give rise to one dimensional datasets for spectroscopic applications that only depend on the frequency of the excitation light. If more than one pulse is used, spectra may be extended to multiple dimensions akin to multi-dimensional nuclear magnetic resonance (NMR) experiments. For example, a two-pulse experiment may be used to probe the transient absorption of a sample.19 In this experiment, a pump pulse resonantly excites molecules while a subsequent probe pulse monitors this excited state population as a function of time and frequency. The excited state population removes ground state absorbers and therefore may give rise to increased probe transmission. Alternatively, the excited state population may absorb at a new frequency which will decrease probe transmission. Additionally, the excited state population may give rise to stimulated emission at a particular probe frequency also resulting in increased probe transmission. Figure 1 summarizes all potential signals that may arise in a transient absorption experiment. The signals also exhibit a time dependence that is a consequence of lifetime, spectral diffusion, photochemical and/or thermal effects. By simultaneously accessing the ground and excited states for a given probe wavelength, different molecules may be resolved even if they exhibit similar absorptivities at the pump wavelength. This effectively stretches absorptive spectral features to multiple dimensions because four variables may be controlled during a transient absorption experiment: pump frequency, probe frequency, inter-pulse delay and the polarization of the light fields. This type of enhanced molecular specificity is ideal for studying melanins which likely consist of a pigment distribution.
Figure 1.
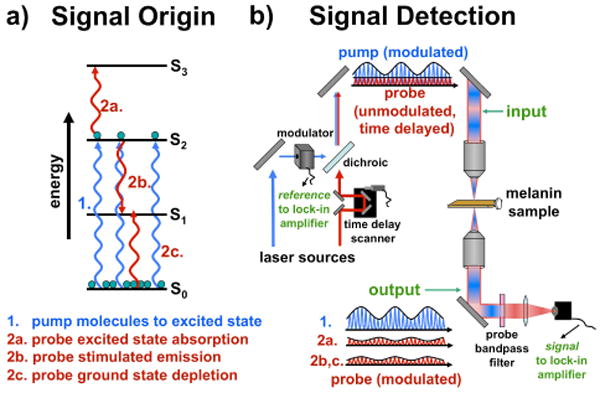
The molecular origin (a) and laboratory detection (b) of transient absorption signals. Two pulses are used to pump molecules to an excited state and probe this population as a function of time as shown in (a). Excited state absorption, stimulated emission and ground state depletion all compete to affect the probe pulse transmission. In the laboratory, two pulses are generated and the pump pulses are modulated using an acousto-optic modulator (AOM) and time delayed relative to the probe pulses before being collinearly focused into the sample as displayed in (b). Probe transmission or backscattering may be detected from the sample by filtering the pump wavelength.
Transient absorption studies of melanins were initially carried out by the Simon group.20,21 They used UV light as a pump source to track the excited state dynamics of Sepia eumelanin and synthetic pheomelanin. Degenerate transient absorption experiments revealed that the excited state relaxes quickly and nonradiatively illustrating the photo-protective nature of melanins. Further broadband experiments revealed a near instantaneous excitation band in a large range spanning the visible and near-IR spectral region for eumelanin and pheomelanin. These data are suggestive of a band type structure for melanin pigments that may result from more extended polymers. Fu et al. have measured the transient absorption of Sepia eumelanin as well as eumelanin/pheomelanin in black and red hairs in the near-IR and have illustrated some differences in human melanin samples.22 A recent transient absorption study was carried out by Piletic et al. in the near-IR spectral region that mapped out the molar absorptivity spectra of Sepia eumelanin and synthetic pheomelanin.23 They observed that pheomelanin absorbs more strongly than eumelanin over a broad spectral range (including the UV) under the assumption that all pigment constituents exhibit broad absorption bands. Estimates of average polymer lengths for eumelanin and pheomelanin were observed to be significantly larger than oligomers. While this study has provided novel insight into the absorptive and structural properties of melanins, it was conducted with a single pump and probe wavelength combination. Multiwavelength studies are warranted to assess the nonlinear response over a broader spectral range in the near IR where scattering contributions are minimized.24
In this work, we apply transient absorption over a broader spectral range in the near-IR with a variety of melanin samples containing either eumelanin or pheomelanin predominantly, including human melanins detected directly in hair without implementing chemical extraction procedures that may affect sample integrity.25,26 We are able to discern between ground state depletion, excited state absorption and stimulated emission contributions by systematically analyzing transient absorptive amplitudes, phase shifts and decay times. This more comprehensive analysis enables us to effectively distinguish between eumelanin and pheomelanin despite their linear absorptive properties being very similar in the near IR spectral range. Stark differences between the two pigment classes are observed when examining their transient absorption wavelength dependence in the near-IR. Furthermore, different types of pheomelanins are optically resolved for the first time to our knowledge. These observations provide important guidance for ongoing work that examines human pigmentation in skin lesions such as melanoma, where we find striking similarities in the dynamics.27
2. Experimental Section
2.1 Materials
Several different synthetic and natural melanin sources were obtained for all optical experiments. Sepia eumelanin which is derived from the ink sac of the cuttlefish Sepia officinalis was purchased from Sigma Aldrich Co. and used without further purification. A dilute suspension of Sepia eumelanin powder was prepared in a phosphate buffer in order to produce an optical quality sample in a 1 mm cuvette. Because Sepia eumelanin is sparingly soluble in aqueous solutions, we used filtration methods to remove large undissolved particles in order to achieve viable clear samples (suspensions) for spectroscopic measurements. Specifically, we mixed 32 mg of Sepia in 10 mL of pH = 7 phosphate buffer solution (50 mM) and sonicated it for 1 hour to break apart larger particles. Because a large amount of the particles still did not dissolve, we filtered the solution using a 1 μm syringe filter (Whatman). The filter was held under vacuum for 18 hours to remove solvent in order to obtain the residual undissolved mass and therefore a final mass concentration. The resulting eumelanin solution had a final mass concentration of 0.84 mg/mL and remained stable for at least several days without the appearance of undissolved particles.
An analogous optical quality solution was made with synthetic pheomelanin because natural pheomelanins are difficult to extract in sufficient quantities.4 We followed the procedure given by Ito.28 All reagents were purchased from Sigma Aldrich Co. and used without further purification. L-dopa (0.1924 g; ∼1 mmol), cysteine (0.1805 g; ∼1.5 mmol) and mushroom tyrosinase (9.2 mg; 5370 units/mg) were allowed to react in a 50 mM sodium phosphate buffer solution (pH = 7) under oxygen for 4 hours. The resulting solution was acidified (pH = 3) and cooled to 4°C for 1 hour giving rise to a red/brown powder that was extracted using ultracentrifugation. We obtained 110 mg (50% yield) of synthetic pheomelanin. 20.5 mg was dissolved and diluted in a pH = 7 phosphate buffer solution (50 mM) to a final mass concentration of 0.84 mg/mL.
