

Abstract
Steady increase in the incidence of atherosclerosis is becoming a major concern not only in the United States but also in other countries. One of the major risk factors for the development of atherosclerosis is high concentrations of plasma low density lipoprotein (LDL), which are metabolic products of very low density lipoprotein (VLDL). VLDLs are synthesized and secreted by the liver. In this review, we discuss various stages through which VLDL particles go from their biogenesis to secretion in the circulatory system. Once VLDLs are synthesized in the lumen of the endoplasmic reticulum (ER), they are transported to the Golgi. The transport of nascent VLDLs from the ER to Golgi is a complex multi-step process, which is mediated by a specialized transport vesicle, the VLDL transport vesicle (VTV). The VTV delivers VLDLs to the cis-Golgi lumen where nascent VLDLs undergo a number of essential modifications. The mature VLDL particles are then transported to the plasma membrane and secreted in the circulatory system. Understanding of molecular mechanisms and identification of factors regulating the complex intracellular VLDL trafficking will provide insight into the pathophysiology of various metabolic disorders associated with abnormal VLDL secretion and identify potential new therapeutic targets.
Introduction
The biogenesis of very low density lipoproteins (VLDLs) and their secretion into the circulatory system by the liver is a complex but highly controlled process that plays an important role in overall lipid homeostasis. Enhanced production of VLDLs and their eventual secretion into the circulatory system constitute one of the major risk factors for the development of atherosclerosis.1,2 Higher concentrations of VLDLs in the blood are often translated into higher levels of atherogenic particles, low density lipoproteins (LDLs). Interestingly, the production of VLDLs needs to be synchronized with their secretion to avoid adverse consequences such as hepatic steatosis.3 These two clinical scenarios arising from abnormal assembly and secretion of VLDLs underscore their importance. From their production to secretion, VLDLs go through several complex intracellular processes that determine the rate of secretion in the circulation. A number of factors have been well documented in literature, which affect VLDL assembly and secretion.12,89 Of particular interest is insulin resistance, which is generally described as decreased physiological response to circulating insulin by the liver, skeletal muscle and adipose tissue.90 Insulin resistance leads to enhanced VLDL synthesis and secretion; 12,89 however, its effects on intracellular VLDL trafficking have not been investigated.
In this review, we focus on intracellular trafficking of nascent VLDLs, particularly their movement from their site of synthesis, the endoplasmic reticulum (ER) to the cis-Golgi because this step is crucial for their secretion and abnormalities associated with this process lead to various clinical disorders. Also, we highlight differences in the intracellular transports of VLDLs and their small intestinal counterparts, the chylomicrons.
Biogenesis of VLDL
One of the major functions of the liver is to synthesize VLDLs and this process primarily relies on the availability of triglycerides.4–7 Triglycerides utilized for VLDL assembly are synthesized in the ER lumen in a preventive response to free fatty acids influxes. There are mainly three sources that supply free fatty acids to the liver: (i) free fatty acids from adipocytes; (ii) chylomicron remnants and; (iii) from the intestine via the portal vein. Nascent VLDLs produced in the liver are composed of a core of neutral lipids (mostly triglycerides), which is surrounded by phospholipids, cholesterol, cholesteryl esters and specific apolipoproteins.8 The size of newly synthesized VLDL particle ranges between 35 – 100 nm in diameter.9 Of several apolipoproteins, apolipoprotein B (apoB) is the most important and provides structural stability to the nascent VLDL particle.10
ApoB is an essential structural component of VLDL, intermediate-density lipoprotein (IDL), LDL and chylomicrons.11,12 It is a large amphipathic glycoprotein and exists in two forms: apoB100 and apoB48. ApoB100 is the fully translated protein consisting of 4536 amino acids and has a molecular mass of ~515 kDa.13 ApoB48 is the truncated form (48 percent) of the full-length apoB and contains 2152 amino acid residues. ApoB48 is synthesized as a result of apobec-1 mediated site-specific mRNA editing that produces a stop codon.14,15 In humans, apoB100 is expressed in liver and is the key structural protein of VLDL, IDL and LDL, whereas apoB48 is primarily synthesized by the intestine and is a major structural protein of chylomicrons.16–18 However, rodent liver synthesizes both apoB100 and apoB48.93
Using a computer program, Segrest et al proposed that apoB100 has a pentapartite structure (NH2-βα1−β1−α2−β2−α3−COOH) consisting of two amphipathic α-helices and two amphipathic β-sheet domains.19 The N-terminal sequence (1000 amino acids) of apoB100, in conjunction with microsomal triglyceride transfer protein, forms a lipid binding pocket.20 Like other secretory proteins, apoB is synthesized at the ER surface and is targeted to the ER by its N-terminal 27-amino acids sequence, which acts as a signal-peptide. Translocation of apoB into the ER lumen occurs through translocons simultaneously with its translation, however, there is evidence that suggest translocation of apoB is not continuous.91,92
The biogenesis of VLDL occurs in the lumen of the ER and this process is accomplished in two steps.21,22 The formation of VLDL starts with the translocation of newly translated apoB100 across the rough ER membrane. In the first step, nascent apoB100 is partially lipidated to form a lipid-poor primordial VLDL particle. This step is facilitated by microsomal triglyceride transfer protein (MTP).23 MTP is a lipid transfer protein and is shown to have three domains: (i) an apoB-binding domain; (ii) a lipid transfer domain and; (iii) a membrane association domain.24–26 MTP is able to transfer both neutral and polar lipids to the developing VLDL particle, however, recent data from the Hussain’s group clearly indicate that the phospholipid transfer domain of MTP can alone generate VLDL particles.27 Lipidation of apoB depends on the availability of triglycerides; without lipidation a significant amount of nascent apoB gets degraded.7,28,29 Independent of its translational status, nascent apoB can undergo degradation by different processes.29,30 In the absence of sufficient lipids, it can undergo degradation co-translationally in the ER lumen where cytosolic chaperons like hsp70 and hsp90 direct it to the proteasome for degradation.31,32 Additionally, it can undergo a proteasome-independent post-translation degradation, which can be stimulated by n-3 fatty acids and insulin.7 In addition to MTP, Hsp110 has been shown to prevent apoB from degradation.33 In the second step of VLDL formation, the primordial VLDL particle fuses with triglyceride-rich particles already present in the cytosol. The role of MTP activity at this step is debated.8,34 Recently, it has been proposed that cideB (a homolog of cell death-inducing DFF45-like effector) protein also plays a role in the lipidation of the apoB100-containing poorly lipidated primordial VLDL particle.35