In addition to these samples, we also obtained several human hair samples from four young adults (aged 20 – 30 years old) for nonlinear optical experiments. Each of the four individuals possessed hair colors belonging to the following generalized pigment classes: black, brown, red and blond. All hair sources were not previously chemically treated or modified. Several hairs from each individual were collected and cleaned with a dilute soap and water solution and rinsed prior to spectroscopic measurements.
2.2 Experimental Apparatus
Our transient absorption spectrometer consists of a mode-locked Ti:Sapphire laser (Tsunami; Spectra Physics; 80 MHz repetition rate) coupled to an optical parametric oscillator (Opal; Spectra Physics) that are used to generate two tunable femtosecond laser pulses (725 - 810 nm). Longer wavelength pulses are provided by the residual fundamental from the Tsunami laser that is coupled out of the Opal cavity. Shorter wavelengths are accessible by frequency doubling the output of the Opal. The resulting pulses have bandwidths of approximately 9 nm and pulse durations of ∼150 fs as determined by cross-correlation measurements in a BBO crystal. These pulses are used to resonantly excite a small fraction of the ground state population at a particular wavelength and then probe the transient excited state population at another wavelength as a function of time. In order to detect these small absorbance changes, we use a pulse train modulation technique coupled with a sensitive lock-in amplifier detection scheme.22,29 The pump pulses were amplitude modulated at 2 MHz using an acousto-optic modulator (AOM) and time delayed with respect to the probe pulses using a translation stage. Both pulse pathways contain AOMs so that either arm can act as a pump in the transient absorption experiments. Both pulses are then collinearly combined with a beam splitter (Chroma) and focused into the sample with an objective (Olympus; 10×) as shown Figure 1. Typical laser powers for experiments were ∼5 mW for the cuvettes and ∼0.5 mW for the hairs while the laser spot size was ∼3 μm. Lower powers were necessary in the hair experiments to avoid damage associated with absorptive heating in the focal volume. With such high photon and pigment density in the focal volume, it was necessary to arbitrarily move the hair sample while maintaining signal intensity between time delay scans for signal averaging. Prolonged exposure (minutes) at these power levels on the same spot in the hair would cause the transient absorption signals to significantly decrease due to thermal damage. For all the data presented in this work, the pump and probe pulses were polarized parallel with respect to one another. A lock-in amplifier (Stanford Research Systems) is used to detect the amplitude and phase of the small modulation that has been transferred to the probe pulse train. For time delay scans, a 100 ms lock-in time constant was typically used and multiple scans (1 - 20) were averaged to achieve a good signal to noise ratio. For these experiments, a 1 mm cuvette containing a 30 mM rhodamine 6G solution in methanol is used to set the phase of the lock-in amplifier (30 ms time constant; X channel – positive; Y channel – 0) because two-photon absorption occurs at these wavelengths.19 Figure 1b suggests that only two phases will be observed in a typical transient absorption experiment: 180° for excited state absorption (positive signal in the X channel by the convention adopted in this work) and 0° for stimulated emission and ground state depletion (negative signal in the X channel). The same processes can also give arbitrary phase shifts (signals in the X and Y channels) if the dynamics are comparable or longer than the modulation period (500 ns). As will be discussed in detail in the following section, all melanins give rise to short and long time dynamics thereby yielding signals with arbitrary phase shifts.
3. Results and Discussion
Figure 2 displays the transient absorption data for different melanin samples with pump and probe wavelengths centered at 810 nm and 750 nm respectively. The datasets were normalized for pump and probe intensities as well as the absorbance at the pump wavelength. The latter was more difficult to estimate for the hair samples because the absorption spectrum of the hair samples was unavailable and hair scattering contributions also hamper accurate calibrations. Despite this difficulty, estimates for the optical density of hair at 810 nm were based on averaging the laser pulse transmission through different locations in red and black hairs. Scattering contributions to the optical density were partially mitigated by recording the laser pulse transmission through a white hair. This normalization procedure allowed us to not only compare the time dependent transient absorption decays but also their relative signal amplitudes. In addition to these observables, we also recorded the phase shifts of the transient absorption signals as was initially demonstrated by Fu et al. for hair samples.22 Small phase shifts relative to the large transient signals are present in all melanin samples. These phase shifts are a consequence of long lived signals that persist or are comparable to the amplitude modulation period (500 ns).30 All three experimental observables enable the chemical resolution of different melanins including human derived specimens as we demonstrate below.
Figure 2.
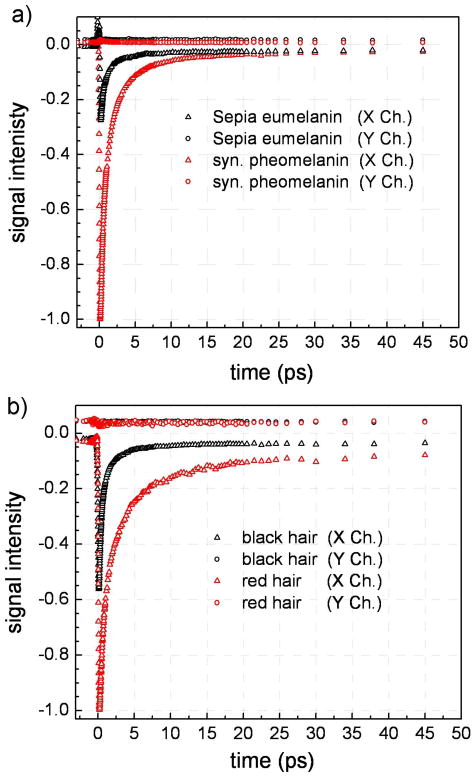
Normalized transient absorption signals for non-human melanins (a) and human melanins (b) detected using 810 nm pump and 750 nm probe pulses. The signals were normalized for input laser intensities and pump absorbance. All samples exhibit rapid non-exponential decays as well as small phase shifts (signal in X and Y channels of the lock-in amplifier – see text). The decays are similar yet distinct in intensity and time dependence when comparing melanins within the same subclass (eumelanins or pheomelanins).