ER-exit of VLDL
The movement of nascent VLDLs from the site of their biogenesis, the ER, to their next destination, the cis-Golgi, is prerequisite for their ultimate secretion from hepatocytes. Because of their large size (average diameter > 80 nm), it is highly unlikely that VLDL particles can move across the ER membrane due to physical restraints. Moreover, VLDLs need to be directed to the cis-Golgi to avoid their delivery to a random intracellular compartment. This suggests that VLDLs may require specialized transport machinery for their export from the ER to the cis-Golgi. Transport of nascent proteins from the ER to the cis-Golgi along the secretory pathway involves their exit from the ER and delivery to the Golgi.36 These newly synthesized proteins are packaged into protein transport vesicles (PTVs), which bud off from the ER membrane, traverse the ER-Golgi intra-cellular space, and fuse with the Golgi. The size of these vesicular carriers, however, ranges between 55 – 70 nm in diameter, limiting their ability to accommodate VLDL-sized particles.37
The biogenesis of PTV that transports nascent proteins from the ER to the Golgi is mediated by the orderly recruitment of a specific set of cytosolic proteins to the ER membrane. These proteins form a protein-complex, known as coat protein complex II (COPII) on the ER membrane, which leads to cargo selection and vesicle formation.36,38 The assembly of the COPII coat occurs at distinct sites on the ER membrane, called ER exit sites (ERES).39,40 The formation of COPII begins with the conversion of Sar1-GDP to Sar1-GTP, which is facilitated by Sec12, a guanine nucleotide exchange factor (GEF) for Sar1, localized to the ER membrane.37,41–44 Sec12-mediated conversion of Sar1 triggers the exposure of its α-helical element, which in turn inserts into the ER membrane.45,46 Once attached to the ER membrane, Sar1 recruits Sec23 and Sec24 as a heterodimer to the ER membrane. The Sec23–Sec24 complex forms the inner layer of the COPII coat, which plays a major role in cargo selection.47–49 The Sar1–Sec23–Sec24 complex attached to the ER membrane is generally referred as the pre-budding complex and recruits Sec13 and Sec31 as a heterotetramer to the ER membrane. The Sec13–Sec31 heterotetramer forms the outer layer of the COPII coat and triggers membrane deformation leading to vesicle formation.50,51
In an elegant study using cryo-electron microscopy, Balch and his colleagues have delineated the structure of the COPII coat and suggested that the geometry of the COPII coat possesses sufficient flexibility to form enlarged vesicles which can accommodate cargoes of up to ~100 nm in size.50 One interpretation of their data could be that VLDL can travel in PTVs on their way to the cis-Golgi. Although PTVs have been suggested to be able to accommodate VLDL-sized particles, recent studies suggested that VLDLs exit the ER differently than newly synthesized proteins. Independent studies from the Fisher’s group and our laboratory clearly indicate that VLDL does not exit the ER in canonical COPII vesicles.52,53
Formation of VTV
Unlike chylomicrons, the lipoproteins produced in small intestine, VLDLs depart the ER in COPII-dependent fashion.53,54 In an effort to find out how nascent VLDLs leave the ER in primary hepatocytes, an in vitro ER-budding assay was established to follow their transport from the ER. This cell-free in vitro system allowed us to monitor the ER-exit of both nascent VLDLs and newly synthesized proteins simultaneously.53 These studies revealed that the newly synthesized hepatic secretory protein, albumin and VLDL exit the hepatic ER separately in two different vesicles (Figure 1). The transport of albumin utilizes the classical COPII-coated PTVs whereas VLDL departs the ER in a specialized vesicle, the VLDL transport vesicle (VTV). The size of VTV ranges between 100 – 120 nm in diameter – suitable to comfortably contain VLDL-sized cargo.53 A detailed biochemical characterization of the VLDL-containing vesicle suggests that VTV is an authentic transport compartment which (i) concentrates the VLDL marker protein, apoB100, indicating the presence of VLDL inside the vesicle (ii) is not broken ER membranes as confirmed by proteinase K data which shows that VLDL- apoB100 within VTV is protected from proteolysis (iii) excludes ER-resident proteins calnexin or calreticulin (iv) concentrates COPII proteins, marker proteins for the ER-to-Golgi intermediate compartment (v) is able to fuse with hepatic cis-Golgi to deliver its cargo, the VLDL. Gusarova et al. suggested that newly synthesized apoB100 is exported from the ER to the Golgi in a COPII-dependent manner in rat hepatoma cells, McA-RH7777; however, vesicles containing apoB100 appeared to be different than bona fide COPII vesicles.52 The size of these vesicles was relatively smaller; one possibility is that McA-RH7777 cells produce relatively smaller or lipid-poor VLDL particles that can be accommodated in smaller vesicles. Despite the two cargoes being transported in different vesicles simultaneously, the same initiator of ER vesicle budding, Sar1, is utilized by both types of vesicles.52,53 It is possible that different proteins are involved with the selection of cargo for each of the two types of transport vesicles. Proteins mediating VLDL-selection into VTVs remain to be identified.
Figure 1.
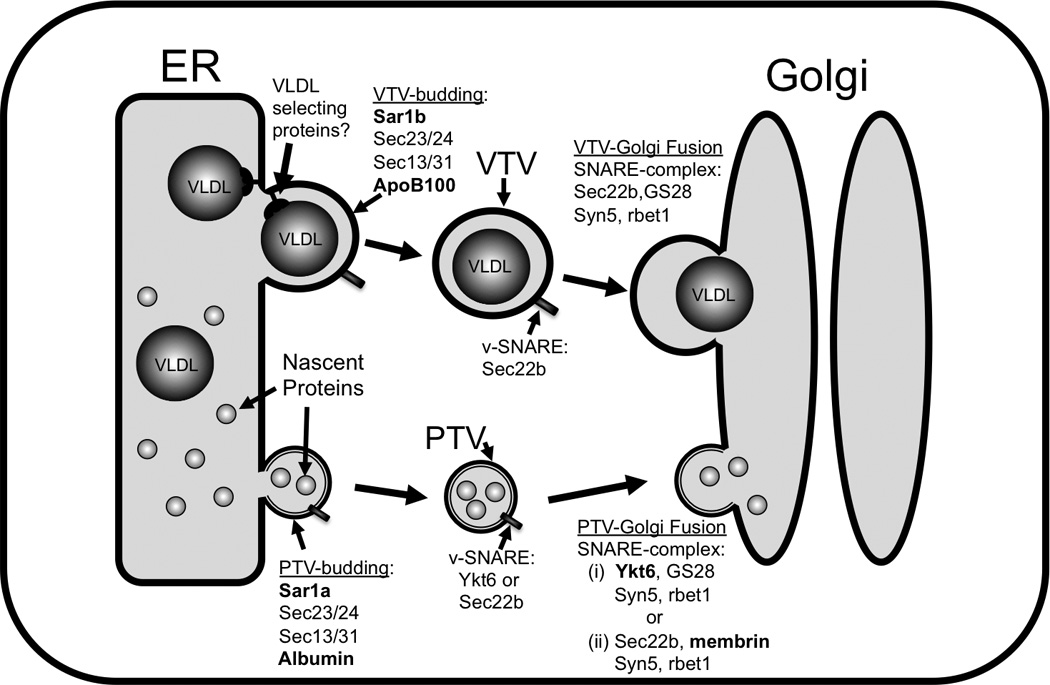
VLDL transport from the ER to the Golgi in hepatocytes. The assembly of VLDLs occurs in the lumen of the endoplasmic reticulum (ER). After their biogenesis in the ER lumen, VLDLs are packaged into specialized vesicles known as VLDL transport vesicles (VTVs). The average diameter of the VTVs is ~110 nm, which is sufficient to enclose VLDL-sized particles. VTVs bud off the ER membrane and move to and fuse with the cis-Golgi, delivering their VLDL cargo to the Golgi lumen. Proteins involved in VLDL-selection into VTV and VTV-Golgi docking are not known yet. Nascent proteins are transported from the ER to the Golgi protein transport vesicles (PTVs). Their size ranges between ~55 and 70 nm. Although biogenesis of both VTVs and PTVs from the ER membrane requires coat protein complex II (COPII) machinery, different homologs of Sar1 are required for their budding process (shown in bold). VTVs are, however, different from PTVs in their size, buoyant density, cargo, protein composition and require a unique set of SNARE proteins for fusion-complex formation.