There are notable similarities and differences when comparing the cuvette and hair melanin samples. First of all, all melanins exhibit negative transient signals (X channel) which indicates increased probe transmission subsequent to pump excitation. This sign convention for the transient absorption signals for melanins was initially adopted by Simon and coworkers.21 Figure 1 illustrates that the signals are dominated by either ground state depletion or stimulated emission. Stimulated emission is unlikely to feature in data where the pump wavelength is longer than the probe wavelength because of inaccessible (filled) energy levels. Therefore all melanins exhibit a ground state depletion signal. The data also clearly show that pheomelanins exhibit larger ground state depletion relative to eumelanins for these excitation wavelengths. Piletic et al. have shown that this may be correlated with absorptivity for synthetic pheomelanin and Sepia eumelanin implying that the former absorbs more strongly than the latter.23 This work illustrates that this feature is more general as it also occurs in human derived melanins. Furthermore, these observations may be carried over to the UV regime because melanin pigment constituents are calculated to exhibit broad UV-visible absorption spectra.31-33 These data indicate that significantly less pheomelanin is required to absorb the same amount of UV light relative to eumelanin. Because recent studies have suggested that pheomelanins act as photosensitizers,34,35 their enhanced UV absorptivity highlights the potential significance of an indirect mechanism for the development of skin cancer. In addition to direct UV absorption by DNA and/or proteins in fair skinned individuals, UV light may be absorbed by pheomelanins to yield reactive oxygen species that may subsequently cause cellular damage. Further investigation will unravel the relative importance of these pathways for skin cancer development.
Some significant differences also appear in the transient absorption decays for the melanin samples. All samples exhibit differing phase shifts (in the X and Y channels) in the transient absorption signal. This phase shift arises from dynamics that occur on timescales longer than the pump pulse train modulation period. The lag in molecular response causes increased/decreased probe pulse transmission at times when the pump pulses may be attenuated because they were due to earlier and more intense pump pulses in the modulated pulse train. The tangent of the phase shift is proportional to the time constant of the process (tan(φ) = ωτ)30 and therefore arbitrary phases are possible other than the two depicted in Figure 1. Because the phase shift is a result of long time dynamics, the signal will not depend on the interpulse time delay, which spans a very small time range (ps) relative to the long time signals (ns). Effectively, this translates to time invariant signals in the X and Y channels although the magnitude in the X channel may only be determined before time zero where it is not masked by the large transients that occur when the two pulses have a positive time delay (pump precedes probe). The data in Figure 2 indicate long dynamical timescales are present in all the melanins under study whether they were derived from natural, synthetic, human or nonhuman samples. The amplitude of the long lived signals is larger for the human hair melanins and Sepia eumelanin in comparison with synthetic pheomelanin where it is nearly zero (although non negligible) on the scale shown in Figure 2. Further analysis and discussion of this difference are provided below where data fits are described.
The transient absorption signals present throughout the time delay sequence in Figure 2 were subsequently examined to determine their laser intensity and concentration dependences. Only a concentration dependent study of synthetic pheomelanin was carried out because all hair melanins were studied in situ and it was difficult to manipulate the concentration of Sepia eumelanin because it is sparingly soluble in aqueous solutions and it possessed low absolute signal intensities. Figure 3 displays the concentration and power dependences of the pheomelanin transient absorption signals at different interpulse delays for synthetic pheomelanin. All timescales reveal a linear concentration dependence and quadratic total input laser power dependence. This is characteristic of a transient absorption signal,36 and therefore all signals from all samples were interpreted to arise from similar transient absorption processes.
Figure 3.
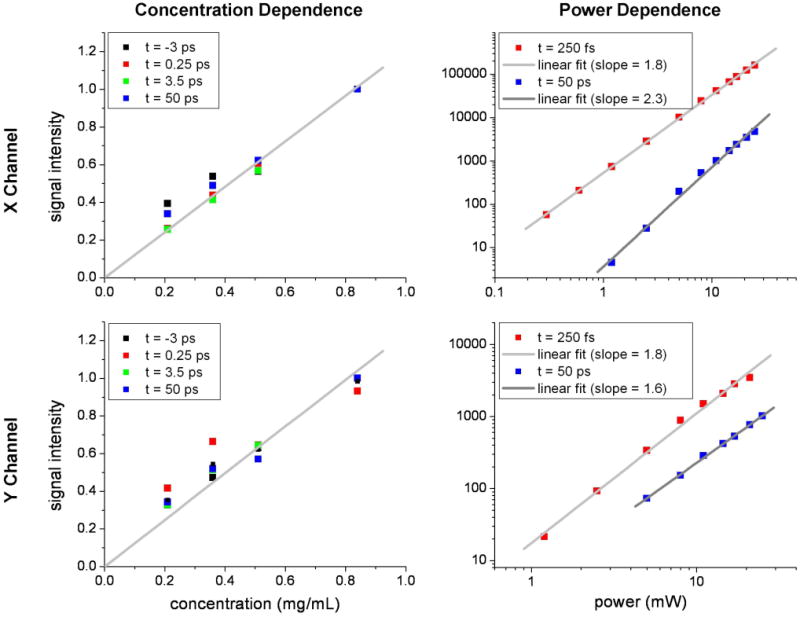
Transient absorption concentration and power dependent studies (810 nm pump, 750 nm probe) of synthetic pheomelanin at various inter-pulse time delays in the X and Y lock-in amplifier channels. All signals exhibit a linear concentration dependence and a quadratic total input power dependence as expected for transient absorption.
In order to understand the differences in the transient absorption dynamics for the different melanins, all of the data in Figure 2 were fit using several models. All fits started at delay times longer than the cross correlation of the pump and probe pulses in order to avoid coherent interactions and nonresonant signals37 occurring while the pulses were overlapped. In the overlap region, the two laser pulses may coherently interact with the molecular system making it extremely difficult to deconvolute molecular dynamics from pulse interactions particularly when the dephasing times are comparable to the pulse duration.38 This cross correlation was typically measured in a cuvette containing rhodamine 6G solution in methanol (30 mM) and was determined to be ∼210 fs. Consequently, all data fits started at 300 fs. The data in Figure 2 appear nonexponential with a fast initial decay followed by a slower process and therefore a biexponential model was initially used to fit all the decays. Figure 4 displays a biexponential fit through pheomelanin data that involved four floating parameters (two amplitudes and two time constants). It inadequately fits intermediate timescales (5 – 50 ps) and more complicated models were introduced. Figure 4 also displays an exponential plus stretched exponential fit (five parameters) as well as a triexponential fit (six parameters). These models necessarily introduce more parameters for better accuracy. The stretched exponential has been used for analyzing dielectric relaxation behavior in polar materials.39 The exponential plus stretched exponential model improves the fit although it still does relatively poorly at intermediate timescales. A triexponential fit introduces six parameters and does the best job in fitting most data points from 300 fs – 50 ps. As a result, this function was used for all subsequent fitting. However, this function only models transients on the picosecond timescale and does not account for the nanosecond components that give rise to a phase shift of the transient absorption data. The amplitude and time constant of the nanosecond components were determined by the amplitude of the X channel component and the ratio of the Y and X channels before time zero respectively (tan(φ) = ωτ = Y/X). Effectively this amounted to a multi-exponential relaxation model whereby four amplitudes and time constants were determined from the data shown in Figure 2.