Sar1, a COPII component that initiates the process of vesicle generation, is required for VTV formation because Sar1-depleted systems fail to generate VTVs.53 The clinical relevance of COPII components specifically Sar1, and its role in intracellular lipid trafficking has recently been demonstrated by several groups.55–57 Using proteomic approach, the Adeli’s group has found that the expression of Sar1 associated with PCTV is increased in insulin resistant mice, which is consistent with increased secretion of intestinal lipoproteins, chylomicrons.94,95 It is likely that Sar1 expression is up-regulated in liver under insulin resistant state because VLDL secretion is greatly enhanced under this pathophysiological condition, however, further studies are required to substantiate this thesis.
Several mutations have been reported in Sar1b in patients suffering from the rare condition, chylomicron retention disease (CMRD).55–57 Interestingly, the loss of Sar1b function by described mutations led to the enhanced expression of Sar1a in these patients.57 More recently overexpression of Sar1b has been demonstrated to up-regulate a number of genes, which are directly involved in lipid metabolism. Data emanating from the Levy’s group show that overexpression of Sar1b in Caco2 cells, a human colon cancer cell line, induces the expression of several proteins including apoB, apoAIV, SREBP-1c and MTP resulting in an increased production of triglycerides and chylomicrons.58
Little is known about the regulation of VTV biogenesis; however, the requirement of ATP in this process indicates a role for a kinase and/or phosphorylation event. Studies using H89, a non-selective inhibitor of protein kinase A (PKA) revealed that H89 significantly abrogated VTV budding.53 Hence, it is possible that PKA regulates VTV formation; more studies are required to unravel the factors regulating VTV biogenesis.
That the VTV is different from protein transport vesicle is revealed by detailed biochemical and morphological studies.53 As defined by their cargo, VTVs were expected to have a lighter buoyant density as compared to protein transport vesicle because of VLDL-triglycerides. Our data showing that VTV distributed in light density fractions whereas protein transport vesicle appeared in relatively denser fractions in the same continuous sucrose density gradient substantiated this speculation. Electron microscopic studies revealed that VTVs were larger in size (~100 –120 nm in diameter) than protein transport vesicles (~55 – 70 nm in diameter).53 Other biochemical data suggested differences in protein composition of both types of ER-derived vesicles. Figure 1 depicts the current model for the ER-to-Golgi transport of proteins and lipoproteins in primary hepatocytes.
Fusion of VTV with Golgi
The next task of newly formed VTVs present in the cytosol is to dock and fuse with their target membranes, the cis-Golgi, in order to deliver their special cargo, the VLDL. How are VTVs targeted to and fuse with their cognate cis-Golgi membranes? In their seminal work Rothman and associates proposed a general mechanism in which specific soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins facilitate the targeting, docking and fusion of transport vesicles with their specific target membranes.59–64 These SNAREs are type II integral membrane proteins, which are generally present on transport vesicles (v-SNAREs) and their target membranes (t-SNAREs).59 Structurally, each SNARE protein has a characteristic SNARE motif, which is composed of heptad repeats of approximately 60–70 amino acid stretch.65 Based on the crystal structure obtained for the SNARE-complex involved in the synaptic exocytosis, Fasshauer and coworkers suggested the presence of a “0” layer or iconic layer of amino acids present at the center of the fusion complex.66 The iconic or “0” layer contains one arginine (R) and three glutamine (Q) residues. Based on the presence of R or Q residues at the center of the fusion complex, SNAREs were re-classified as R-SNARE (localized to vesicles) and Q-SNAREs (localized to target membranes).66 The Q-SNAREs are further classified into Qa-, Qb- and Qc-SNARE proteins. Interaction of one R-SNARE and three Q-SNAREs (Qa, Qb and Qc) leads to the formation of a SNARE complex also termed as SNAREpin.67 A SNAREpin can bring the two membranes into close proximity and initiate the fusion process.67
In humans, there are 36 members in the SNARE super family, which are involved in a variety of intracellular transport events.63 Different combinations of SNARE proteins form a number of four-membered α-helix coiled-coil structures or SNAREpins that facilitate the docking and fusion of different types of vesicles with their cognate target membranes. The composition of a SNAREpin or SNARE complex varies in diverse intracellular transport events and thus provides specificity. In mammalian cells, there are three v-SNARE proteins (Sec22b, VAMP7 and Ykt6), which have been actively involved in ER-to-Golgi transport of lipoproteins and proteins.68–73 These three v-SNARE proteins form different SNAREpins (or SNARE complexes) with their cognate t-SNAREs localized to the cis-Golgi membranes. Since VTVs appeared to be different from other ER-derived vesicles in their morphology and biochemical composition, a distinct fusion module was expected to be involved in their targeting to and fusion with hepatic cis-Golgi. Studies to delineate the fusion event between VTV and cis-Golgi at the molecular level suggested that Sec22b serves as v-SNARE for the VTV.74 To observe the SNARE complex formation, an in vitro VTV-Golgi docking assay was developed that furnished favorable conditions for SNARE complex formation but prevented the fusion event.74 Using Sec22b as bait, a SNARE complex required for VTV-Golgi fusion was isolated and all components of the complex were identified. Sec22b on VTV forms a distinct SNARE complex with three t-SNAREs GOS28, Syntaxin5 and rBet1 present on the surface of hepatic cis-Golgi.74 This SNARE complex represents the fourth physiologically active complex so far identified for ER-to-Golgi transport of proteins and lipoproteins. Each component of this SNARE complex was demonstrated to be functionally active as the blocking of any component of the complex resulted in significant inhibition of VTV-Golgi fusion and thus VLDL delivery to the Golgi. Lipoproteins of small intestinal origin, chylomicrons, utilize VAMP7 as a v-SNARE,72,73 which forms a unique SNARE complex for their export from the ER to the Golgi supporting the notion that it is the composition of a SNARE complex or SNAREpin that provides specificity.
The clinical significance of SNARE proteins has recently been shown by the increased expression of VAMP7 in PCTV in insulin resistant state.94 However, nothing is known so far how insulin resistance affects the SNARE proteins required for the VTV-mediated delivery of nascent VLDLs to the Golgi.
Intra-Golgi processing of VLDL and post-Golgi trafficking
Once nascent VLDLs are delivered to the Golgi lumen by VTVs, several indispensible modifications occur to VLDL particles. Their structural protein apoB100 gets further glycosylated as demonstrated by the Yao group.75 ApoB100 immunoprecipitated from the Golgi has been shown to be endoglycosidase H (EndoH) resistant indicating the addition of complex sugar moieties.53,75 Additionally, phosphorylation of nascent VLDL-apoB100 occurs in the Golgi lumen.96 Interestingly, nascent VLDL particles exiting the ER in VTVs do not have apoAI,98 however, new unpublished data from our laboratory suggest that post-Golgi VLDL particles contain apoAI indicating a possibility of apoAI acquisition by VLDL in Golgi lumen. If this is the case, a question arises why apoAI is added to VLDL in the Golgi? Further investigation is required to address this issue. Current data, showing apoAI is not present in VTV, indicate the possibility of an alternative transport mechanism for apoAI, which is consistent with previous reports suggesting that apoB and apoAI are transported from the ER to the Golgi separately.82,97,98 Although the exact mechanism of how apoAI is being transported to the Golgi is not known, it is likely that newly synthesized apoAI is exported from the ER to the Golgi in PTV.
Another, perhaps the most important yet most debatable, intra-Golgi modification of VLDLs is their additional lipidation. Elegant studies by several independent groups support this transformation, however, equally excellent data from other groups challenge additional VLDL lipidation in Golgi lumen.9,76–84 Obviously this important process demands further investigation. Not much is known about the post-Golgi transport of VLDL. Our laboratory has just begun investigating this crucial step. It is believed that another specialized post-Golgi vesicle carries the VLDL to the plasma membrane. One possibility is that this secretory post-Golgi vesicle contains several VLDL particles since electron microscopic data suggest that in small intestine, the post-Golgi vesicles transport several chylomicrons to basolateral membrane.85
VTV vs PCTV
In the mammalian system, liver and small intestine are the principal organs that produce apoB-containing lipoproteins - VLDL and chylomicrons, respectively. These lipoproteins are synthesized in the ER and transported to the Golgi. This transport is the rate-determining step in their secretion. Both VLDL and chylomicrons possess substantial similarities but are exported to the Golgi in distinct vesicles (VTV and PCTV, respectively) and their ER-exit follows entirely different paradigms.53,54 The biogenesis of VTV and PCTV are fundamentally different in their requirement for GTP and COPII proteins. As discussed above, the formation of VTVs from the hepatic ER membranes is GTP and COPII-dependent, whereas PCTVs do not require either GTP or COPII proteins for their generation from intestinal ER membranes.54,86 Immuno-depletion of Sar1 from the hepatic cytosol does not support VTV formation.53 In contrast, Sar1-depleted intestinal cytosol enhances PCTV-budding several fold. Since PCTVs do not utilize COPII proteins for their generation, it was immediately proposed that they might use a novel budding-complex of cytosolic proteins. Using sequential column-chromatography and fast protein liquid chromatography (FPLC) techniques, our group isolated a multi-protein complex that has been demonstrated to be sufficient to form bona fide PCTV. This unique budding-complex contains nine proteins namely, vesicle-associated membrane protein 7 (VAMP7), apoB48, liver fatty acid-binding protein (L-FABP), and CD36 in addition to the five COPII proteins.87 L-FABP has been shown to be the minimal requirement to form the PCTV from the small intestinal ER; however, these PCTVs were unable to fuse with small intestinal Golgi.86,88
The differences between VTV and PCTV are not confined to their biogenesis but are extended to the next step of vesicle-Golgi fusion.73,74 It appears that both VTV and PCTV utilize different sets of SNARE proteins to form distinct SNARE-complexes for their fusion with the cis-Golgi.73,74 The PCTVs employ VAMP7, a protein heavily involved in post-Golgi protein transport event, as their v-SNARE. In contrast, VTVs do not contain VAMP7 but utilize Sec22b instead as their v-SNARE. A number of differences between VTV and PCTV are summarized in Table 1.
Table 1.
Differences between VTV and PCTV
| VLDL transport vesicle (VTV) |
Pre-chylomicron transport vesicle (PCTV) |
References |
|---|---|---|
| Transports VLDL from ER to Golgi in liver | Transports Chylomicrons from the ER to the Golgi in small intestine | 52–54 |
| VTV-budding requires Sar1 | PCTV-budding requires L-FABP | 52–54,86 |
| GTP is required for VTV-budding | GTP is not required for PCTV- budding | 52–54 |
| ~100 nm –120 nm in size | ~142 nm – 500 nm in size | 53,54 |
| VAMP7 is absent on VTV | VAMP7 is present on PCTV | 53,54 |
| Sec22b is present on VTV | Sec22b is absent on PCTV | 53,54 |
| VTVs are denser than PCTVs consistent with their proportionally smaller amount of TAG | PCTVs are of lighter density than VTVs consistent with their proportionally higher amount of TAG | 53,54 |
Concluding Remarks
The liver experiences intermittent fluxes of free fatty acids from a variety of sources and much of these are converted to triglycerides at the level of the ER. A major portion of triglycerides synthesized de novo is utilized for the formation of liver specific lipoproteins, the VLDL, in the lumen of the ER. The site of VLDL maturation, however, remains a subject of debate and necessitates further investigations. The movement of nascent VLDLs from the site of their genesis, the ER, to the Golgi is imperative for their eventual secretion and is facilitated by a specialized transport vesicle, the VTV. Several questions remain to be answered at each step of the intracellular transport of the VLDL. For example, how is VLDL sorted for packaging into VTV? Also, are there un-identified homologs of COPII proteins involved in VTV-formation that form a larger vesicle? It is likely that the liver utilizes vesicle-associated proteins in unique ways to bud VTV from the ER membrane and to select its specific cargo. It has been previously proposed that diverse signals or adapter proteins are required for inclusion of specific cargo into transport vesicles. Why should VTV vesicle generation require a unique system for its formation? One possibility is that VTV generation varies with the availability of triglycerides for transport in VLDL, whereas protein vesicle formation is more constant. In this event, by having a unique mechanism for VTV budding, there might be less competition for the proteins required for protein transport vesicle formation and thus their formation would remain uninterrupted.