Figure 4.
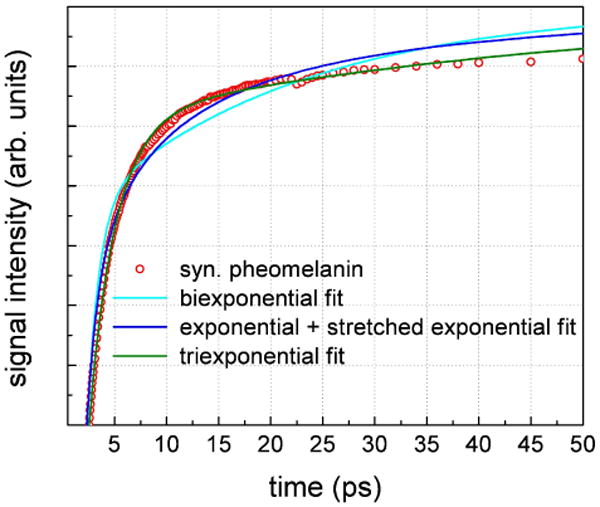
Fits to pheomelanin transient absorption data (810 nm pump, 750 nm probe) using three models: biexponential, exponential plus stretched exponential and triexponential decays. The triexponential model consistently gave the best results and was used to fit other datasets.
The fitting parameters for the different melanin samples are specified in Table 1. Error estimates were included to assess the accuracy of the fits. Pearson's cumulative test statistic, X2 = Si (Yi – Mi)2/Mi (Yi data at point i, Mi model fit at point i) was used as a measure of fit accuracy for each time-delay scan. Each parameter was then systematically varied one at a time (while letting all other parameters float) to record the range over which X2 varied conservatively by 20%. All amplitudes in Table 1 are expressed as percentages of the total fit amplitude. The amplitude of the nanosecond component was fixed to the value in the X channel before time zero. Table 1 illustrates that all melanins exhibit similar time constants: a fast ∼400 femtosecond (fs) component, a ∼2.5 picosecond (ps) component, a ∼50 ps component and a ∼200 ns component. The eumelanins have slightly faster femtosecond decays relative to the pheomelanins and similar intermediate decay times. The large differences in the ∼50 ps component between the eumelanins and pheomelanins may be more subtle as there is a larger error associated in extracting a time constant whose decay extends significantly beyond the data acquired (50 ps). Larger differences are observed in the signal amplitudes, particularly for the pheomelanins. They possess larger amplitude picosecond components relative to the femtosecond component. In particular, the pheomelanins in red hair have extended decays because of this amplitude shift. Sepia eumelanin and eumelanins from black hair have virtually identical decays. Pigment aggregation effects in eumelanins therefore do not seem to affect the transient absorption data because Sepia eumelanin granules are expected to be smaller than 150 nm in size via based on the absorption spectrum reported by Simon and coworkers, while hair eumelanosomes are approximately 1 μm in size.26 One other major difference is that synthetic pheomelanin has an extremely small amplitude in the long time (ns) component. In this instance, aggregation effects may determine the amplitude of the long lived signal because only synthetic pheomelanin is expected to be relatively dispersed in aqueous solution relative to the other samples where pigments are packaged in granules or melanosomes.
Table 1.
Multi-exponential fit parameters to transient absorption data (810 nm pump, 750 nm probe).
| parameters | Sepia eumelanin | black hair | synthetic pheomelanin | red hair |
|---|---|---|---|---|
| A1 | 0.68 +/− 0.10 | 0.73 +/− 0.05 | 0.60 +/− 0.04 | 0.49 +/− 0.04 |
| τ1 (ps) | 0.38 +/− 0.10 | 0.36 +/− 0.06 | 0.47 +/− 0.07 | 0.53 +/− 0.10 |
| A2 | 0.24 +/− 0.10 | 0.20 +/− 0.04 | 0.35 +/− 0.05 | 0.38 +/− 0.04 |
| τ2 (ps) | 2.2 +/− 0.9 | 2.2 +/− 0.6 | 2.4 +/− 0.4 | 3.3 +/− 0.6 |
| A3 | 0.06 +/− 0.02 | 0.04 +/− 0.01 | 0.048 +/− 0.01 | 0.11 +/− 0.02 |
| τ3 (ps) | 64 +/− 35 | 80 +/− 60 | 38+/− 13 | 41 +/− 20 |
| A4 | 0.02 +/− 0.005 | 0.03 +/− 0.004 | 0.002 +/− 0.001 | 0.02 +/− 0.006 |
| τ4 (ns)* | 200 +/− 80 | 160 +/− 30 | 220 +/− 190 | 100 +/− 70 |
τ4 was determined by examining the phase shift of data before time zero analogous to frequency domain fluorescence lifetime measurements.
Long-lived signals have been observed in melanins by Ye et al.21 when using a UV pump source and by Fu et al. using near IR pump pulses.22 In the work by Ye et al., a long lived absorptive signal (ns) was observed when probing at visible wavelengths while Fu et al. observed long lived ground state depletion signals when probing at near IR wavelengths. Both works ascribe the long-lived signals to certain intermediate states in melanins although this would have to be verified using detailed molecular structure and energy relaxation calculations. Fu et al. investigated the possibility that this signal may also be due to thermal effects associated with depositing energy in the laser focal volume.22 Two thermal effects may occur subsequent to the absorption of the pump pulse: a thermal lens may form which will distort the transmitted probe beam or a temperature gradient may give rise to spectral changes at the probe wavelength. Thermal lensing was effectively ruled out as a likely contributor by placing an aperture before the detector and observing no changes to the shape of the spectral decays. Fu et al. deemed spectral changes due to increased temperature insignificant because the ratio of the long lived signal to the short transients was independent of beam overlap. In this instance, it is assumed that the thermal signals should not change appreciably at or near the pump focal volume. This explanation is expected to hold in a sample with a small thermal gradient whereby the hot spot has diffused significantly beyond the laser focal volume thereby creating a fairly uniform temperature distribution. Additionally, the spatial distribution of pigments should not change appreciably when tuning away from the focal volume. Both assumptions may not hold for human hair samples because melanins have slow diffusion times relative to the size of single melanosome (1 μm)40 and there is a nonuniform spatial pigment distribution in the hairs. Thermal diffusion in melanin was therefore modeled to determine whether the calculated timescales correspond to the observed long time dynamics.