Acknowledgements
This study was supported by NIH’s DK-81413 (SAS) from the National Institute of Diabetes And Digestive and Kidney Diseases The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes And Digestive and Kidney Diseases or the National Institutes of Health. The authors sincerely thank Dr. Charles Mansbach (UTHSC, Memphis) and Dr. Deborah Altomare (UCF, Orlando) for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None
References
- 1.Ginsberg HN. New perspectives on atherogenesis: role of abnormal triglyceride-rich lipoprotein metabolism. Circulation. 2002;106:2137–2142. doi: 10.1161/01.cir.0000035280.64322.31. [DOI] [PubMed] [Google Scholar]
- 2.Sehayek E, Eisenberg S. The role of native apolipoprotein B-containing lipoproteins in atherosclerosis: cellular mechanisms. Curr Opin Lipidol. 1994;5:350–353. doi: 10.1097/00041433-199410000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Minehira K, Young SG, Villanueva CJ, Yetukuri L, Oresic M, Hellerstein MK, Farese RV, Jr, Horton JD, Preitner F, Thorens B, Tappy L. Blocking VLDL secretion causes hepatic steatosis but does not affect peripheral lipid stores or insulin sensitivity in mice. J Lipid Res. 2008;49:2038–2044. doi: 10.1194/jlr.M800248-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher EA, Ginsberg HN. Complexity in the secretory pathway: the assembly and secretion of apolipoprotein B-containing lipoproteins. J Biol Chem. 2002;277:17377–17380. doi: 10.1074/jbc.R100068200. [DOI] [PubMed] [Google Scholar]
- 5.Yao Z, McLeod RS. Synthesis and secretion of hepatic apolipoprotein B-containing lipoproteins. Biochim Biophys Acta. 1994;1212:152–166. doi: 10.1016/0005-2760(94)90249-6. [DOI] [PubMed] [Google Scholar]
- 6.Olofsson SO, Stillemark-Billton P, Asp L. Intracellular assembly of VLDL: two major steps in separate cell compartments. Trends Cardiovasc Med. 2000;10:338–345. doi: 10.1016/s1050-1738(01)00071-8. [DOI] [PubMed] [Google Scholar]
- 7.Fisher EA, Pan M, Chen X, Wu X, Wang H, Jamil H, Sparks JD, Williams KJ. The triple threat to nascent apolipoprotein B. Evidence for multiple, distinct degradative pathways. J Biol Chem. 2001;276:27855–27863. doi: 10.1074/jbc.M008885200. [DOI] [PubMed] [Google Scholar]
- 8.Pan M, Liang Js JS, Fisher EA, Ginsberg HN. The late addition of core lipids to nascent apolipoprotein B100, resulting in the assembly and secretion of triglyceride-rich lipoproteins, is independent of both microsomal triglyceride transfer protein activity and new triglyceride synthesis. J Biol Chem. 2002;277:4413–4421. doi: 10.1074/jbc.M107460200. [DOI] [PubMed] [Google Scholar]
- 9.Alexander CA, Hamilton RL, Havel RJ. Subcellular localization of B apoprotein of plasma lipoproteins in rat liver. J Cell Biol. 1976;69:241–263. doi: 10.1083/jcb.69.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olofsson SO, Boren J. Apolipoprotein B: a clinically important apolipoprotein which assembles atherogenic lipoproteins and promotes the development of atherosclerosis. J Intern Med. 2005;258:395–410. doi: 10.1111/j.1365-2796.2005.01556.x. [DOI] [PubMed] [Google Scholar]
- 11.Ginsberg HN, Fisher EA. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J Lipid Res. 2009;50(Suppl):S162–S166. doi: 10.1194/jlr.R800090-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundaram M, Yao Z. Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr Metab (Lond) 2010;7:35. doi: 10.1186/1743-7075-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lusis AJ, West R, Mehrabian M, Reuben MA, LeBoeuf RC, Kaptein JS, Johnson DF, Schumaker VN, Yuhasz MP, Schotz MC, Elovson J. Cloning and expression of apolipoprotein B, the major protein of low and very low density lipoproteins. Proc Natl Acad Sci U S A. 1985;82:4597–4601. doi: 10.1073/pnas.82.14.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen SH, Li XX, Liao WS, Wu JH, Chan L. RNA editing of apolipoprotein B mRNA. Sequence specificity determined by in vitro coupled transcription editing. J Biol Chem. 1990;265:6811–6816. [PubMed] [Google Scholar]
- 15.Chen SH, Habib G, Yang CY, Gu ZW, Lee BR, Weng SA, Silberman SR, Cai SJ, Deslypere JP, Rosseneu M, Gotto AM, Li WH, Chan L. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science. 1987;238:363–366. doi: 10.1126/science.3659919. [DOI] [PubMed] [Google Scholar]
- 16.Kane JP, Hardman DA, Paulus HE. Heterogeneity of apolipoprotein B: isolation of a new species from human chylomicrons. Proc Natl Acad Sci U S A. 1980;77:2465–2469. doi: 10.1073/pnas.77.5.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lo CM, Nordskog BK, Nauli AM, Zheng S, Vonlehmden SB, Yang Q, Lee D, Swift LL, Davidson NO, Tso P. Why does the gut choose apolipoprotein B48 but not B100 for chylomicron formation? Am J Physiology Gastrointest Liver Physiol. 2008;294:G344–G352. doi: 10.1152/ajpgi.00123.2007. [DOI] [PubMed] [Google Scholar]
- 18.Mansbach CM, Siddiqi SA. The biogenesis of chylomicrons. Annu Rev Physiol. 2010;72:315–333. doi: 10.1146/annurev-physiol-021909-135801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Segrest JP, Jones MK, De Loof H, Dashti N. Structure of apolipoprotein B-100 in low density lipoproteins. J Lipid Res. 2001;42:1346–1367. [PubMed] [Google Scholar]
- 20.Dashti N, Gandhi M, Liu X, Lin X, Segrest JP. The N-terminal 1000 residues of apolipoprotein B associate with microsomal triglyceride transfer protein to create a lipid transfer pocket required for lipoprotein assembly. Biochemistry. 2002;41:6978–6987. doi: 10.1021/bi011757l. [DOI] [PubMed] [Google Scholar]
- 21.Swift LL. Assembly of very low density lipoproteins in rat liver: a study of nascent particles recovered from the rough endoplasmic reticulum. J Lipid Res. 1995;36:395–406. [PubMed] [Google Scholar]
- 22.Rustaeus S, Lindberg K, Stillemark P, Claesson C, Asp L, Larsson T, Boren J, Olofsson SO. Assembly of very low density lipoprotein: a two-step process of apolipoprotein B core lipidation. J Nutr. 1999;129:463S–466S. doi: 10.1093/jn/129.2.463S. [DOI] [PubMed] [Google Scholar]
- 23.Shelness GS, Ingram MF, Huang XF, DeLozier JA. Apolipoprotein B in the rough endoplasmic reticulum: translation, translocation and the initiation of lipoprotein assembly. J Nutr. 1999;129:456S–462S. doi: 10.1093/jn/129.2.456S. [DOI] [PubMed] [Google Scholar]
- 24.Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003;44:22–32. doi: 10.1194/jlr.r200014-jlr200. [DOI] [PubMed] [Google Scholar]
- 25.Hussain MM, Bakillah A, Nayak N, Shelness GS. Amino acids 430–570 in apolipoprotein B are critical for its binding to microsomal triglyceride transfer protein. J Biol Chem. 1998;273:25612–25615. doi: 10.1074/jbc.273.40.25612. [DOI] [PubMed] [Google Scholar]
- 26.Liang J, Ginsberg HN. Microsomal triglyceride transfer protein binding and lipid transfer activities are independent of each other, but both are required for secretion of apolipoprotein B lipoproteins from liver cells. J Biol Chem. 2001;276:28606–28612. doi: 10.1074/jbc.M100294200. [DOI] [PubMed] [Google Scholar]
- 27.Khatun I, Zeissig S, Iqbal J, Wang M, Curiel D, Shelness GS, Blumberg RS, Hussain MM. The phospholipid transfer activity of MTP produces apoB-lipoproteins and reduces hepatosteatosis while maintaining low plasma lipids. Hepatology. 2011 doi: 10.1002/hep.25504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakata N, Wu X, Dixon JL, Ginsberg HN. Proteolysis and lipid-facilitated translocation are distinct but competitive processes that regulate secretion of apolipoprotein B in Hep G2 cells. J Biol Chem. 1993;268:22967–22970. [PubMed] [Google Scholar]
- 29.Yao Z, Tran K, McLeod RS. Intracellular degradation of newly synthesized apolipoprotein B. J Lipid Res. 1997;38:1937–1953. [PubMed] [Google Scholar]
- 30.Liao W, Yeung SC, Chan L. Proteasome-mediated degradation of apolipoprotein B targets both nascent peptides cotranslationally before translocation and full-length apolipoprotein B after translocation into the endoplasmic reticulum. J Biol Chem. 1998;273:27225–27230. doi: 10.1074/jbc.273.42.27225. [DOI] [PubMed] [Google Scholar]
- 31.Gusarova V, Caplan AJ, Brodsky JL, Fisher EA. Apoprotein B degradation is promoted by the molecular chaperones hsp90 and hsp70. J Biol Chem. 2001;276:24891–24900. doi: 10.1074/jbc.M100633200. [DOI] [PubMed] [Google Scholar]
- 32.Zhou M, Fisher EA, Ginsberg HN. Regulated Co-translational ubiquitination of apolipoprotein B100. A new paradigm for proteasomal degradation of a secretory protein. J Biol Chem. 1998;273:24649–24653. doi: 10.1074/jbc.273.38.24649. [DOI] [PubMed] [Google Scholar]
- 33.Hrizo SL, Gusarova V, Habiel DM, Goeckeler JL, Fisher EA, Brodsky JL. The Hsp110 molecular chaperone stabilizes apolipoprotein B from endoplasmic reticulum-associated degradation (ERAD) J Biol Chem. 2007;282:32665–32675. doi: 10.1074/jbc.M705216200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raabe M, Veniant MM, Sullivan MA, Zlot CH, Bjorkegren J, Nielsen LB, Wong JS, Hamilton RL, Young SG. Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J Clin Invest. 1999;103:1287–1298. doi: 10.1172/JCI6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye J, Li JZ, Liu Y, Li X, Yang T, Ma X, Li Q, Yao Z, Li P. Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab. 2009;9:177–190. doi: 10.1016/j.cmet.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 36.Barlowe C. COPII and selective export from the endoplasmic reticulum. Biochim Biophys Acta. 1998;1404:67–76. doi: 10.1016/s0167-4889(98)00047-0. [DOI] [PubMed] [Google Scholar]
- 37.Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93:263–275. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- 38.Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 39.Budnik A, Stephens DJ. ER exit sites--localization and control of COPII vesicle formation. FEBS Lett. 2009;583:3796–3803. doi: 10.1016/j.febslet.2009.10.038. [DOI] [PubMed] [Google Scholar]
- 40.Forster R, Weiss M, Zimmermann T, Reynaud EG, Verissimo F, Stephens DJ, Pepperkok R. Secretory cargo regulates the turnover of COPII subunits at single ER exit sites. Curr Biol. 2006;16:173–179. doi: 10.1016/j.cub.2005.11.076. [DOI] [PubMed] [Google Scholar]
- 41.Kuge O, Dascher C, Orci L, Rowe T, Amherdt M, Plutner H, Ravazzola M, Tanigawa G, Rothman JE, Balch WE. Sar1 promotes vesicle budding from the endoplasmic reticulum but not Golgi compartments. J Cell Biol. 1994;125:51–65. doi: 10.1083/jcb.125.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barlowe C, Schekman R. SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature. 1993;365:347–349. doi: 10.1038/365347a0. [DOI] [PubMed] [Google Scholar]
- 43.Barlowe C, d'Enfert C, Schekman R. Purification and characterization of SAR1p, a small GTP-binding protein required for transport vesicle formation from the endoplasmic reticulum. J Biol Chem. 1993;268:873–879. [PubMed] [Google Scholar]
- 44.Aridor M, Balch WE. Kinase signaling initiates coat complex II (COPII) recruitment and export from the mammalian endoplasmic reticulum. J Biol Chem. 2000;275:35673–35676. doi: 10.1074/jbc.C000449200. [DOI] [PubMed] [Google Scholar]
- 45.Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- 46.Huang M, Weissman JT, Beraud-Dufour S, Luan P, Wang C, Chen W, Aridor M, Wilson IA, Balch WE. Crystal structure of Sar1-GDP at 1.7 A resolution and the role of the NH2 terminus in ER export. J Cell Biol. 2001;155:937–948. doi: 10.1083/jcb.200106039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuehn MJ, Herrmann JM, Schekman R. COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature. 1998;391:187–190. doi: 10.1038/34438. [DOI] [PubMed] [Google Scholar]