In a rudimentary thermal diffusion model, the absorption of pump photons leads to a local hot spot in the pigment aggregate (granule or melanosome), which may affect the local pigment absorption spectrum that is subsequently detected. Thermal diffusion carries the thermal energy away from the focal volume and will give rise to a decay in the temperature induced signal. Figure 5 displays the one dimensional spatial and temporal temperature profile of an excited melanin aggregate (1 μm in size) using the heat equation. The thermal diffusivity of water is 140,000 μm2/s41 while it is 300,000 μm2/s for eumelanin.40 The former diffusion coefficient is valid for the melanin aqueous solutions while the latter is more applicable to the hair samples which contain concentrated melanin pigments (aggregated melanosomes). The calculation in Figure 5 implemented the thermal diffusivity of water which represents an upper bound to the calculated time constant shown in Figure 5b because water's thermal diffusivity is smaller than melanin's. Figure 5 illustrates that a substantial temperature gradient exists on the nanosecond timescale while a significant portion of the thermal energy leaves the micron spot within 1.4 microseconds. This long component is within an order of magnitude of the measured timescales in the various samples. The model also accounts for the suppressed long-time signal in synthetic pheomelanin because the thermalization process will lead to heating of the local solvent (water) instead of other pigments in the more aggregated samples and will therefore not contribute to the transient absorption signal because of the minimal absorptivity of water in the near-IR. Consequently, thermal diffusion must be taken into account when interpreting the nanosecond transient absorption signals.
Figure 5.
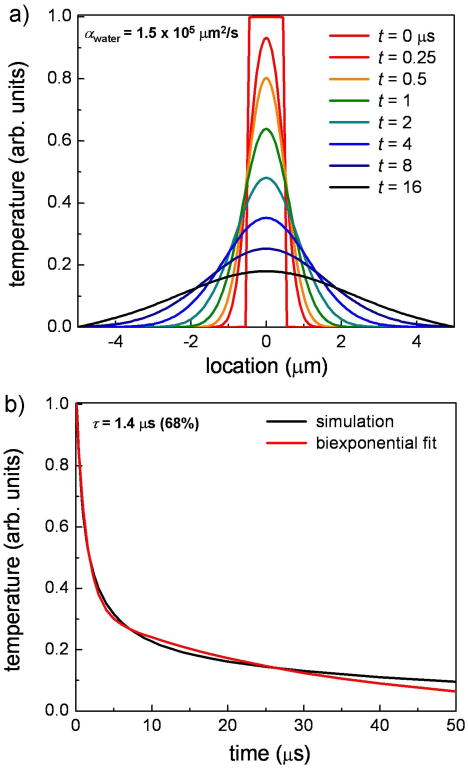
Solution to the one-dimensional heat equation for an initial temperature distribution localized to 1 μm (approximate size of a human melanosome). The model assumes that the absorbed pump radiation converts to heat and gives rise to a localized ‘hot spot’ that affects the local absorption spectrum. Water's thermal diffusion constant was used in the calculations which is valid for melanin aggregates surrounded by water (cuvette samples). In (a), substantial thermal gradients exist on the microsecond timescale which also corresponds to the time it takes for the hot spot to dissipate as shown in (b). The calculated signal in this no adjustable parameter simple model is within an order of magnitude of the measured long time signals for melanins.
Several effects need to be considered to enable more accurate estimates of the thermal diffusion times given the simplicity of the model. They include thermal impedance mismatch at melanosome boundaries, non-uniform melanosome excitation and sample heterogeneity. Incorporating all effects in a more comprehensive model would be extremely challenging considering that pigment structures, concentrations and distribution within melanosomes are not well known. However, estimations regarding the impact of each effect on the thermal diffusion model may be made readily. Thermal impedance effects due to a mismatch of diffusivities will increase the thermal diffusion time. However, this will more significantly affect the Sepia melanin solution since granules are suspended in water. In hair, melanosomes are more aggregated and the thermal diffusivity of melanin would be an appropriate estimate for thermal diffusion. Non-uniform melanosome excitation will have an opposing effect on the thermal diffusion signal timescale and amplitude. In this case, the ‘shell’ of the aggregate is preferentially excited and therefore the thermal energy will dissipate away more quickly because it is closer to the solvent. This may also affect dynamics occurring at shorter timescales due to the close proximity of near-IR absorbing and non-absorbing species. We have observed picosecond dynamics being affected with increased excitation intensity (tens of milliwatts). This trend of decreasing signal amplitude and timescale is also expected for an inhomogeneous distribution of pigments within a melanosome because excited pigments will dissipate absorbed energy to proteins and lipids that do not appreciably absorb in the near-IR. When taking all these effects into account, the calculated timescale is expected to be shorter in general akin to the long timescale data measured for all melanins (see Table 1).
Thermal effects may be evaluated in the laboratory by conducting temperature dependent UV-visible spectroscopy. Preliminary measurements of melanins show extremely small spectral changes (∼0.001 O.D.), which are highly susceptible to baselining errors making it difficult to determine the sign of the associated change. However, because our transient absorption spectrometer is sensitive to 10−6 absorption changes,19 temperature associated spectral changes are more easily detected. Other laboratory measurements such as the power dependence shown in Figure 2 do not preclude temperature dependent thermal effects for the long time signals because a temperature dependent spectral change will also have a quadratic total power dependence. It will depend linearly on the pump pulse intensity due to linear absorption and linearly on the probe pulses because the signal scales with the probe intensity difference between preceding high and low intensity pump pulses. Thermal effects must therefore be carefully considered in comprehensive investigations involving the nonlinear excitation of melanins.