- 48.Aridor M, Fish KN, Bannykh S, Weissman J, Roberts TH, Lippincott-Schwartz J, Balch WE. The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J Cell Biol. 2001;152:213–229. doi: 10.1083/jcb.152.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allan BB, Weissman J, Aridor M, Moyer B, Chen CD, Yoo JS, Balch WE. Stage-specific assays to study biosynthetic cargo selection and role of SNAREs in export from the endoplasmic reticulum and delivery to the Golgi. Methods. 2000;20:411–416. doi: 10.1006/meth.2000.0954. [DOI] [PubMed] [Google Scholar]
- 50.Stagg SM, Gurkan C, Fowler DM, LaPointe P, Foss TR, Potter CS, Carragher B, Balch WE. Structure of the Sec13/31 COPII coat cage. Nature. 2006;439:234–238. doi: 10.1038/nature04339. [DOI] [PubMed] [Google Scholar]
- 51.Lederkremer GZ, Cheng Y, Petre BM, Vogan E, Springer S, Schekman R, Walz T, Kirchhausen T. Structure of the Sec23p/24p and Sec13p/31p complexes of COPII. Proc Natl Acad Sci U S A. 2001;98:10704–10709. doi: 10.1073/pnas.191359398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gusarova V, Brodsky JL, Fisher EA. Apolipoprotein B100 exit from the endoplasmic reticulum (ER) is COPII-dependent, and its lipidation to very low density lipoprotein occurs post-ER. J Biol Chem. 2003;278:48051–48058. doi: 10.1074/jbc.M306898200. [DOI] [PubMed] [Google Scholar]
- 53.Siddiqi SA. VLDL exits from the endoplasmic reticulum in a specialized vesicle, the VLDL transport vesicle, in rat primary hepatocytes. Biochem J. 2008;413:333–342. doi: 10.1042/BJ20071469. [DOI] [PubMed] [Google Scholar]
- 54.Siddiqi SA, Gorelick FS, Mahan JT, Mansbach CM., 2nd COPII proteins are required for Golgi fusion but not for endoplasmic reticulum budding of the pre-chylomicron transport vesicle. J Cell Sci. 2003;116:415–427. doi: 10.1242/jcs.00215. [DOI] [PubMed] [Google Scholar]
- 55.Shoulders CC, Stephens DJ, Jones B. The intracellular transport of chylomicrons requires the small GTPase, Sar1b. Curr Opin Lipidol. 2004;15:191–197. doi: 10.1097/00041433-200404000-00012. [DOI] [PubMed] [Google Scholar]
- 56.Charcosset M, Sassolas A, Peretti N, Roy CC, Deslandres C, Sinnett D, Levy E, Lachaux A. Anderson or chylomicron retention disease: molecular impact of five mutations in the SAR1B gene on the structure and the functionality of Sar1b protein. Mol Genet Metab. 2008;93:74–84. doi: 10.1016/j.ymgme.2007.08.120. [DOI] [PubMed] [Google Scholar]
- 57.Georges A, Bonneau J, Bonnefont-Rousselot D, Champigneulle J, Rabes JP, Abifadel M, Aparicio T, Guenedet JC, Bruckert E, Boileau C, Morali A, Varret M, Aggerbeck LP, Samson-Bouma ME. Molecular analysis and intestinal expression of SAR1 genes and proteins in Anderson's disease (Chylomicron retention disease) Orphanet J Rare Dis. 2011;6:1. doi: 10.1186/1750-1172-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levy E, Harmel E, Laville M, Sanchez R, Emonnot L, Sinnett D, Ziv E, Delvin E, Couture P, Marcil V, Sane AT. Expression of Sar1b enhances chylomicron assembly and key components of the coat protein complex II system driving vesicle budding. Arterioscler Thromb Vasc Biol. 2011;31:2692–2699. doi: 10.1161/ATVBAHA.111.233908. [DOI] [PubMed] [Google Scholar]
- 59.Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 60.Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sollner TH, Rothman JE. Molecular machinery mediating vesicle budding, docking and fusion. Cell Struct Funct. 1996;21:407–412. doi: 10.1247/csf.21.407. [DOI] [PubMed] [Google Scholar]
- 62.Hay JC, Scheller RH. SNAREs and NSF in targeted membrane fusion. Curr Opin Cell Biol. 1997;9:505–512. doi: 10.1016/s0955-0674(97)80026-9. [DOI] [PubMed] [Google Scholar]
- 63.Jahn R, Scheller RH. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 64.Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 65.Kloepper TH, Kienle CN, Fasshauer D. An elaborate classification of SNARE proteins sheds light on the conservation of the eukaryotic endomembrane system. Mol Biol Cell. 2007;18:3463–3471. doi: 10.1091/mbc.E07-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fasshauer D, Sutton RB, Brunger AT, Jahn R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci U S A. 1998;95:15781–15786. doi: 10.1073/pnas.95.26.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 68.Hay JC, Chao DS, Kuo CS, Scheller RH. Protein interactions regulating vesicle transport between the endoplasmic reticulum and Golgi apparatus in mammalian cells. Cell. 1997;89:149–158. doi: 10.1016/s0092-8674(00)80191-9. [DOI] [PubMed] [Google Scholar]
- 69.Zhang T, Wong SH, Tang BL, Xu Y, Hong W. Morphological and functional association of Sec22b/ERS-24 with the pre-Golgi intermediate compartment. Mol Biol Cell. 1999;10:435–453. doi: 10.1091/mbc.10.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang T, Hong W. Ykt6 forms a SNARE complex with syntaxin 5, GS28, and Bet1 and participates in a late stage in endoplasmic reticulum-Golgi transport. J Biol Chem. 2001;276:27480–27487. doi: 10.1074/jbc.M102786200. [DOI] [PubMed] [Google Scholar]
- 71.Hasegawa H, Zinsser S, Rhee Y, Vik-Mo EO, Davanger S, Hay JC. Mammalian ykt6 is a neuronal SNARE targeted to a specialized compartment by its profilin-like amino terminal domain. Mol Biol Cell. 2003;14:698–720. doi: 10.1091/mbc.E02-09-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Siddiqi SA, Mahan J, Siddiqi S, Gorelick FS, Mansbach CM., 2nd Vesicle-associated membrane protein 7 is expressed in intestinal ER. J Cell Sci. 2006;119:943–950. doi: 10.1242/jcs.02803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Siddiqi SA, Siddiqi S, Mahan J, Peggs K, Gorelick FS, Mansbach CM., 2nd The identification of a novel endoplasmic reticulum to Golgi SNARE complex used by the prechylomicron transport vesicle. J Biol Chem. 2006;281:20974–20982. doi: 10.1074/jbc.M601401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siddiqi S, Mani AM, Siddiqi SA. The identification of the SNARE complex required for the fusion of VLDL-transport vesicle with hepatic cis-Golgi. Biochem J. 2010;429:391–401. doi: 10.1042/BJ20100336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tran K, Thorne-Tjomsland G, DeLong CJ, Cui Z, Shan J, Burton L, Jamieson JC, Yao Z. Intracellular assembly of very low density lipoproteins containing apolipoprotein B100 in rat hepatoma McA-RH7777 cells. J Biol Chem. 2002;277:31187–31200. doi: 10.1074/jbc.M200249200. [DOI] [PubMed] [Google Scholar]