Interestingly, the multi-exponential fits described above very closely resemble those obtained by Ye et al.21 Using a UV pump source, they also found that four relaxation timescales adequately fit the data (ranging from femtoseconds to nanoseconds). The similarity of the observed relaxation time constants is highly suggestive of similar molecules being targeted in both experiments. This may only occur if the pigment molecules exhibit broad spectral absorptions. This was calculated to be the case for eumelanin oligomers32 and observed in pheomelanin precursors42 and therefore larger polymers are also expected to exhibit this feature. This observation further supports the claim that pheomelanin absorbs UV more strongly than eumelanin even when measurements were made at near IR wavelengths.23 Because energy relaxation processes depend on how specific energy levels couple to the surrounding bath of modes,36 identical relaxation timescales likely imply that similar energy levels are being excited and subsequently probed. The only way this may occur in two experiments that pump melanins with drastically different wavelengths is if there is a fast internal relaxation (< 300 fs) event that brings an excitation (UV or near IR) to the same energy level(s) before it goes on to relax further. Depending on the probe pulse wavelength, this intermediate state may absorb photons or give rise to stimulated emission in addition to depleting the ground state. In the discussion that follows, we assess the relative importance of these contributions to the transient absorption signals.
Figure 6 displays transient absorption decays for two wavelength combinations (810 nm and 750 nm) where the pump and probe were interchanged. The data were once again normalized for pump absorbance as well as the input laser intensities. For both Sepia eumelanin and synthetic pheomelanin, the decays are more rapid and lower in intensity when pumping at higher energy. The signal intensity for Sepia eumelanin is almost completely extinguished when pumping at 750 nm. As mentioned previously, stimulated emission processes are unlikely when pumping at lower energy because the lower energy states are filled. When the pump is centered at 810 nm, only ground state depletion and excited state absorption contribute to the detected response. The large negative intensities reveal that ground state depletion dominates the signal. However, when pumping at 750 nm, all three mechanisms may affect the probe transmission. Blanchard et al. have shown that in the limit of ground state depletion, the normalized signals will be proportional to the molar absorptivity at the probe wavelength at time zero (before relaxation takes place).43 Therefore if the ratio of the two signals at early time is similar to the ratio of the absorbances at 750 nm and 810 nm, then ground state depletion is solely responsible for the observed signals. The absorbance ratios (Abs750nm/Abs810nm) for Sepia eumelanin and synthetic pheomelanin are 1.5 and 1.6 respectively while the transient absorption early time signal ratios are 2.8 and 1.2. It is evident that pheomelanin more closely tracks the ground state absorbance indicating that ground state depletion is dominant at both wavelengths. However, Sepia eumelanin possesses a significantly larger nonlinear absorptive ratio that is caused by the vanishingly small signals occurring with 750 nm pump pulses. While excited state absorption may affect both wavelengths, it is more dramatically influencing the dynamics when 750 nm pump pulses are used by effectively competing with ground state depletion and canceling out the transient absorption signal because both mechanisms give rise to signals of opposite phase (see Figure 1). Excited state absorption cross sections must therefore be more significant when pumping at higher energy relative to the ground state. This may account for why only excited state absorption was observed by Simon and coworkers when pumping with UV photons in their nondegenerate experiments.21
Figure 6.
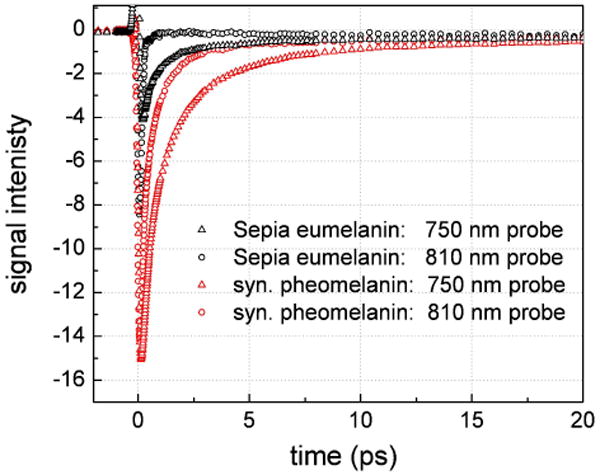
Normalized transient absorption decays for Sepia eumelanin and synthetic pheomelanin at two different wavelength combinations: 810 nm pump, 750 nm probe and 750 pump, 810 nm probe. The signals were normalized for input laser intensities and pump absorbance. The Sepia eumelanin signal nearly vanishes when using 750 nm pump and 810 nm probe excitations indicating that excited state absorption is beginning to cancel out the effect of ground state depletion.
The presence of excited state absorption prompted further wavelength dependent studies to determine the pump wavelengths that cause the transient absorption signals to switch phase (from ground state depletion to excited state absorption). Figure 7 portrays pump wavelength dependent transient absorption decays for Sepia eumelanin and synthetic pheomelanin. It is readily apparent that Sepia eumelanin exhibits a change in the sign of the signal between 735 and 750 nm. Therefore, at approximately 745 nm Sepia eumelanin transitions from ground state depletion (at longer wavelengths) to excited state absorption (at shorter wavelengths) when probing in the near-IR. For a fixed probe wavelength, pheomelanin too exhibits a similar trend to eumelanin although it lags in pump wavelength and never goes positive for the wavelengths studied. If the data are extrapolated, the crossover point is estimated to occur at ∼700 nm for pheomelanin. Ye et al. also observed this trend although at longer wavelengths when pumping with UV photons.21 They observed that pheomelanin signals vanish at ∼800 nm while eumelanin signals are still substantial there implying a longer crossover wavelength. Recent studies have examined the optical band gap for synthetic eumelanins and have determined them to be 1.39 eV (892 nm)44 and 1.7 eV (730 nm)40 using optical absorption and photothermal methods respectively. Our crossover wavelength at 745 nm lies in this range and may very well correspond to a HOMO-LUMO transition that yields excited state absorption when pumping at higher energies. We have now observed this phenomenon in a natural eumelanin sample (Sepia) as well as a pheomelanin sample.
Figure 7.
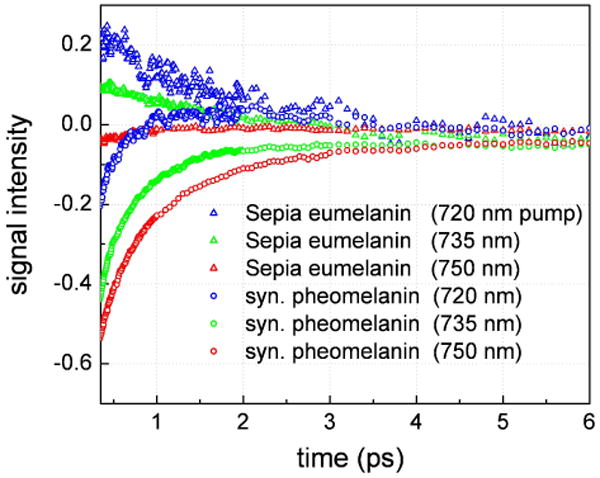
Normalized transient absorption decays for Sepia eumelanin and synthetic pheomelanin with fixed probe wavelength (810 nm). The signals were normalized for input laser intensities and pump absorbance. By varying the pump wavelength, the Sepia eumelanin signals revert from ground state depletion to excited state absorption at ∼ 745 nm while they are estimated to approach this point at ∼700 nm for pheomelanin.