- 76.Claude A. Growth and differentiation of cytoplasmic membranes in the course of lipoprotein granule synthesis in the hepatic cell. I. Elaboration of elements of the Golgi complex. J Cell Biol. 1970;47:745–766. doi: 10.1083/jcb.47.3.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morre DJ, Ovtracht L. Structure of rat liver Golgi apparatus: relationship to lipoprotein secretion. J Ultrastruct Res. 1981;74:284–295. doi: 10.1016/s0022-5320(81)80119-0. [DOI] [PubMed] [Google Scholar]
- 78.Rusinol A, Verkade H, Vance JE. Assembly of rat hepatic very low density lipoproteins in the endoplasmic reticulum. J Biol Chem. 1993;268:3555–3562. [PubMed] [Google Scholar]
- 79.Yamaguchi J, Gamble MV, Conlon D, Liang JS, Ginsberg HN. The conversion of apoB100 low density lipoprotein/high density lipoprotein particles to apoB100 very low density lipoproteins in response to oleic acid occurs in the endoplasmic reticulum and not in the Golgi in McA RH7777 cells. J Biol Chem. 2003;278:42643–42651. doi: 10.1074/jbc.M306920200. [DOI] [PubMed] [Google Scholar]
- 80.Borchardt RA, Davis RA. Intrahepatic assembly of very low density lipoproteins. Rate of transport out of the endoplasmic reticulum determines rate of secretion. J Biol Chem. 1987;262:16394–16402. [PubMed] [Google Scholar]
- 81.Kulinski A, Rustaeus S, Vance JE. Microsomal triacylglycerol transfer protein is required for lumenal accretion of triacylglycerol not associated with ApoB, as well as for ApoB lipidation. J Biol Chem. 2002;277:31516–31525. doi: 10.1074/jbc.M202015200. [DOI] [PubMed] [Google Scholar]
- 82.Bamberger MJ, Lane MD. Possible role of the Golgi apparatus in the assembly of very low density lipoprotein. Proc Natl Acad Sci U S A. 1990;87:2390–2394. doi: 10.1073/pnas.87.7.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Higgins JA. Evidence that during very low density lipoprotein assembly in rat hepatocytes most of the triacylglycerol and phospholipid are packaged with apolipoprotein B in the Golgi complex. FEBS Lett. 1988;232:405–408. doi: 10.1016/0014-5793(88)80780-4. [DOI] [PubMed] [Google Scholar]
- 84.Gusarova V, Seo J, Sullivan ML, Watkins SC, Brodsky JL, Fisher EA. Golgi-associated maturation of very low density lipoproteins involves conformational changes in apolipoprotein B, but is not dependent on apolipoprotein E. J Biol Chem. 2007;282:19453–19462. doi: 10.1074/jbc.M700475200. [DOI] [PubMed] [Google Scholar]
- 85.Sabesin SM, Clark SB, Holt PR. Ultrastructural features of regional differences in chylomicron secretion by rat intestine. Exp Mol Pathol. 1977;26:277–289. doi: 10.1016/0014-4800(77)90055-7. [DOI] [PubMed] [Google Scholar]
- 86.Neeli I, Siddiqi SA, Siddiqi S, Mahan J, Lagakos WS, Binas B, Gheyi T, Storch J, Mansbach CM., 2nd Liver fatty acid-binding protein initiates budding of pre-chylomicron transport vesicles from intestinal endoplasmic reticulum. J Biol Chem. 2007;282:17974–17984. doi: 10.1074/jbc.M610765200. [DOI] [PubMed] [Google Scholar]
- 87.Siddiqi S, Saleem U, Abumrad NA, Davidson NO, Storch J, Siddiqi SA, Mansbach CM., 2nd A novel multiprotein complex is required to generate the prechylomicron transport vesicle from intestinal ER. J Lipid Res. 2010;51:1918–1928. doi: 10.1194/jlr.M005611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Siddiqi S, Siddiqi SA, Mansbach CM., 2nd Sec24C is required for docking the prechylomicron transport vesicle with the Golgi. J Lipid Res. 2010;51:1093–1100. doi: 10.1194/jlr.M002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab. 2011;22:353–363. doi: 10.1016/j.tem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Petersen KF, Shulman GI. Etiology of insulin resistance. Am J of Med. 2006;119:S10–S16. doi: 10.1016/j.amjmed.2006.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chuck SL, Yao Z, Blackhart BD, McCarthy BJ, Lingappa VR. New variation on the translocation of proteins during early biogenesis of apolipoprotein B. Nature. 1990;346:382–385. doi: 10.1038/346382a0. [DOI] [PubMed] [Google Scholar]
- 92.Chuck SL, Lingappa VR. Pause transfer: a topogenic sequence in apolipoprotein B mediates stopping and restarting of translocation. Cell. 1992;68:9–21. doi: 10.1016/0092-8674(92)90202-n. [DOI] [PubMed] [Google Scholar]
- 93.Tennyson GE, Sabatos CA, Higuchi K, Meglin N, Brewer HB., Jr Expression of apolipoprotein B mRNAs encoding higher- and lower-molecular weight isoproteins in rat liver and intestine. Proc Natl Acad Sci U S A. 1989;86:500–504. doi: 10.1073/pnas.86.2.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wong DM, Webb JP, Malinowski PM, Xu E, Macri J, Adeli K. Proteomic profiling of intestinal prechylomicron transport vesicle (PCTV)-associated proteins in an animal model of insulin resistance (94 char) J Proteomics. 2010;73:1291–1305. doi: 10.1016/j.jprot.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 95.Taskinen MR. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia. 2003;46:733–749. doi: 10.1007/s00125-003-1111-y. [DOI] [PubMed] [Google Scholar]
- 96.Swift LL. Role of the Golgi apparatus in the phosphorylation of apolipoprotein B. J Biol. Chem. 1996;271:31491–31495. [PubMed] [Google Scholar]
- 97.Bamberger MJ, Lane MD. Assembly of very low density lipoprotein in the hepatocyte. Differential transport of apoproteins through the secretory pathway. J Biol. Chem. 1988;263:11868–11878. [PubMed] [Google Scholar]
- 98.Rahim A, Nafi-valencia E, Siddiqi S, Basha R, Runyon CC, Siddiqi SA. Proteomic. Analysis of the Very Low Density Lipoprotein (VLDL) Transport Vesicles. J Proteomics. 2012 doi: 10.1016/j.jprot.2012.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]