The question arises as to why the crossover transition lies at higher energy for pheomelanin? If this transition is correlated to a HOMO-LUMO transition, then conjugation may play an important role. Eumelanins have been modeled as biopolymers containing indole units which are aromatic and conjugated over two rings. Furthermore, bonding in planar arrangements may extend this conjugation significantly. In pheomelanins, benzothiazines have been proposed as intermediates which possess one conjugated benzene ring. The extent of further conjugation upon polymerization appears unlikely and therefore may give rise to a higher energy HOMO-LUMO transition. Stacking of constituent polymers may also play an important role in determining the electronic structures.
Different types of eumelanin and pheomelanin pigments such as those derived from different human hairs will also exhibit distinct electronic structures. It remains a great experimental challenge to discern between pigments within the eumelanin and pheomelanin classes because of the low sensitivity afforded by chemical and spectroscopic assays and the difficulty in extracting the insoluble pigments. Nonlinear transient absorption spectroscopy offers the advantages of enhanced molecular specificity as well as the capability of making high spatial resolution measurements of melanins in situ. Figure 8 displays the transient absorption decays of melanins from six sources: Sepia, synthetic pheomelanin, black, brown, red and blond hairs. All data were acquired in transmission mode. In Figure 8, all the data were baselined before time zero and normalized in order to readily observe differences in the decays. Using 810 nm as a pump source, a single wavelength measurement was sufficient to distinguish between the synthetic pheomelanin and the pheomelanin present in the red and blond hairs. Synthetic pheomelanin possesses the fastest decay while red hair pheomelanin has the slowest decay. This is somewhat surprising considering that there is some eumelanin present in the red hairs.45 The blond hair transient absorption data more closely resembles the synthetic pheomelanin decay suggesting a similarity in their chemical structures. All the eumelanin transient absorption data was virtually super imposable indicating that concentration effects dominate the visible appearance of these melanins and not different pigments or aggregation states. Further transient absorption experiments over a broader wavelength range and a larger variety of melanin samples derived from different natural sources will enhance our understanding of the melanin pigment family.
Figure 8.
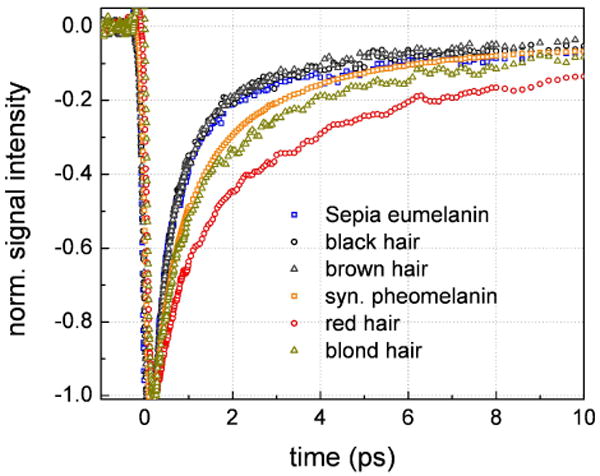
Baselined and normalized transient absorption decays for various non-human and human melanins. In this case the signals were normalized to have the same value at early time (200 fs) in order to compare the short-lived transients. The signals exhibit variability for different pheomelanins while they are virtually identical for different eumelanins.
In addition to optical experiments, sensitive structural and chemical techniques need to be developed and applied to melanins in order to further progress the current knowledge of melanin structure. Conventional techniques like infrared spectroscopy, nuclear magnetic resonance spectroscopy and elemental analysis do provide important molecular details but are of limited value when trying to analyze the transient absorption data of melanins presented here. Only calculations based on accurate molecular electronic structures will aid the comprehensive analysis of melanin transient absorption spectra because it will enable energy relaxation processes to be effectively modeled. Further development in these areas will benefit the field of melanin chemistry tremendously.
4. Concluding Remarks
Human and nonhuman melanins were studied with nonlinear near-IR transient absorption spectroscopy. Relaxation dynamics exhibit similar timescales when pumping in the UV and the near IR. The relative contributions of excited state absorption, stimulated emission and ground state depletion were determined. Eumelanin exhibits a transition from ground state depletion to excited state absorption at ∼745 nm while the transition for pheomelanin occurs at ∼700 nm. This transition corresponds closely to recent reports for the band gap of eumelanins. Varying transient absorption decays for six different melanin samples allowed us to decipher different types of pheomelanins. All the information presented above was obtained from transient absorption experiments conducted at only several wavelengths. This work in conjunction with previous calibrated measurements have helped to address some long standing challenges in melanin chemistry; namely pigment sizes, photo-relaxation dynamics and molecular detection in situ. The latter application holds significant promise for analyzing pigmented skin diseases such as malignant melanoma, which are frequently accompanied by pigment proliferation.
Acknowledgments
We thank the National Institutes of Health (NIH: 1RC1CA145105-01) for the financial support of this work.
References
- 1.Nofsinger JB, Forest SE, Simon JD. J Phys Chem B. 1999;103:11428. [Google Scholar]
- 2.Simon JD, Peles D, Wakamatsu K, Ito S. Pigm Cell Melanoma R. 2009;22:563. doi: 10.1111/j.1755-148X.2009.00610.x. [DOI] [PubMed] [Google Scholar]
- 3.Ito S. Pigm Cell Res. 2003;16:230. doi: 10.1034/j.1600-0749.2003.00037.x. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y, Hong L, Wakamatsu K, Ito S, Adhyaru B, Cheng CY, Bowers CR, Simon JD. Photochem Photobiol. 2005;81:135. doi: 10.1562/2004-08-03-RA-259.1. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Simon JD. Pigm Cell Res. 2005;18:42. doi: 10.1111/j.1600-0749.2004.00197.x. [DOI] [PubMed] [Google Scholar]
- 6.Ito S, Fujita K. Anal Biochem. 1985;144:527. doi: 10.1016/0003-2697(85)90150-2. [DOI] [PubMed] [Google Scholar]
- 7.Meredith P, Sarna T. Pigm Cell Res. 2006;19:572. doi: 10.1111/j.1600-0749.2006.00345.x. [DOI] [PubMed] [Google Scholar]
- 8.Riesz J, Sarna T, Meredith P. J Phys Chem B. 2006;110:13985. doi: 10.1021/jp054869l. [DOI] [PubMed] [Google Scholar]
- 9.Zonios G, Dimou A, Carrara M, Marchesini R. Photochem Photobiol. 2010;86:236. doi: 10.1111/j.1751-1097.2009.00630.x. [DOI] [PubMed] [Google Scholar]
- 10.McGinness J, Corry P, Proctor P. Science. 1974;183:853. doi: 10.1126/science.183.4127.853. [DOI] [PubMed] [Google Scholar]
- 11.Tran ML, Powell BJ, Meredith P. Biophys J. 2006;90:743. doi: 10.1529/biophysj.105.069096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng J, Moss SC, Eisner M. Pigm Cell Res. 1994;7:263. doi: 10.1111/j.1600-0749.1994.tb00061.x. [DOI] [PubMed] [Google Scholar]
- 13.Clancy CMR, Simon JD. Biochemistry-US. 2001;40:13353. doi: 10.1021/bi010786t. [DOI] [PubMed] [Google Scholar]
- 14.Watt AAR, Bothma JP, Meredith P. Soft Matter. 2009;5:3754. [Google Scholar]
- 15.Ye T, Fu D, Warren WS. Photochem Photobiol. 2009;85:631. doi: 10.1111/j.1751-1097.2008.00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denk W, Strickler JH, Webb WW. Science. 1990;248:73. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 17.Fejer MM, Magel GA, Jundt DH, Byer RL. IEEE J Quantum Elect. 1992;28:2631. [Google Scholar]
- 18.Fischer MC, Liu HC, Piletic IR, Warren WS. Opt Express. 2008;16:4192. doi: 10.1364/oe.16.004192. [DOI] [PubMed] [Google Scholar]
- 19.Fu D, Ye T, Matthews TE, Yurtsever G, Warren WS. J Biomed Opt. 2007;12:054004. doi: 10.1117/1.2780173. [DOI] [PubMed] [Google Scholar]
- 20.Nofsinger JB, Ye T, Simon JD. J Phys Chem B. 2001;105:2864. [Google Scholar]
- 21.Ye T, Simon JD. J Phys Chem B. 2003;107:11240. [Google Scholar]
- 22.Fu D, Ye T, Matthews TE, Grichnik J, Hong L, Simon JD, Warren WS. J Biomed Opt. 2008;13:054036. doi: 10.1117/1.2976424. [DOI] [PubMed] [Google Scholar]
- 23.Piletic IR, Matthews TE, Warren WS. J Chem Phys. 2009;131:181106. doi: 10.1063/1.3265861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riesz J, Gilmore J, Meredith P. Biophys J. 2006;90:4137. doi: 10.1529/biophysj.105.075713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Kempf VR, Nofsinger JB, Weinert EE, Rudnicki M, Wakamatsu K, Ito S, Simon JD. Pigm Cell Res. 2003;16:355. doi: 10.1034/j.1600-0749.2003.00059.x. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Simon JD. Pigm Cell Res. 2003;16:606. doi: 10.1046/j.1600-0749.2003.00098.x. [DOI] [PubMed] [Google Scholar]
- 27.Matthews TE, Piletic IR, Selim MA, Simpson MJ, Warren WS. Sci Transl Med. 2010 doi: 10.1126/scitranslmed.3001604. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito S. Pigm Cell Res. 1989;2:53. doi: 10.1111/j.1600-0749.1989.tb00158.x. [DOI] [PubMed] [Google Scholar]
- 29.Fu D, Matthews TE, Piletic IR, Warren WS. J Biomed Opt. 2008;13:0405031. doi: 10.1117/1.2968260. [DOI] [PubMed] [Google Scholar]
- 30.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3rd. Springer; New York: 2006. [Google Scholar]
- 31.Kaxiras E, Tsolakidis A, Zonios G, Meng S. Phys Rev Lett. 2006;97:218102. doi: 10.1103/PhysRevLett.97.218102. [DOI] [PubMed] [Google Scholar]
- 32.Zonios G, Dimou A, Bassukas I, Galaris D, Tsolakidis A, Kaxiras E. J Biomed Opt. 2008;13:014017. doi: 10.1117/1.2844710. [DOI] [PubMed] [Google Scholar]
- 33.Stark KB, Gallas JM, Zajac GW, Eisner M, Golab JT. J Phys Chem B. 2003;107:11558. [Google Scholar]
- 34.Takeuchi S, Zhang WG, Wakamatsu K, Ito S, Hearing VJ, Kraemer KH, Brash DE. P Natl Acad Sci USA. 2004;101:15076. doi: 10.1073/pnas.0403994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wenczl E, Van der Schans GP, Roza L, Kolb RM, Timmerman AJ, Smit NPM, Pavel S, Schothorst AA. J Invest Dermatol. 1998;111:678. doi: 10.1046/j.1523-1747.1998.00357.x. [DOI] [PubMed] [Google Scholar]
- 36.Yan YJ, Mukamel S. Phys Rev A. 1990;41:6485. doi: 10.1103/physreva.41.6485. [DOI] [PubMed] [Google Scholar]
- 37.Lebedev MV, Misochko OV, Dekorsy T, Georgiev N. J Exp Theor Phys. 2005;100:272. [Google Scholar]
- 38.Hamm P. Chem Phys. 1995;200:415. [Google Scholar]
- 39.Shlesinger MF, Montroll EW. P Natl Acad Sci USA. 1984;81:1280. doi: 10.1073/pnas.81.4.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Albuquerque JE, Giacomantonio C, White AG, Meredith P. Appl Phys Lett. 2005;87:061920. [Google Scholar]
- 41.James DW. J Mater Sci. 1968;3:540. [Google Scholar]
- 42.Napolitano A, Di Donato P, Prota G, Land EJ. Free Radical Bio Med. 1999;27:521. doi: 10.1016/s0891-5849(99)00098-2. [DOI] [PubMed] [Google Scholar]
- 43.Blanchard GJ, Wirth MJ. Anal Chem. 1986;58:532. [Google Scholar]
- 44.Ligonzo T, Ambrico M, Augelli V, Perna G, Schiavulli L, Tamma MA, Biagi PF, Minafra A, Capozzi V. J Non-Cryst Solids. 2009;355:1221. [Google Scholar]
- 45.Ito S, Wakamatsu K. Pigm Cell Res. 2003;16:523. doi: 10.1034/j.1600-0749.2003.00072.x. [DOI] [PubMed] [Google Scholar]